Received: November 24, 2013
Accepted: January 17, 2014
Meditter J Hematol Infect Dis 2014, 6(1): e2014012, DOI 10.4084/MJHID.2014.012
This article is available on PDF format at:

Ariel Koren12, Lora Profeta3, Luci Zalman4, Haya Palmor4, Carina Levin1,2, Ronit Bril Zamir5, Stavit Shalev2,5 and Orna Blondheim2,3
1
Pediatric Dept B and Pediatric Hematology Unit, Emek Medical Center,
Afula.
2 The Ruth and Baruch Rappaport School of
Medicine, Technion, Israel Institute of Technology, Haifa.
3 Emek Medical Centre, Afula, Israel, Affiliated
with the Technion School of Medicine, Haifa, Israel.
4 Hematology Laboratory, Emek Medical Centre,
Afula, Israel.
5 Unit of Genetics, Emek Medical Centre, Afula,
Israel.
|
This
is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract Background: β Thalassemia major is characterized by hemolytic anemia, ineffective erythropoiesis and hemosiderosis. About 4% of the world population carries a Thalassemia gene. Management includes blood transfusions and iron chelation. However, this treatment is costly, and population screening may be significantly more cost beneficial.Purpose: The purpose of the current study is to analyze the cost of running a prevention program for β Thalassemia in Israel and to compare it to the actual expenses incurred by treating Thalassemia patients. Methods: Three cost parameters were analyzed and compared: the prevention program, routine treatment of patients and treatment of complications. An estimation of the expenses needed to treat patients who present with complications was calculated based on our ongoing experience in treating deteriorating patients. Results and Conclusions: The cost of preventing one affected newborn was $63,660 compared to $1,971,380 for treatment of a patient during 50 years (mean annual cost: $39,427). Thus, the prevention of 45 affected newborns over a ten year period represents a net saving of $88.5 million to the health budget. Even after deducting the cost of the prevention program ($413.795/year), the program still represents a benefit of $76 million over ten years. Each prevented case could pay the screening and prevention program for 4.6 years. |
Introduction
β
Thalassemia major is a hemoglobinopathy characterized by chronic
hemolytic anemia, ineffective erythropoiesis and progressive
hemosiderosis. Thalassemia is considered the most frequent genetic
disease in the world, about 4% of the world population carries a
Thalassemia gene.[1] In northern
Israel the mean carrier frequency for
β Thalassemia is about 2.4%; however, in villages with a primarily Arab
population the frequency rises to 9%.[2]
Treatment of β Thalassemia
major includes regular blood transfusions and iron chelation, to
preclude early death.[3,4] Disease
management, which includes regular
follow-up visits, is costly and carries a significant financial burden
to health services.[5]
Approaches to Thalassemia prevention include general population
screening at school age, as reported in Thailand;[6]
premarital
screening, as described in Middle East countries such as North Cyprus,
where abortion is not generally accepted;[7,8]
and the screening of
couples at marriage or in early pregnancy, as performed in Sardinia.[9]
In Israel β Thalassemia prevention is based on the screening of young
couples at early pregnancy for carrier detection and subsequent
prenatal diagnosis. This approach may be significantly less expensive
and more cost effective than others.[10]
In recent years more couples
in Israel attend community health clinics in request of premarital
genetic counseling.
Fifteen years ago the cost-benefit of a national Thalassemia prevention
program in Israel was calculated.[11]
The cost of caring for
Thalassemia patients with a life expectancy of 25-35 years was
assessed, and the cost of a prevention program estimated; the real cost
of the latter was not known then, as such a program had not yet been
implemented. Changes in the approach to treatment have since
significantly increased the life expectancy of individuals with
Thalassemia, by 50 years and even more.
The purpose of the current study is to analyze the cost of prevention
of β Thalassemia based on data from a prevention program that has been
carried out in northern Israel since 1987,[2]
and to compare this cost
to the actual expenses incurred by treating patients in the Pediatric
Hematology Unit at the Emek Medical Centre, Israel.
Materials and Methods
Prevention Program. Since 1987, a screening program for the prevention of β Thalassemia and other hemoglobinopathies has been carried out in northern Israel. Pregnant women are screened at their first visit to community Mother and Child Health Clinics. Subsequently, husbands of affected women are screened, and couples identified as being at risk of having an affected offspring are referred for genetic counseling and prenatal testing (Figure 1).[2] In the screening program all blood samples are analyzed by HPLC (High Performance Liquid Chromatography, cation exchange, Variant Hgb Testing, Biorad Co, USA), irrespective of the red blood cell indexes, in order to detect carriers with a MCV >78μ3 and carriers of Hgb S.[11]
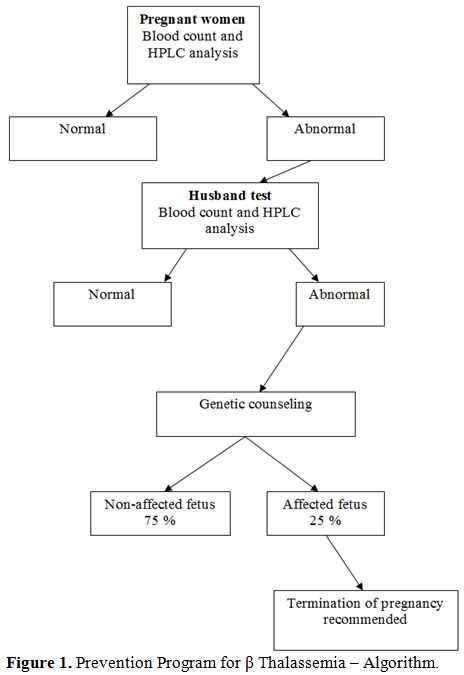 |
Figure 1. Prevention Program for β Thalassemia – Algorithm. |
Since the inception
of the screening program, 71 β Thalassemia major
and 25 β Thalassemia intermedia patients were diagnosed and treated at
the Pediatric Hematology Unit in the Emek Medical Centre. Twenty-six β
Thalassemia major patients succumbed to disease complications, most
frequently iron overload, and died in the second or third decade of
life.
Cost
Estimations: Three cost parameters were analyzed and
compared:
A. The prevention program.
B. Routine treatment of patients with β Thalassemia major.
C. Treatment of complications of β Thalassemia major.
A. The annual cost of the prevention program was measured and calculated during the year 2011, including laboratory expenses with the HPLC method, expenses for laboratory personnel and expenses for a project manager (Table 1). We included the cost of the initial blood count, even though it is part of the routine tests of the first visit of pregnant women to Mother and Child Health Clinics.
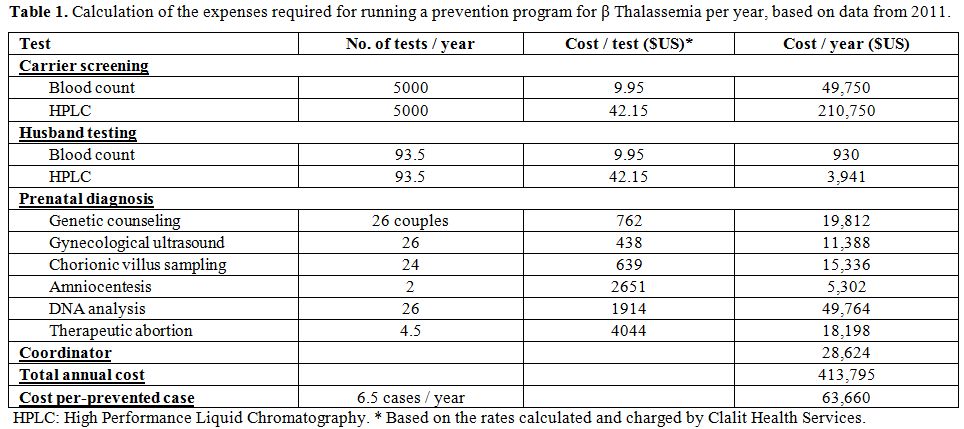 |
Table 1. Calculation of the expenses required for running a prevention program for β Thalassemia per year, based on data from 2011. |
B. The cost for routine treatment of patients with β Thalassemia major was computed according to the actual management applied in the Pediatric Hematology Unit, based on the rates calculated and charged by Clalit Health Services, including the price of iron chelator Desferrioxamine (Desferal®, Novartis, provided by Teva, Home treatment Pharmaceuticals, Israel) or Deferasirox (Exjade®, Novartis, Israel) or Deferiprone (L1® - Ferrirprox® - ApoPharma – Canada provided by Luxemburg, Israel). In addition to the cost of treating β Thalassemia major patients, we included the cost of diagnosing new patients, including blood analysis of their parents (Blood count and HPLC analysis), blood transfusions [usually every three weeks], iron chelation (Desferal® or Exjade® or Deferiprone®) from age 2 years and the cost of annual follow-ups (Tables 2-4). The proportion of patients treated by Desferrioxamine, Deferiprone, combination therapy or Deferasirox was calculated according to the proportion of patients treated by each chelator during the year 2011.
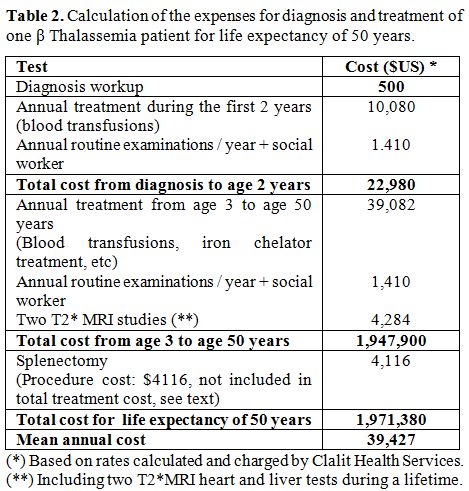 |
Table 2. Calculation of the expenses for diagnosis and treatment of one β Thalassemia patient for life expectancy of 50 years. |
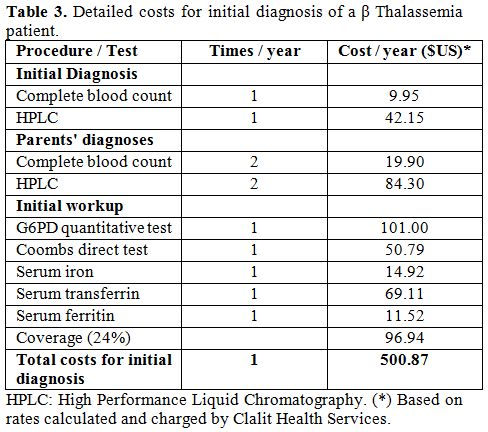 |
Table 3. Detailed costs for initial diagnosis of a β Thalassemia patient. |
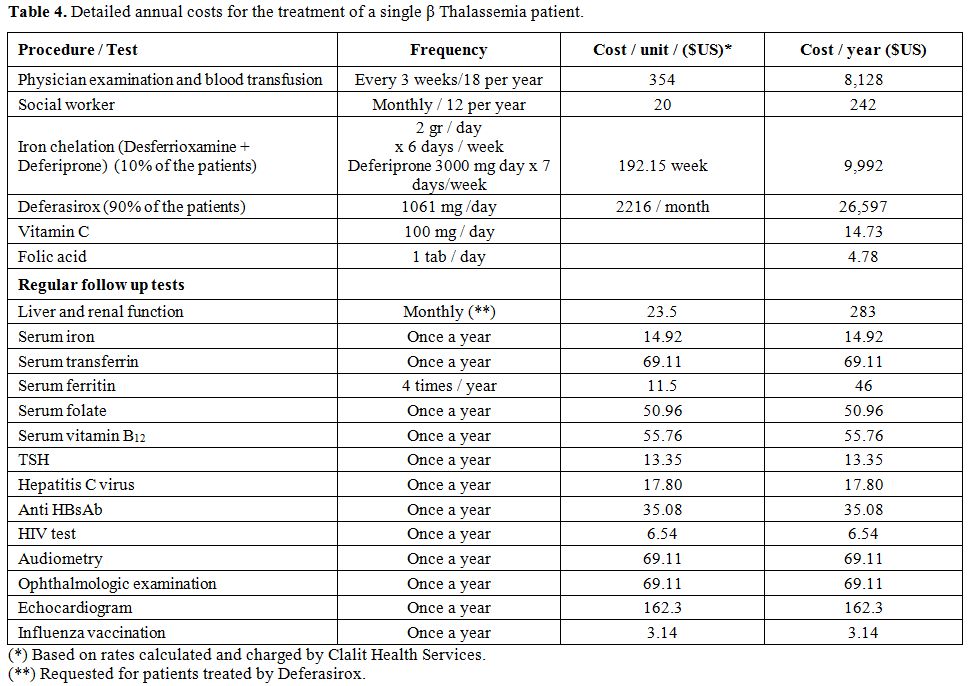 |
Table 4.Detailed annual costs for the treatment of a single β Thalassemia patient. |
C. An estimation of
the expenses of treating the complications of β
Thalassemia was calculated based on our ongoing experience in treating
deteriorating β Thalassemia patients in the Pediatric Hematology Unit.
It is difficult to estimate the proportion of deteriorating patients
since compliance with chelation treatment correlates inversely with the
incidence of complications.
In our analysis, we did not include expenses for treatment of
deteriorating patients due to poor compliance with chelation therapy,
heart failure, endocrine workup and replacement therapy, diagnosis and
treatment of osteoporosis and treatment of blood-related acquired
infections. These expenses are presented in Table 5,
with the proportion of patients presenting with each complication,
based on our experience and compared with the rates reported in the
literature.[12] Also, expenses
incurred directly by the families and
direct compensation payments to the patients from the National
Insurance Fund were not included in our analysis. Indirect costs
related to poor quality of life and lost productivity are also not
included in this analysis because of the wide variations between
patients in these parameters.
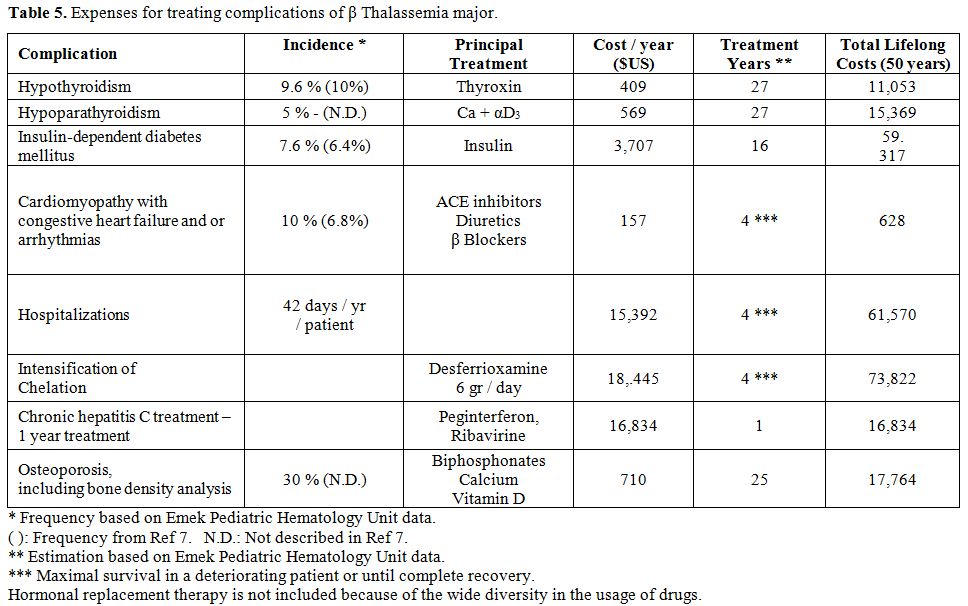 |
Table 5. Expenses for treating complications of β Thalassemia major. |
The estimated costs
are based on the fares calculated by the Health
Insurance Services in Israel and include laboratory test and imaging
costs, laboratory personnel, administrative staff and overhead for the
general expenses that cannot be specifically calculated.
For the purpose of this study an exchange rate of 3.82 New Israel
Shekel (NIS) for 1 US dollar was used, based on the average NIS/Dollar
conversion rate for the year 2011.
Results
During a 24 year period (1987 – 2011), about 75.000 women were screened and more than 500 couples at risk of having an affected offspring were detected, representing a prevalence of 6.25 per 1000. Of 600 prenatal tentative diagnoses, about 110 affected fetuses were diagnosed and therapeutically aborted. During the screening period, 32 new β Thalassemia patients were diagnosed. All except one were born to women who were detected as carriers by the screening program but refused genetic counseling or prenatal diagnosis after counseling, or preferred not to perform a therapeutic abortion despite the diagnosis of an affected fetus (Figure 2). Of those fetuses, 22 were born in the first ten years that the program was implemented, while only four were born in the latter ten years.
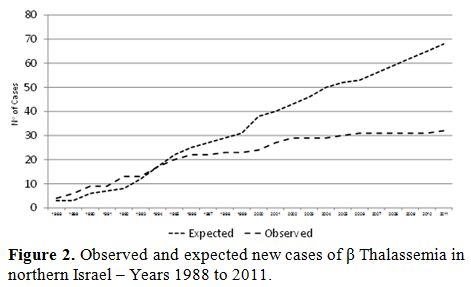 |
Figure 2. Observed and expected new cases of β Thalassemia in northern Israel – Years 1988 to 2011. |
As part of the
screening program, about 5000 blood tests were performed
annually. In the last ten years a mean of 26 couples/year were
identified as being at risk of having an affected offspring, and
prenatal diagnosis was subsequently performed. Assuming a 25% incidence
of affected fetuses i.e. 6.5 affected fetuses/year, an average of four
or five therapeutic abortions would be performed each year. The overall
annual cost of running the prevention program was $413,795 (Table 1). The
calculated cost for each affected fetus diagnosed was $63,660.
The cost of the diagnostic workup of a new β Thalassemia patient was
$500 (Table 3).
The routine follow-up for each patient was $1,410 annually. The yearly
expenditure for treatment during the first two years of life without
chelation therapy was $11,490. The introduction of chelation therapy
from the third year increased annual expenses to $40,581. The overall
mean cost per treatment-year was $39,427 including 2 T2*MRI heart and
liver tests. The cost of performing splenectomy was about $4,116, but
since only a few patients underwent splenectomy in recent years, the
mean cost for this procedure in the whole cohort was negligible (Table 2). The total
expenditure per patient for 50 years of life was $1,971,380, or an
average of $39,427 per year (Tables
2 and 4).
The costs of treating a patient with cardiac, endocrine or other
complications were not included in the cost-effect calculations but are
presented in Table 5.
The rates of those complications are based on the experience in our
Unit. Table 6
presents a method for calculation of treatment costs for a single β
Thalassemia patient in different countries.
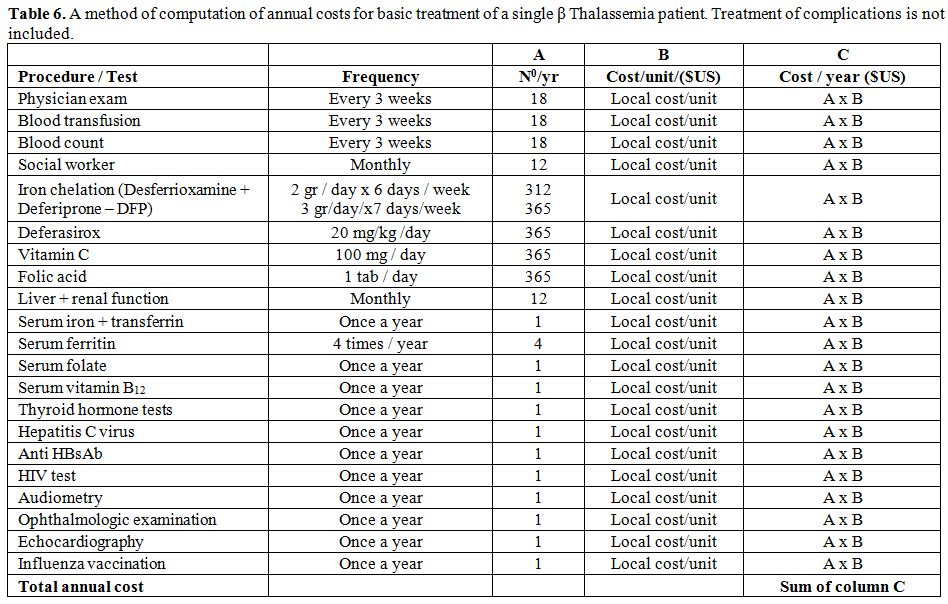 |
Table 6. A method of computation of annual costs for basic treatment of a single β Thalassemia patient. Treatment of complications is not included. |
Discussion
The burden of treating β Thalassemia major patients is substantial and
the prevention program implemented in our region is of low cost
compared to screening programs that employ molecular analysis for the
detection of carriers.
Our assumption of life expectancy for β Thalassemia major is less than
the 60 years estimated by Karnon et al.[5]
Modell et al.[13] failed to
show improvement in life expectancy in patients born with Thalassemia
in the United Kingdom after 1965, or in those born after 1975. The
authors estimated that not more than 50% of patients survive more than
35 years.[13] However, 68% of β
Thalassemia patients in Italy were
alive at age 35 years.[12] This
may serve as evidence of the longevity
that can be expected in developed countries that provide quality
treatment and where compliance with chelation treatments is high. Our
calculations are based on 100% compliance, which is an optimistic
assumption since reported compliance rates range from 64 to 90%.[14]
Our calculated costs did not include treatment of deteriorating
patients due to poor chelation compliance, such as cardiac or endocrine
treatment or treatment of osteoporosis and blood related acquired
infections. The expenses for such treatments can be easily calculated
for a single patient, but the proportion of patients that present with
such complications may differ considerably from country to country,
depending on the quality of available medical treatment and compliance
with chelation treatment. Other expenses not included in our cost
analysis are the expenses paid directly by the families of the
patients, such as travel to the hospital, loss of work days and
compensation by the National Welfare Fund. Indirect costs related to
the poor quality of life of such individuals were also not included in
this analysis.
Healthcare expenses for a new Thalassemia patient in our study were 4
fold higher than calculated in Israel by Ginsberg et al. 15 years
ago.[15] They calculated a total
expenditure of $283,154 for life
expectancy of 25-35 years, compared to our calculation of $1,971,380
for life expectancy of 50 years (ie: $9,438 and $39,427 per year,
respectively). Since that study did not include a detailed calculation
for treating a β Thalassemia patient, we cannot determine the source of
the discrepancy between our calculations and theirs. Increased expenses
following introduction of the new oral iron chelator, Deferasirox,
significant improvements in quality of life and almost double life
expectancy of patients with β Thalassemia are factors that may explain
the differences.
Our calculations of treatment costs are higher than those estimated by
Karnon et al. for treating one β Thalassemia patient in the United
Kingdom, $1,245,030, assuming a 60 year life expectancy (ie: $20,750
per year), and considerably higher than the annual cost of $9,168, as
calculated by Cronin et al., also from the United Kingdom.[16] Other
recent studies from the United States calculated the annual cost of
treating β Thalassemia patients with combination therapy of
Desferrioxamine and Deferiprone at $22,199 in uncomplicated patients
and $55,690 in patients with complications related to iron
overload.[17] When Deferasirox was
used as the chelator treatment, the
annual cost ranged from $24,400 to $53,095 in patients without
complications[18] and the total
treatment costs for the life expectancy
of 34.4 years was $1.8 million. This figure is similar to the one that
we calculated ($1.97 million) for a 50 year life expectancy, and to
that calculated in 1984 in Quebec, Canada.[19]
The purchase power in
Israel and the United Kingdom are similar ($77.70 and $89.96,
respectively), while higher in Canada and the United States ($112.10
and $140.80, respectively).
In Thailand, expenses to treat β Thalassemia patients are significantly
lower than in the western world, probably due to less frequent blood
transfusions and to the administration of low chelator doses. The costs
reported were only $7,604 per year.[20]
The difference can be explained
by a significantly lower purchase power, $35.56 compared to western
countries.
The systematic screening of pregnant women at the Mother and Child
Health Care Clinics in Israel precluded the need to establish a new
system of clinics dedicated to screening. This reduced program costs. A
similar screening program is currently implemented on a national basis,
but the database for information analysis is not in operation, thus the
data presented herein is based on the experience in northern Israel
only.
Several means exist for instituting a prevention program of genetic
disease. The least effective is the public distribution of
information.[1,21]
Compulsory screening is the most effective method
but not ethical in democratic countries. In countries where prenatal
diagnosis is not permitted, compulsory screening and prevention of
marriages between detected carriers is often utilized to reduce the
incidence of genetic disease.[8,22] Ginsberg et al.[15]
estimated at
$291,000 the cost of instituting an educational program for β
Thalassemia prevention among the non-Jewish population in Israel, which
is the population at highest risk in this country; the prevention of
one affected fetus each year could cover this investment. However,
educational programs alone cannot be cost beneficial and efficient
enough to provide timely results to populations with a high incidence
of carriers, especially when religious beliefs and traditions conflict
with screening and intervention. In the program instituted in northern
Israel, a significant decrease in the rate of affected newborns during
the recent years, reflects the population's collaboration
with
the program. In the last ten years (2001 – 2011), only four new
patients with β Thalassemia major were born, only one of them in the
last five years.
We showed that the cost of preventing one affected newborn is $63,660
compared to $1,971,380 for treatment of a β Thalassemia patient for 50
years (annual cost of $39,427). Thus, the prevention of 45 affected
newborns over a ten year period represents a net saving of $88.5
million to the health budget. Even after deducting the costs of
operating the prevention program in our region ($413.795/year, $20
million for 50 years) and the costs of treating the four newborn
patients for 50 years ($8 million), the program still represents a
substantial benefit of $60 million. Each prevented case could cover the
costs of the screening and prevention program for 4.6 years. A study
from Canada calculated the cost for each prevented case at $6700;
however, that study was based on a small cohort of 6,748 persons in a
high risk community.[19] Our
program provides universal screening; more
than 80,000 pregnant women were screened since 1987. Increased costs
over the last thirty years and the large experience of our medical
center and healthcare system may explain the differences between the
studies.
Our calculations are based on the social characteristics, gene
frequencies and birth rate of the high risk population in northern
Israel. Similar carrier rates were found in other non-Jewish
populations, in the country. Specifically, tailored screening
programs,[6-9,23]
including educational and voluntary pre-marital or
pre-conception screening for populations of high socioeconomic levels,
can reduce the cost of universal screening. We have shown the cost
benefit of a national program for the screening of pregnant women for
prevention of β Thalassemia in Israel, which may be applicable to
countries with similar social settings. An additional benefit of such
programs is the provision of prenatal counseling for future pregnancies
without the need for further screening.
The total expense for treatment of deteriorating β Thalassemia patients
is difficult to estimate. Incidence rates of complications have
decreased significantly due to improvement in compliance with chelation
treatment and to the high quality of blood units transfused.
Nevertheless, the rates for such complications in our population are
similar to those reported in 2004 from Italy.[12]
Because of the
difficulty in computing the expenses of complications, we did not take
them into account. Stem cell transplant is capable of curing
thalassemia, but it is limited to eligible patients. The reported cost
for each transplant varies from $100.000 to 200.000 in the United
States and in Thailand[24-26] to
about $50.000, as is estimated in
Israel, though no detailed data were calculated in Israel. Considering
that the cost of one transplant is equivalent to one year of treatment,
a successful transplant can certainly be cost effective.[24] Since only
a small proportion of thalassemia patients have been transplanted
around the world, and calculation of the impact of stem cell transplant
on the overall expenses of treating a cohort of patients is difficult,
we did not include these calculations in our study.
The calculations presented herein also did not take into account the
burden to the national economy in terms of welfare payments; loss of
work capacity and educational support needed. According to one
estimation, published 15 years ago, less than 10% of Thalassemia
patients in Israel are employed.[15]
Adding the expenses of treatment
for deteriorating patients and welfare payments would further raise the
cost-effectiveness of the prevention program.
Two factors, the introduction of the oral chelator, Deferasirox,
(Exjade ®, Novartis, Switzerland) and the improvement in survival
beyond age 50, while raising the costs for treating Thalassemia
patients, increase the attractiveness of the preventive
program.
Conclusions
This study showed that each new β Thalassemia patient born incurred an
excessive budget of about $2 million for a life expectancy of 50 years.
Such a budget could fund a prevention program for 4.6 years and prevent
at least 31 affected patients.
Based on our calculations, and according the nationwide Mother and
Child Health Care Clinics in Israel, implementation of a national β
Thalassemia prevention program appears to have a high benefit- cost
ratio. Benefits to society include, in addition to the direct financial
savings of millions of dollars, the saving of hundreds of blood units,
work power, compensation fees, treatment of endocrinological and
cardiac complications and treatment of intercurrent complications and
expenses during the phase of terminal deterioration.
Conflict of Interest Declaration
Koren A. received support for the preparation of the paper from
Novartis, Pharma Israel and, he also acts as a medical consultant for
Novartis, Pharma Israel and Apopharma – Lapidot Israel.
All the other authors declare that they have no conflicts of interest
related to the preparation of this manuscript.
References

[TOP]