Received: March 28, 2014
Accepted: July 4, 2014
Meditterr J Hematol Infect Dis 2014, 6(1): e2014054, DOI 10.4084/MJHID.2014.054
This article is available on PDF format at:

Antonella Isgro'1, Pietro Sodani1, Marco Marziali1, Javid Gaziev1, Daniela Fraboni2, Katia Paciaroni1, Cristiano Gallucci1, Gioia De Angelis1, Cecilia Alfieri1, Michela Ribersani1, Daniele Armiento1, Andrea Roveda1, Marco Andreani1, Manuela Testi1 and Guido Lucarelli1
1
International Center for Transplantation in Thalassemia and Sickle Cell
Anemia, Mediterranean Institute of Hematology, Policlinic of the
University of Roma Tor Vergata., Rome, Italy.
2
Laboratory of Oncohematology, Department of Laboratory Medicine,
Policlinic of the University of Roma Tor Vergata, Rome, Italy.
|
This
is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract Background and Purpose:
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only
curative treatment for sickle cell anemia (SCA). We report our
experience with transplantation in children with the Black African
variant of SCA and the effects of transplant on erythroid compartment
in bone marrow (BM).
Patients and Methods: Twenty-seven consecutive patients who underwent BM transplantation from HLA-identical donors following a myeloablative conditioning regimen were included. Using both CD71 and FSC parameters, we obtained three erythroid populations: EryA–C. Ery A (CD71high FSChigh) are basophilic; Ery B (CD71high FSClow) are late basophilic and polychromatic; and Ery C (CD71low FSClow) are orthochromatic erythroblasts and reticulocytes. To analyze the effect of transplantation on intramedullary apoptosis, we studied Fas (CD95+) and caspase-3 expression in erythroblast subpopulations. Results: All patients experienced sustained engraftment, and all surviving patients remained free of SCA-related events after transplantation. The erythroid population showed expansion in the BM at baseline. After transplant, levels decreased, especially of Ery C, in parallel to reduced Fas expression and an initial caspase 3 increase in erythroid population, similar to reported later steps of “normal” erythroid maturation. Conclusions: The results suggest a good chance of cure for children with SCA, with an excellent survival rate. We also observed “normalization” of erythroid populations in parallel with a decreased intramedullary apoptosis rate, suggesting normal erythroid maturation in ex-SCA patients after HSCT. |
Introduction
Sickle
cell disease is a group of genetic conditions in which pathology
results from the inheritance of the sickle cell gene variant either
homozygously or as a double heterozygote with another interacting gene.
The spectrum of resulting conditions is influenced by the geography of
individual hemoglobin genes, but in most populations, the commonest
genotype at birth is homozygous sickle cell (SS) disease. Since this
genotype involves a greater mortality, the relative proportion of
sickle cell genotypes is influenced by age as well as by the
geographical distribution of individual genes.[1]
The sickle cell trait is widespread throughout Africa. Frequencies are
low (<1%–2%) in the north and south of the continent but high
with
variable frequencies throughout much of equatorial Africa. SCA is
characterized by a cascade of events that begin with the polymerization
of hemoglobin S and the sickling of red blood cells. Following these
events is occlusion of small and larger vessels because of the
adherence of the sickled cells to the vascular endothelium, which leads
to pain crises, stroke, acute chest syndrome, and multi-organ failure
as the most frequent complications.[2]
In patients with the Black
African variant, painful crises, chest syndrome, and stroke are more
frequent and appear earlier in life. The causes of death are strongly
influenced by the prevalence of malaria and other infections, and
almost certainly by the availability and sophistication of medical and
other services. In sub-Saharan Africa, survival is markedly shortened,
and median survival may be as short as 5 years.
Hematopoietic stem cell transplantation (HSCT) is the only radical cure
for this genetic disorder,[3] and
to date, several hundred patients
have undergone gene-identical HSCT.[4-9]
In accordance with data
recently published,[25] our
experiences confirm that it is possible to
offer a good chance of cure to children with SCA, and we thus have made
the following recommendation: “HSCT
should be considered the standard
of care for SCA children with a human leukocyte antigen-identical
donor, before complications result from the sickling of red blood
cells.”
Less well established is the potential contribution of ineffective
erythropoiesis to the pathophysiology of this hemoglobinopathy. As in
thalassemia patients, an expansion of erythroid precursors is observed
in SCA patients at the bone marrow (BM) level but is less severe than
in thalassemia.[10-11] Normal
homeostasis of the erythropoietic system
requires an appropriate balance between the rate of erythroid cell
production and red blood cell destruction. Growing evidence indicates
that apoptotic mechanisms play a relevant role in the control of
erythropoiesis under physiologic and pathologic conditions.[10] Death
receptors of the TNF receptor superfamilies (Fas-Ligand (Fas-L), TNF-α,
TRAIL) activate the extrinsic apoptotic pathway. Fas and Fas-L are
expressed in cultured erythroblasts, but there are controversies
regarding the level and differentiation stage at which they are
expressed. Some studies suggest the existence of a negative regulatory
feedback operating at low erythropoietin (Epo) levels in a paracrine
pathway. In this system, Fas-L–expressing mature erythroblasts display
cytotoxicity against immature erythroblasts expressing Fas.[12,13] Epo
can partially protect immature erythroid cells from Fas-mediated
apoptosis; thus, Fas and Fas-L are major regulators of erythropoiesis.
Both proteins are downregulated in BM or spleen in proerythroblast and
basophilic cells in β-thalassemic mice compared to control mice in
vivo. This downregulation of Fas/Fas-L expression might be a marker of
erythropoietic stress and explain, at least in part, erythroid
expansion in thalassemia.[14]
We hypothesized that Fas might contribute to the cell death of SS
erythroid precursors at the BM level, but that transplant may be
corrective. Here we report our experience with transplantation in a
group of pediatric patients with Black African variant SCA, who
received transplantations from HLA-identical siblings. We analyzed the
effect of transplant on erythropoiesis and intramedullary apoptosis,
studying Fas (CD95+) and caspase-3 expression in erythroblast
subpopulations before and after transplant. We also used this
opportunity to directly compare the differentiation and survival of SCA
and donor (AA or AS trait carrier)-derived erythropoiesis in vivo.
Patients and Methods
This study included 27 consecutive SCA patients who underwent BM
transplantation from HLA-identical sibling donors between January 2010
and June 2013. Twenty-seven patients with the Black African SCA variant
were treated with a modification of our Protocol 26, which was in use
for Class 3 thalassemia patients[15]
(here identified as Protocol 28).
The institutional review board approved the treatment protocol, and all
parents of patients provided written informed consent in accordance
with the Declaration of Helsinki.
Patient
characteristics.
The median patient age was 10 years (range 2–17 years), and the median
donor age was 11 years (range 1–26 years). Patient characteristics at
the time of transplantation are summarized in Table 1. All
patients
showed good performance status (Lansky/Karnofsky 100) before
transplantation. No patient had a splenectomy before transplantation,
and only two received chronic blood transfusions; the serum ferritin
level before transplantation was 278 +/- 231 ng/mL (mean +/- SD).
Before
transplantation, 11 patients had recurrent, painful, vaso-occlusive
crisis; nine patients had recurrent painful crisis in association with
acute chest syndrome; three patients experienced ischemic stroke and
recurrent vaso-occlusive crisis; two patients experienced ischemic
stroke; one patient exhibited leukocytosis, and one patient exhibited
priapism. HLA typing at the
molecular level was performed, and all donors for both groups were
fully matched.
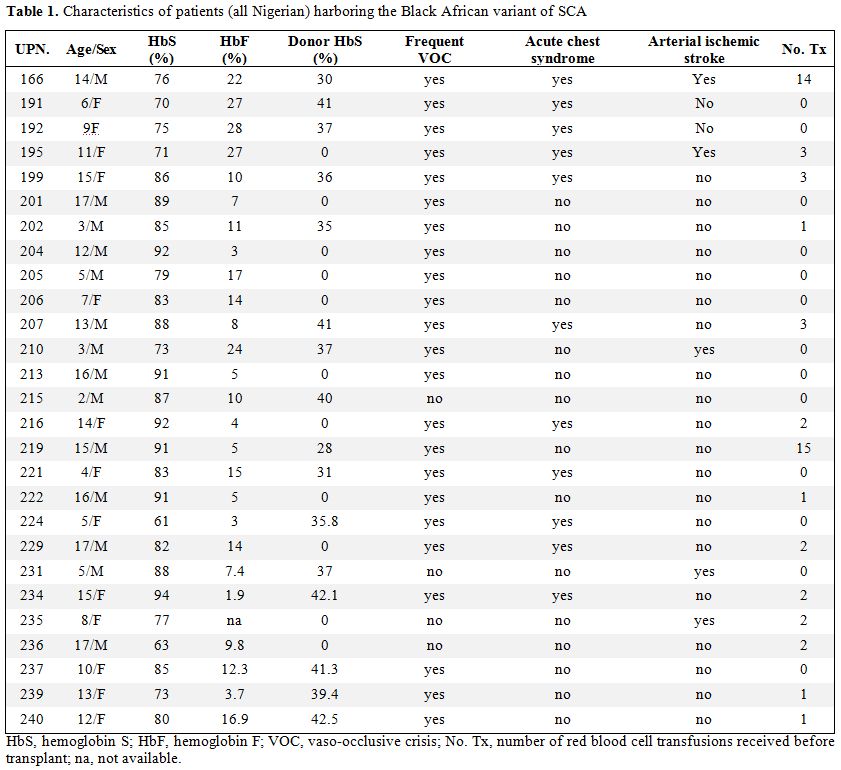 | Table 1. Characteristics of patients (all Nigerian) harboring the Black African variant of SCA |
Transplantation
procedure. Patients received fludarabine (30 mg/m2/day)
for 5 days and a conditioning regimen including targeted intravenous
busulfan (14 mg/kg total dose) and cyclophosphamide (200 mg/kg total
dose). All patients received cyclosporine A, low-dose
methylprednisolone, and a short course of methotrexate as GVHD
prophylaxis. Among the patients, six had cyclosporine A-related
neurotoxicity with seizures. All patients received valproic acid
(Depakin; Sanofi-Aventis) at a dose of 30 mg/kg/day in 3 divided doses
starting at 24 hours before the first busulfan administration. Many
risk factors for the development of CSA-related neurotoxicity have been
investigated in our patients, including arterial hypertension, fluid
overload, hypercholesterolemia, hypomagnesaemia and pre-existing brain
disease. In the screening examinations of these patients the brain
magnetic resonance imaging (MRI) showed gliosis in 11/27 stroke free
Black African SCA patients (manuscript in preparation). The brain MRI
finding, usually associated to CSA neurotoxicity, was posterior
reversible leukoencephalopathy syndrome (PRES), typically distributed
in the posterior regions of the white matter of the brain. We cannot
rule out the pre-existing brain disease in these patients might
predispose to seizures during CSA treatment. In general, the prognosis
of CSA neurotoxicity has been good and posterior leukoencephalopathy
usually resolved completely with dose reduction or drug withdrawal. As
alternative GVHD prophylaxis, we opted for tacrolimus. This calcineurin
inhibitor, although similar to CSA in mechanism and metabolism, did not
produce neurological side effects in these patients.
Children with Black African variant SCA were prone to invasive
infections caused by S. pneumonia, H. influenzae and Plasmodium
falciparum (in malarial areas). Malaria is more endemic in Black
African areas and therefore malaria is more common in Black SCA
patients. In Africa, malaria contributes substantially to the early
mortality of patients with SCA. For these reasons we preferred in this
population fludarabine-based preparative protocols, well tolerated,
with less immunosuppression and minimal toxicity.
All patients received BM from HLA-identical sibling donors 36 h after
the final dose of cyclophosphamide, and all donors with sickle cell
trait received hyperhydration and blood transfusion before the multiple
marrow aspirations. The median number of total nucleated cells infused
was 4.08 x 108/kg
(range 1.7 x 108/kg
to 10.0 x 108/kg),
and the median number of CD34 cells was 5.8 x 106/kg
(range 1.2 x 106/kg
to 11.2 x 106/kg).
The diagnosis and degree of acute and chronic GVHD were assessed
according to standard criteria.[16,17]
All patients were given
prophylactic broad-spectrum antibiotics and antifungal drugs until the
neutrophil level exceeded 1.0 x 109/L,
and also received acyclovir as herpes virus prophylaxis and
trimethoprim/sulfamethoxazole as Pneumocystis jiroveci prophylaxis.
Patients were monitored weekly for the presence of Epstein-Barr virus,
cytomegalovirus (CMV), adenovirus, and BK virus in the blood and/or
urine using sensitive reverse transcriptase polymerase chain reaction
(PCR), from the beginning of transplant preparation until at least 100
days post-transplant.
Assessment of chimerism. The first chimerism analysis was performed on BM samples obtained 20 days after transplant to determine the percentage of donor/recipient DNA using PCR-based analysis of short tandem repeats. Subsequently, at 60, 90, 180, and 365 days post-transplant, lineage-specific chimerism analysis was performed by PCR using fluorescent primers flanking a single informative short tandem repeat (AmpFLSTR Profiler Plus; Applera, CA, USA) previously identified to be polymorphic between the patient and donor.
Cytometric
assay for erythroid cell precursors.
We previously developed a flow cytometric assay to identify
stage-specific erythroblasts directly in hematopoietic tissue (BM)
based on their expression of the transferrin receptor (CD71), which
declines with erythroblast maturation.[18]
However, the decline in CD71
appeared to be gradual, without the formation of well-resolved
subpopulations. In this study, we distinguished well-resolved
erythroblast subpopulations by considering, in addition to CD71, the
forward scatter (FSC) parameter. FSC is a function of cell size and has
been used previously to assess erythroblast maturation independently of
cell surface marker expression. When the cells were analyzed using both
CD71 and FSC parameters, they consistently resolved into three
principal subpopulations, which we labeled Ery A, Ery B, and Ery C
erythroblasts. Ery A (CD71high
FSChigh)
are basophilic; Ery B (CD71high
FSClow)
are late basophilic and polychromatic; and Ery C (CD71low
FSClow)
are orthochromatic erythroblasts and reticulocytes.[11,14]
Bone marrow specimens of patients and donors were obtained to evaluate
Fas and caspase 3 expression in erythroblasts: anti-CD95 PE, anti-CD71
FITC, and anti-CD45 PercP Cy5.5 were mixed in a tube. A volume of 10 μL
of these MoAb cocktails (BD, Becton Dickinson, San Diego, CA, USA) was
combined with 100 μL of bone marrow mononuclear cells for 10 minutes at
room temperature, then lysed with BD Pharm Lyse 1x for 20 minutes at
room temperature and washed with 2% phosphate-buffered saline plus
bovine serum albumin. Samples were analyzed with BD FACS Canto II and
the software, BD FACSDiva.
Statistical
analysis. The probabilities of survival, SCA-free
survival, rejection, and mortality were calculated using Kaplan–Meier
curves.[19]
Non-parametric statistics was used (Mann-Whitney,
Wilcoxon test) for
unpaired and paired comparisons between the parameters analysed in
patients and healthy individuals. A p-value less than 0.05 was
considered significant. Statistical analyses were performed by using
Stat View 5.0 software (SAS Institute, Cary, NC, USA).
Results
Clinical
assessment post-transplant. The median time to neutrophil
recovery (absolute neutrophil count ≥ 500 x 109/L
on 3 consecutive days) was 16 days (range 11–23 days). Platelet
recovery ≥20 x 109/L
was observed at a median of 17 days (range, 11-22 days) after
allo-HSCT. In terms of platelet transfusion needs, the median number of
platelet units transfused in the first 100 days after HSCT was 15 U
(range, 0–53 U) but it increased when complications as severe acute
GVHD or sepsis appeared. In our cohort of patients we observed a lower
rate of platelet transfusion and faster platelet recovery kinetics
after HSCT, but also highlighted the negative effect of severe acute
GVHD as a risk factor for increased need for platelet transfusions.
At 2 months after transplantation 3 patients had donor chimerism
between 95% and 98%, and all the remaining patients had full donor
chimerism. At the last control, all patients experienced sustained
engraftment with 100% donor chimerism. All patients and donors except
one had positive serology for CMV before transplantation. Asymptomatic
CMV reactivation occurred in 26 of 27 patients. All patients were
provided pre-emptive antiviral therapy, and none developed CMV disease.
Seven patients developed grade 2 acute GVHD of the skin, and five
patients developed grade 3-4 GVHD, principally after 30 days post
transplant. All patients responded promptly to the steroid treatment
administered to control acute GVHD (1-2 mg/kg/day prednisone). At
present, all patients except one are off immunosuppressive medication.
Chronic GVHD was observed in four patient: one patient developed
bronchiolitis obliterans, and one patient had severe chronic GVHD with
intestinal and hepatic involvement until death, as a result of
multi-organ failure at day +190 post-transplantation. Cumulative
incidence of grade 3-4 acute GVHD was 18%. Cumulative incidence of
persistent severe chronic GVHD was 14%.
One patient died at 77 days post-transplantation from complications of
severe GVHD of the gut. One patients died from multiorgan failure at
190 days post-transplantation. He had no steroid responsive grade 4
acute GVHD of the gut and developed sepsis, which led to multiorgan
failure and death. One patient died from complications of bronchiolitis
obliterans at 445 days post-transplantation.
After transplantation, no patients experienced complications typical of
SCA, such as pain, stroke, or acute chest syndrome. The probabilities
of survival, SCA-free survival, and transplant-related mortality after
transplant were 89%, 89%, and 11%, respectively.
Expression
of Fas and caspase-3 on erythroid population at the BM level.
We observed an expansion of the BM erythroid population at baseline,
probably as an essential process needed to maintain a constant red cell
production in SCA patients (Fig.
1).
Average percentages of CD71+CD45-
were
4.6 +/- 3.7% in
normal AA donors, 7 +/- 2.8% in AS trait
carrier donors, 18.7 +/- 14.6% in SCA patients at
baseline, and 8.1 +/-
5.6% in SCA patients at 60 days after transplant. After HSCT, decreased
levels were observed in all three erythroid subpopulations (average 40 +/-
20% vs. 45.3 +/- 16.7% at baseline for Ery A; 38.4 +/-
20.5% vs. 46.2 +/-
16.4% at baseline for Ery B), especially for Ery C (2 +/-
5% vs. 13.2 +/-
25.4% at baseline; p= 0.0028) (Fig.
2)
in parallel to a reduction in Fas expression (Fig. 3) in the BM
(average CD95+CD34+, 3.7 +/-
1.7% in normal AA donors; 4.9 +/-
3.3% in AS
trait carrier donors; 9.6 +/-
7.3% in SCA patients at baseline, p= 0.004
vs. healthy controls; and 7.3 +/- 4% at 60 days
after transplant, p=
0.007 vs. healthy controls) and specifically in the erythroid
compartment (average CD95+CD71+CD45-, 1.6 +/- 1.9%
in normal AA donors;
1.8 +/- 3.2% in AS trait carrier donors; 2.9 +/-
2.9% in SCA patients at
baseline; and 2 +/-
2.7% at 60 days after transplant) (Fig. 3).
After transplant, a tendency to a normalization of erythropoiesis has
been observed in our patients, with a reduction of Fas expression on
three erythroid population but especially on more mature erythroid
precursors Ery C (Fig. 4).
An initial increase in caspase 3 was observed after HSCT, as has been
reported for later steps of “normal” erythroid cell maturation. Average
percentages of caspase 3+CD71+CD45- were 1.8 +/-
1.8% in normal AA
donors, 2.7 +/- 2.2% in AS trait carriers donors, 2
+/-
1.5% in SCA
patients at baseline, and 3.7 +/- 4.3% in SCA
patients at 60 days after
transplant (Fig. 5).
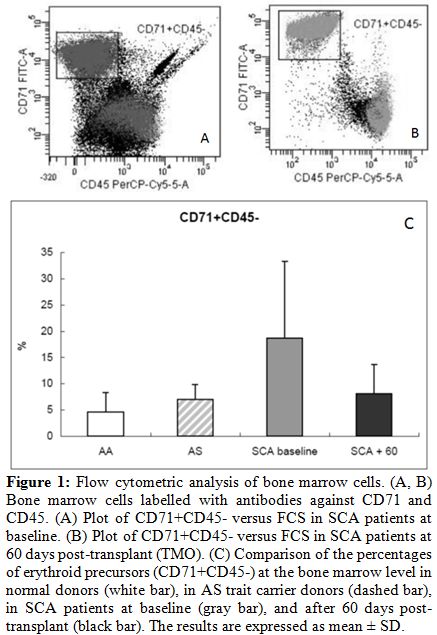 | Figure 1. Flow cytometric analysis of bone marrow cells. (A, B) Bone marrow cells labelled with antibodies against CD71 and CD45. (A) Plot of CD71+CD45- versus FCS in SCA patients at baseline. (B) Plot of CD71+CD45- versus FCS in SCA patients at 60 days post-transplant (TMO). (C) Comparison of the percentages of erythroid precursors (CD71+CD45-) at the bone marrow level in normal donors (white bar), in AS trait carrier donors (dashed bar), in SCA patients at baseline (gray bar), and after 60 days post-transplant (black bar). The results are expressed as mean +/- SD. |
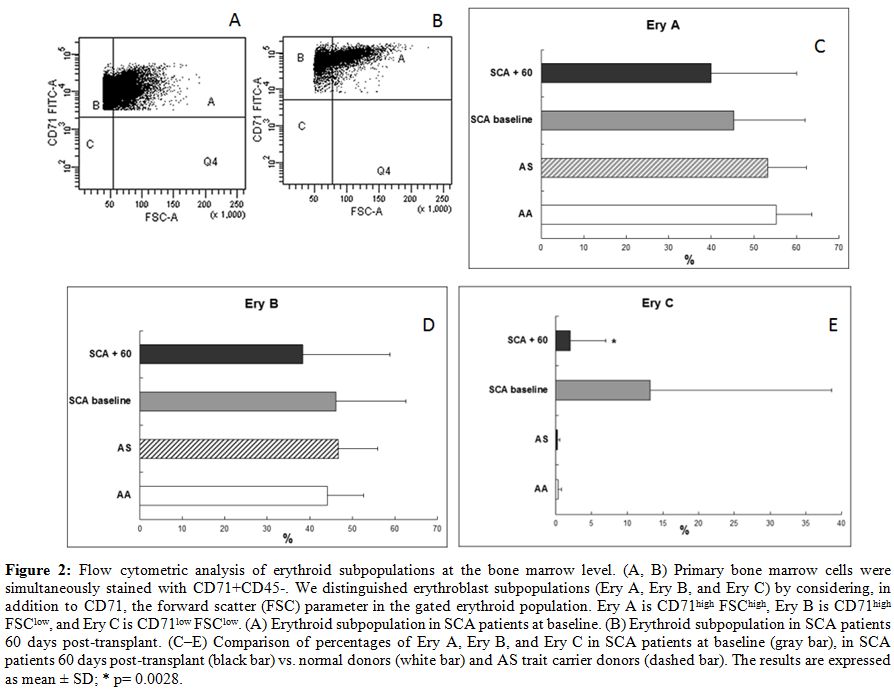 | Figure 2. Flow cytometric analysis of erythroid subpopulations at the bone marrow level. (A, B) Primary bone marrow cells were simultaneously stained with CD71+CD45-. We distinguished erythroblast subpopulations (Ery A, Ery B, and Ery C) by considering, in addition to CD71, the forward scatter (FSC) parameter in the gated erythroid population. Ery A is CD71high FSChigh, Ery B is CD71high FSClow, and Ery C is CD71low FSClow. (A) Erythroid subpopulation in SCA patients at baseline. (B) Erythroid subpopulation in SCA patients 60 days post-transplant. (C–E) Comparison of percentages of Ery A, Ery B, and Ery C in SCA patients at baseline (gray bar), in SCA patients 60 days post-transplant (black bar) vs. normal donors (white bar) and AS trait carrier donors (dashed bar). The results are expressed as mean +/- SD; * p= 0.0028. |
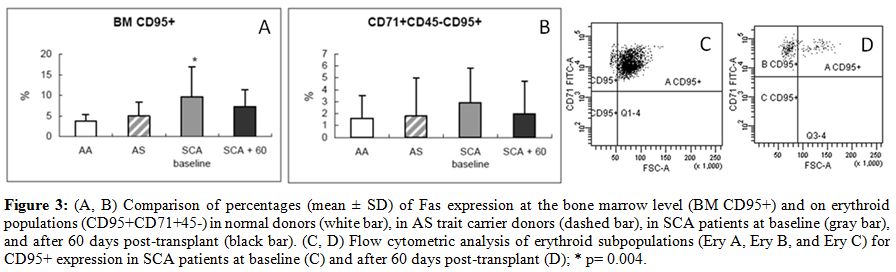 | Figure 3. (A, B) Comparison of percentages (mean +/- SD) of Fas expression at the bone marrow level (BM CD95+) and on erythroid populations (CD95+CD71+45-) in normal donors (white bar), in AS trait carrier donors (dashed bar), in SCA patients at baseline (gray bar), and after 60 days post-transplant (black bar). (C, D) Flow cytometric analysis of erythroid subpopulations (Ery A, Ery B, and Ery C) for CD95+ expression in SCA patients at baseline (C) and after 60 days post-transplant (D); * p= 0.004. |
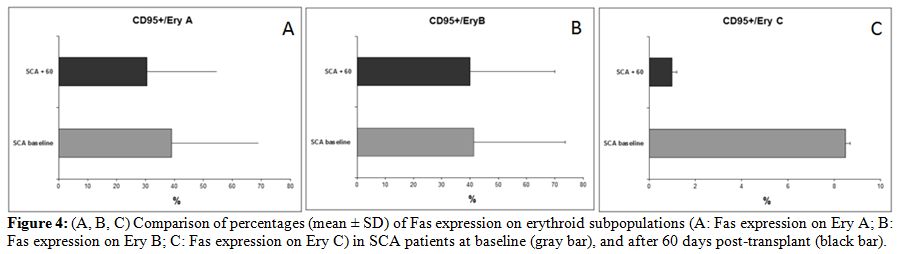 | Figure 4. (A, B, C) Comparison of percentages (mean +/- SD) of Fas expression on erythroid subpopulations (A: Fas expression on Ery A; B: Fas expression on Ery B; C: Fas expression on Ery C) in SCA patients at baseline (gray bar), and after 60 days post-transplant (black bar). |
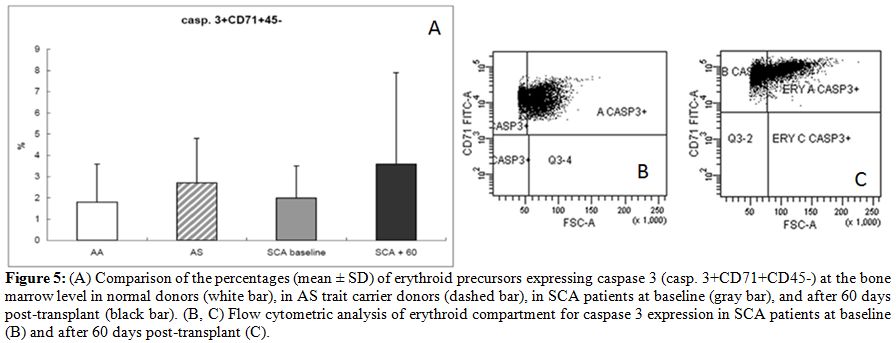 | Figure 5. (A) Comparison of the percentages (mean +/- SD) of erythroid precursors expressing caspase 3 (casp. 3+CD71+CD45-) at the bone marrow level in normal donors (white bar), in AS trait carrier donors (dashed bar), in SCA patients at baseline (gray bar), and after 60 days post-transplant (black bar). (B, C) Flow cytometric analysis of erythroid compartment for caspase 3 expression in SCA patients at baseline (B) and after 60 days post-transplant (C). |
Discussion
The morbidity and mortality associated with SCA are much more frequent
and severe than those associated with thalassemia. The Black African
SCA variant manifests a severe phenotype when compared to the non-Black
African SCA. It is possible that in patients with Black African SCA, a
hyperplasia of the erythroid lineage exists at baseline, probably to
maintain a constant production of erythroid precursors. The high level
of polymerization of the sickle hemoglobin in host RBCs, as well as in
host early and basophilic normoblasts, might also determine mechanical
defects that, in turn, increase host cell susceptibility to clearance
and loss.
The presence of ineffective erythropoiesis in SCA is supported by
previous studies, which have identified structural abnormalities in SS
erythroid precursor cells, thus indirectly indicating the increased
susceptibility of these cells to clearance and loss. Blouin et al.
examined erythropoiesis in the SCA mouse model and found significant
morphological alteration in erythroid lineage late precursors
(polychromatophilic normoblasts) within the marrow.[20]
These
morphological studies identified a high level of hemoglobin polymers
that were associated with increased cell fragmentation occurring during
medullary transendothelial migration of reticulocytes. Older
ultrastructural studies of BM aspirates derived from SCA patients
identified reticulocytes that contain bundles of hemoglobin S polymers
in the absence of intentional deoxygenation, as well as sickling of
nucleated erythroblasts and extensive marrow erythrophagocytosis.[21,22]
Hasegawa et al. also found in an in vitro system that cultured
nucleated erythroid precursors can undergo sickling under deoxygenating
conditions.[23] Recently, our
group observed the presence of sickled
erythrocytes at the BM level in SCA patients, in the absence of
systemic symptoms, as well in AS trait carriers. This condition could
be induced by the cellular stress of the biopsy procedure or
alternatively represents a specific status of the BM of SCA patients,
both the homozygous (SS) and heterozygous (AS) status (manuscript in
preparation).
Apoptosis is an important mechanism by which ineffective erythroblasts
are cleared within the intramedullary space, and our data suggest that
Fas might contribute to the cell death of host erythroid precursors in
SCA. If accelerated apoptosis is not compensated by enhanced
erythropoiesis, however, clinically relevant anemia develops. Our
studies suggest that significant abnormalities in SS erythroid
precursors exist within the intramedullary space and that cells prone
to sickling may be selectively destroyed prior to release from the
erythropoietic compartment.
With HSCT, it is possible to give more than 90% chance of cure for
children with SCA, with excellent survival rate and return to normal
life. We agree with the recommendations of the Haematologica’ s
authors.[25] The young patients
with symptomatic SCA, who have an
HLA-matched sibling donor, should be transplanted as early as possible
before sickling complications appear. The vast majority of our patients
are not regularly transfused/chelated, or highly sensitized due to
receiving RBC transfusions without the use of leukodepletion filters.
Despite the recognized benefits of transfusion therapy, it is not
without the risks of iron overload, alloimmunization, and delayed
hemolytic transfusion reactions. Alloimmunization to RBC antigens is a
major complication associated with RBC transfusions in patients with
SCA. Alloantibodies and autoantibodies complicate RBC cross-matching,
delay provision of transfusions, and increase the labor and cost of
providing compatible RBC units. For these reasons and since patients
had an HLA-identical sibling donor, these patients had indications for
hematopoietic stem cell transplant.
After HSCT, we observed a “normalization” of erythroid populations, in
parallel with a decreased intramedullary apoptosis rate, suggesting
normal erythroid maturation in ex-SCA patients. In fact in the basal
state, the erythropoietic system continuously produces excess numbers
of early erythroblast, which become apoptotic through Fas-mediated
signaling. The principal advantage of a homeostatic mechanism that
relies on negative autoregulation of cell numbers is that it would
self-correct for small perturbations, maintaining a relatively constant
erythroblast population size in the basal state. The major expression
of Fas at the early stage of erythroblast maturation has been observed
at baseline in our patients, contributing to a negative autoregulation
of cell number. After transplant, a tendency to a normalization of
erythropoiesis has been observed in our patients, with a reduction of
Fas expression on three erythroid population but especially on more
mature erythroid precursors Ery C. A progressive maturation advantage
for homozygous hemoglobin A (AA) or heterozygous hemoglobin
S/hemoglobin A (SA) donor erythroid precursor cells resulted in a
greater donor contribution to overall erythropoiesis following stem
cell transplantation and improvement of clinical manifestations.
Conclusions
This study suggests a good chance of cure for children with SCA, with
HLA-identical transplant. We also observed “normalization” of erythroid
populations in parallel with a decreased intramedullary apoptosis rate,
suggesting normal erythroid maturation in ex-SCA patients after HSCT.
HSCT should be considered the standard of care for SCA children with
human leukocyte antigen-identical donor before complications result
from the sickling of red blood cells.
References

[TOP]