Received: 20 May, 2014
Accepted: September 23, 2014
Meditter J Hematol Infect Dis 2014, 6(1): e2014066, DOI 10.4084/MJHID.2014.066
This article is available on PDF format at:

Marco Marziali1, Antonella Isgrò1, Pietro Sodani1, Javid Gaziev1, Daniela Fraboni2, Katia Paciaroni1, Cristiano Gallucci1, Cecilia Alfieri1, Andrea Roveda1, Gioia De Angelis1, Luisa Cardarelli1, Michela Ribersani1, Marco Andreani3 and Guido Lucarelli1
1 International Center for
Transplantation in Thalassemia and Sickle Cell Anemia, Mediterranean
Institute of Hematology, Policlinic of the University of Rome “Tor
Vergata” Italy
2 Department of Biopathology and Diagnostic Images, Polyclinic of Tor Vergata Foundation, Rome, Italy
3
Laboratory of Immunogenetics and Transplant Biology,
Mediterranean Institute of Hematology, Policlinic of the University of
Rome “Tor Vergata”
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Allogeneic cellular gene therapy
through hematopoietic stem cell transplantation is the only radical
cure for congenital hemoglobinopathies like thalassemia and sickle cell
anemia. Persistent mixed hematopoietic chimerism (PMC) has been
described in thalassemia and sickle cell anemia. Here, we describe the
clinical course of a 6-year-old girl who had received bone marrow
transplant for sickle cell anemia. After the transplant, the patient
showed 36% donor hematopoietic stem cells in the bone marrow, whereas
in the peripheral blood there was evidence of 80% circulating donor red
blood cells (RBC). The analysis of apoptosis at the Bone Marrow level
suggests that Fas might contribute to the cell death of host erythroid
precursors. The increase in NK cells and the regulatory T cell
population observed in this patient suggests that these cells might
contribute to the condition of mixed chimerism. |
Introduction
Allogeneic cellular gene therapy through hematopoietic stem cell
transplantation is the only radical cure for congenital
hemoglobinopathies like thalassemia and the sickle cell anemia.[1] Persistent mixed hematopoietic chimerism (PMC) has been described in thalassemia.[2,3] Recently, a split chimerism of the peripheral red blood cells was also described four years after transplantation.[3,4]
PMC provides a unique opportunity to perform a direct side by side
comparison of normal and sickle erythropoiesis. However, the minimum
proportion of donor cells that defines PMC differs in sickle cell
disease (SCD) and thalassemia patients transplanted; and the cell
populations, total leukocytes, mononuclear cells, or lineage-specific
cells assayed for chimerism, also varies. The threshold percentage of
donor cells sufficient to ameliorate the hemoglobin disorders has not
yet been firmly established. In thalassemic patients after
myeloablative HSCT, 10% to 20% of donor cells has been shown to be
curative.[3,5]
Several potential
factors seem to be associated with PMC. Less-intensive conditioning
regimens are associated with a greater proportion of PMC. As just
recently reported, T regulatory cells (Treg) and natural killer (NK)
populations may help to establish persistent mixed chimerism.[6,7]
HLA-mismatched transplants in mice and humans demonstrate that donor NK
cells target host hematopoietic tissue, eliminating host
antigen-presenting cells, host hematopoiesis, and host leukemia. These
effects translate into better engraftment, diminished risk from acute
graft versus host disease (GVHD), reduced relapse from an NK-mediated
graft-versus-leukemia effect and lower rejection rates.[8-10]
Recent
studies suggest that type 1 regulatory cell clones of both donor and
host origin can inhibit the function of effector T cells of either
donor or host origin in vitro.[6] These results suggest that Treg cells could be associated with PMC.
Normal
homeostasis of the erythropoietic system requires an appropriate
balance between the rate of erythroid cell production and red blood
cell destruction. Growing evidence indicates that apoptotic mechanisms
play a relevant role in the control of erythropoiesis under physiologic
and pathologic conditions.[11] We hypothesized that
Fas might contribute to the cell death of SS erythroid precursors. The
two questions, how two different erythroid populations may exist
together during erythropoiesis in the bone marrow of PMC patients and
if T, B, or other lymphocyte subsets, are responsible for allowing this
persistent and stable chimerism, remain to be answered.
Methods
Transplant Protocol. According to the clinical protocol approved by the local institutional review board, the patient received BM from her HLA-matched healthy sister (Hb AA) after a conditioning regimen based on 14 mg/kg busulfan (Bu), 200 mg/kg cyclophosphamide (Cy), and 10 mg/kg anti-thymocyte globulin (ATG). For prophylaxis against GVHD, the patient received cyclosporine (starting on day −2) and short methotrexate (MTX) (10 mg/m2 on post-transplant days 1, 3, and 6 with folinic acid rescue). The course after allogeneic hematopoietic stem cell transplantation was uneventful, with the rapid hematologic engraftment and no signs of acute or chronic GVHD. The clinical characteristics of the patient and donor, and the regimen used in the preparation for the transplant are summarized in Table 1.
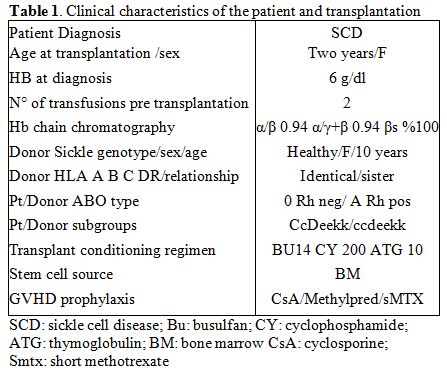 |
Table 1. Clinical characteristics of the patient and transplantation |
Laboratory tests. Chimerism analysis of nucleated cells and burst-forming unit-erythroid colonies.
Peripheral blood and bone marrow samples were collected in EDTA on days
20, 60, and 180 after the transplant, and thereafter during the annual
routine follow-up examinations. DNA samples were extracted using the
QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA) or an automatic
DNA extractor (Promega, Madison, WI, USA). The DNA was typed by short
tandem repeats (STR) and the amelogenin locus using the AmpFISTR
Profiler Plus kit (Applera, Foster City, CA, USA). Amplification
reactions were carried out using 1–2 ng of input DNA following the
manufacturer’s recommendations. Polymerase chain reaction products were
run on an ABI Prism 3130xl Genetic Analyzer (Applera, Foster City, CA,
USA). Informative loci in post-transplant samples were screened to
quantify the percentage of donor cells in mixed chimeras. HSCT
engraftment was quantified using fluorescent polymerase chain reaction
primers for human identity markers based on the ratio between the peak
areas of donor and recipient alleles. The mean value obtained after
performing calculations for each informative STR was taken as the
percentage of mixed chimerism. Burst-forming unit-erythroid (BFU-E)
colonies were grown in agar and picked out singly for STR evaluation.[2]
Clonogenic assay. Assays for clonogenic hematopoietic progenitors were performed in methylcellulose semisolid cultures. Briefly, 1–2×105
low-density bone marrow/peripheral blood cells were plated in duplicate
in 35-mm tissue culture dishes, and suspended in 1 mL methylcellulose
medium supplemented with stem cell factor, granulocyte/macrophage
colony-stimulating factor, interleukin-3, and erythropoietin (Methocult
GFH4434, Stem Cell Technologies, Vancouver, British Columbia, Canada).
Cultures were incubated at 37°C in a fully humidified atmosphere
containing 5% CO2. Plates were scored
for BFU-E growth after 14 days of incubation. Using an inverted
microscope, individual colonies were picked up from the Petri dishes
and dispersed to single cell suspensions in 100 μL saline to assess the
donor/recipient origin of the individual colonies using STR.
Chimerism of red blood cells.
For cytofluorimetric analysis, red blood cells (RBCs) were washed and
diluted in saline (0.5% final dilution). Five microliters of cell
suspension were incubated with anti-ABO and anti-C, -c, -D, -E and -e
monoclonal antibodies, following the manufacturer’s instructions (ABH-
and RH- Erythrokit, Institute Jacque Boy SA, Reims, France). After the
incubation, cells were washed with phosphate-buffered saline (PBS) and
incubated with fluorescein isothiocyanate-conjugated anti-human
immunoglobulin. After the incubation and two additional washes, the
analysis was performed using a FC500 flow cytometer and transferred to
the CXP analysis program (Beckman-Coulter Hialeh, FL, USA).
Evaluation of FAS and Treg in bone marrow and peripheral blood mononuclear cells.
Whole blood and bone marrow specimens were obtained to evaluate T, B,
NK, and FAS on erythroblasts: anti-CD3 FITC, anti-CD4 APC, anti-CD8 PE,
anti-CD45 PercP Cy 5.5, and anti CD45 RA PE Cy7 were mixed in the first
tube; anti-CD16 FITC, anti-CD56 PE, anti-CD45 PercP Cy5.5, anti CD3 PE
Cy7, and anti CD19 APC in the second tube; and anti-CD95 PE, anti-CD71
FITC and anti-CD45 PercP Cy5.5 in the third tube. A volume of 10 µl of
these MoAb cocktails (BD, Becton Dickinson, San Diego, C.A., USA) were
combined with 100 µl of blood for 10 minutes at room temperature, then
lysed with BD Pharm Lyse 1× for 20 minutes at room temperature and
washed with 2% PBS plus bovine serum albumin (BSA). Samples were
analysed with BD FACS Canto II and the software, BD FACSDiva. We
evaluated co-expression of CD39 in CD4+CD25 high T cells for analysis
of T reg cells. In humans, CD39 is a surface marker expressed almost
exclusively by Foxp3+ cells.[12]
We also compared Treg data of the patient with Treg data of not
transplanted SCD patient who was matched for age and sex.
Cytometric assay for erythroid cell precursors.
We previously developed a flow cytometric assay to identify
stage-specific erythroblasts directly in hematopoietic tissue (bone
marrow) based on their expression of the transferrin receptor (CD71),
which declines with erythroblast maturation. However, the decline in
CD71 appeared to be gradual, without the formation of well-resolved
subpopulations. In this study, we distinguished well resolved
erythroblast subpopulations by considering, in addition to CD71, the
forward scatter (FSC) parameter. FSC is a function of cell size and has
been used previously to assess erythroblast maturation independently of
cell surface marker expression. When the cells are analysed using both
CD71 and FSC parameters, they consistently resolve into three principal
subpopulations, which we labelled Ery A, Ery B, and Ery C
erythroblasts. Ery A (CD71high FSChigh) are basophilic; Ery B (CD71high FSClow) are late basophilic and polychromatic; and Ery C (CD71low FSClow) are orthochromatic erythroblasts and reticulocytes.
We
examined potential apoptotic regulators of erythroblasts. Fas (CD95)
has been detected on cultured erythroblasts. We also compared Fas data
of the patient with Fas data of not transplanted SCD patient who
was matched for age and sex.
We examined whether Fas is
coexpressed within the same cells by labelling bone marrow cells
simultaneously with antibodies directed against Fas, as well as CD71.
We also examined CD95 expression in the RBC of the recipient and donor
at bone marrow and peripheral blood levels.
Results
Molecular analysis of sorted cell subgroups revealed mixed chimerism
in nucleated cells, CD34+ progenitors, and RBCs in the PB and BM. Four
years after transplantation, the level of donor nucleated cells (NC)
was 39% in PB and 36% in the BM in parallel with a very high proportion
of donor-derived RBCs (80%) in the PB. The proportion of donor-derived
RBCs and BFU-E in the BM was 40% and 46%, respectively, indicating the
presence of quantitatively different red cell/nucleated cell chimerism (Figure 1).
Hb
electrophoresis on peripheral blood four years post-transplantation
showed HbA1 97,2 % Hb A2 2,8 %, without any traces of HbS.
The same haemoglobin phenotypes were observed on bone marrow examination. The value of HB post-transplant was 13 g/dl.
We
also examined potential apoptotic regulators of erythroblasts,
evaluating Fas (CD95) expression on cultured erythroblasts and red
cells from BM and PB. We found that Fas was expressed by a
significantly higher proportion of host erythroblasts, especially in
late basophilic and polychromatic erythroblasts, and in host RBCs from
BM (Figure 1).
An increase
in Treg was also observed in the patient with respect to the control.
From BM, we observed an increase in cytotoxic CD3-CD16+ NK cells (Figure 2).
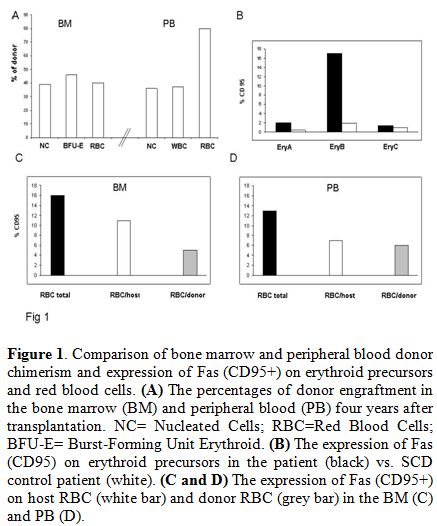 |
Figure 1. Comparison of bone marrow and peripheral blood donor chimerism and expression of Fas (CD95+) on erythroid precursors and red blood cells. (A) The percentages of donor engraftment in the bone marrow (BM) and peripheral blood (PB) four years after transplantation. NC= Nucleated Cells; RBC=Red Blood Cells; BFU-E= Burst-Forming Unit Erythroid. (B) The expression of Fas (CD95) on erythroid precursors in the patient (black) vs. SCD control patient (white). (C and D) The expression of Fas (CD95+) on host RBC (white bar) and donor RBC (grey bar) in the BM (C) and PB (D). |
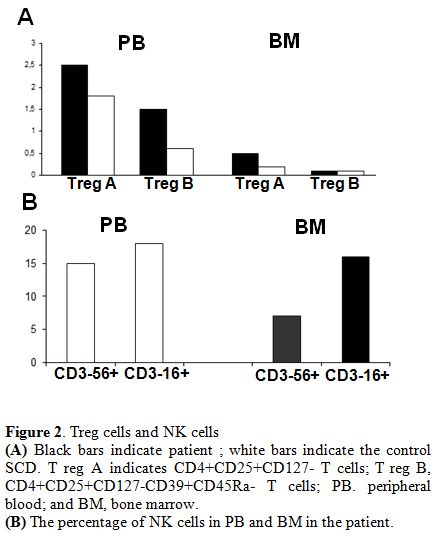 |
Figure 2. Treg cells and NK cells (A) Black bars indicate patient ; white bars indicate the control SCD. T reg A indicates CD4+CD25+CD127- T cells; T reg B, CD4+CD25+CD127-CD39+CD45Ra- T cells; PB. peripheral blood; and BM, bone marrow. (B) The percentage of NK cells in PB and BM in the patient. |
Discussion
In the hematopoietic system, the percentage of donor NCs correlates
with that of erythroid cells, consistent with the current understanding
of myelo- and erythropoiesis deriving from common myelo-erythroid
progenitors.
Furthermore, the percentage of donor RBC in
the PB was much higher, supporting the hypothesis that donor and host
erythroid precursors were at a competitive disadvantage for generating
mature red blood cells. A possible explanation for the presence of a
greater proportion of donor-derived RBC may be the improved survival of
donor erythroid late precursors compared with the host counterparts,
which might be destroyed during ineffective erythropoiesis. Apoptosis
is an important mechanism by which ineffective erythroblasts are
cleared within the intramedullary space, and our data suggest that Fas
might contribute to the cell death of host erythroid precursors. The
high level of polymerization of the sickle hemoglobin in host RBCs as
well as in the host, early and basophilic, normoblasts might also
determine mechanical defects that in turn increase the host cells’
susceptibility to clearance and loss.
The higher percentages of CD3-CD16+
NK cells in a mixed chimerism patient may play a role in control of
host host-cell escape and in maintaining the chimerism condition.
Pioneering studies by Velardi and colleagues revealed that patients
with acute myelogenous leukemia transplanted from an NK alloreactive
donor benefited from higher rates of engraftment and reduced rates of
GVHD.[8] The virtual abrogation of GVHD may be a consequence of NK cell-mediated killing of recipient antigen-presenting cells.[9,10]
The beneficial effects could also be related to depletion of patient
antigen-presenting cells and facilitation of engraftment as a result of
the killing of T cells, removing patient lymphohematopoietic cells, and
production of growth factors required for engraftment and for
accelerating recovery of myelopoiesis.
The increase in Treg
populations, especially in peripheral blood, suggests that these cells
may play an important role in sustaining long-term tolerance in vivo.
Our
observation, confirming the presence of split chimerism between RBCs
and their erythroid precursors, supports the concept that a limited HSC
engraftment can provide a sufficient amount of normal hemoglobin and
mature erythrocytes, and that this limited engraftment can inhibit the
expansion of HbS-erythropoiesis.
Our results support the
evidence that low levels of donor engraftment can result in significant
functional improvement for patients with SCD. The observation that a
few engrafted cells are sufficient to clinically control patients with
SCD is particularly interesting in light of a possible gene therapy
approach or stem cell transplantation in adult patients.
References

[TOP]