Received: June 12, 2014
Accepted: October 18, 2014
Meditter J Hematol Infect Dis 2014, 6(1): e2014072, DOI 10.4084/MJHID.2014.072
This article is available on PDF format at:

Stacy Colaco, Reema Surve, Pratibha Sawant, Anita Nadkarni, Kanjaksha Ghosh and Roshan Colah
National Institute of Immunohaematology, Indian Council of Medical Research, 13th Floor, New Multistoried Building, King Edward Memorial Hospital Campus, Parel, Mumbai – 4000 12.
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Background: Haemoglobinopathies
are the commonest hereditary disorders in India and pose a major health
problem. Both beta thalassaemia and structural haemoglobin variants are
relatively common in northwestern India. Here we report a 29-year-old
Sindhi female who was referred to us for a haemoglobinopathy work up
and genetic counseling since her spouse was a classical beta
thalassaemia carrier. Method: A complete blood count was done on an automated cell counter. Haemoglobin analysis was carried out using HPLC Variant Haemoglobin Testing System. The cellulose acetate electrophoresis was carried out [pH 8.9]. Confirmation of mutations was done by automated DNA sequencing. Results: HPLC analysis showed four major peaks, HbA0, a peak in the HbD window, an unknown peak [retention time 4.74 minutes] and a peak in the HbC window. The HbA2 level was 2.2%, and the HbF level was 0.7%. Cellulose acetate electrophoresis at alkaline pH, a slow moving band was seen at the HbS/D position along with a prominent band at the HbA2 position. DNA sequencing of the β and α genes showed presence of the two hemoglobin variants: Hb D [β 121GAA → CAA] and Hb Q [α 64 AAG → GAG]. The δ globin gene was normal. The additional peak in the HbC window was due to the formation of a heterodimer hybrid. Conclusion: Both HbD Punjab and HbQ India are relatively common in India, but their co-inheritance has not been described in the country. This case is the third report of compound heterozygosity for HbQ India/HbD Punjab haemoglobinopathy globally and the second one from India. |
Introduction
Inherited abnormalities of haemoglobin synthesis include a myriad of disorders ranging from the thalassaemia syndromes to structurally abnormal haemoglobin variants. Identification of these abnormalities is immensely important epidemiologically and aid in the prevention of more severe haemoglobin disorders.[1] Haemoglobinopathies resulting from mutations in the α or β globin gene clusters are the most common inherited disorders in humans. Single nucleotide substitutions can lead to amino acid replacements that cause haemolytic anaemias, such as sickle cell disease, or haemoglobins that are unstable or have altered oxygen affinity.[2] Molecular defects in either regulatory or coding regions of the human α, β or δ globin genes can minimally or drastically reduce their expression, leading to α, β or δ thalassaemia.[3] Other sequence changes have little or no effect on haemoglobin function, but are useful polymorphisms for genetic studies. About 7% of the world population carries a globin gene mutation, and in the vast majority of cases it is inherited as an autosomal recessive trait.[1] To date, over 1,200 different mutant alleles have been characterized at the molecular level4 and each country has its own spectrum of Hb variants and thalassemia mutations.[2] Here, we describe a very rare compound heterozygous haemoglobinopathy, HbD Punjab/HbQ India [ααQ India ββD Punjab], which has been reported only two times in the literature to date, to the best of our knowledge.[5,6]
Materials and methods
Results
The hematological analysis of the patient showed a marginal decrease in the haemoglobin level with normal red cell indices (Table 1). Reticulocyte count was 1.5%. HPLC analysis, using the β thalassaemia short program, showed a peak of 31.5% with a retention time of 4.06 minutes in the Hb D window, followed by an unknown peak of 9.4% with a retention time of 4.74 minutes as well as a peak eluting in the Hb C window [7.4%] with a retention time of 4.98 minutes. The unknown peak at 4.74 minutes was suggestive of Hb Q India (Fig 1A). Cellulose acetate electrophoresis at alkaline pH [8.9] showed three bands viz Hb A, a band at the S/D position and Hb A2 (Fig 1B). Confirmation of absence of HbS was done by sickling test that was negative. α globin gene analysis showed the absence of the common α thalassaemia determinants and absence of α globin gene triplication. Direct DNA Sequencing of the α globin gene revealed the presence of HbQ India [α 64Asp→His] and sequencing of the β globin gene showed the presence of HbD Punjab [β121Glu→Gln]. The δglobin genes showed absence of any mutation.
 |
Table 1. Hematological analysis of the patient |
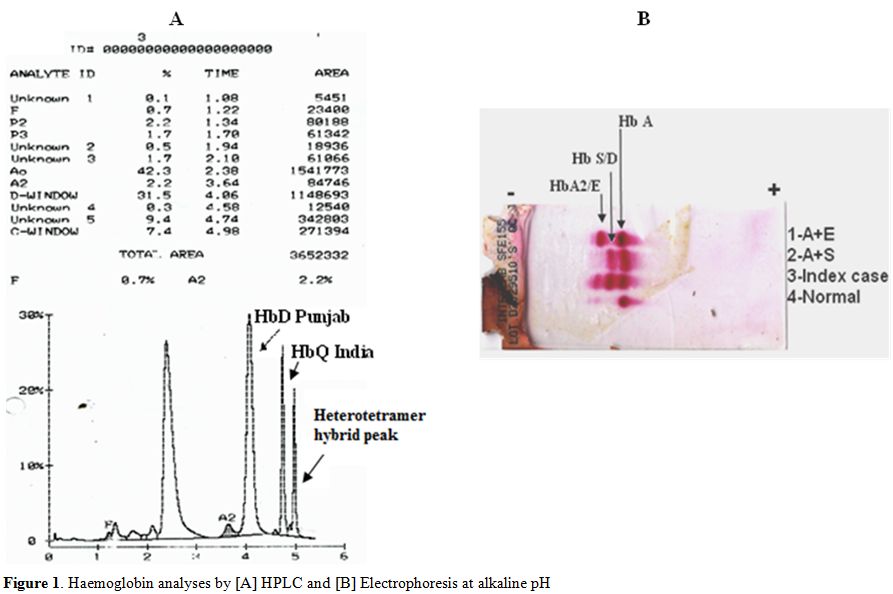 |
Figure 1. Haemoglobin analyses by [A] HPLC and [B] Electrophoresis at alkaline pH |
Discussion
Genomic research has rapidly evolved during the past decades, and
the scientific approach to genetic-based understanding of human disease
has changed along with technological development. This led to the
discovery of a large list of Hb variants. Here we report a very rare
and unusual case of double heterozygosity of the α chain variant HbQ
India with the non-α chain variant HbD Punjab. The relative
electrophoretic mobility of HbD Punjab and HbQ India are practically
indistinguishable during routine screening on alkaline electrophoresis.[9]
Higgins et al.,[5]
reported in 2012 the first case of a HbD Punjab/HbQ India compound
heterozygosity in female of Indian origin. Her investigation for
haemoglobinopathies showed four major peaks Hb A, HbD Punjab, HbQ India
and HbD Punjab/HbQ India. The heterotetramer hybrid peak eluting in the
HbC window reported by them was also seen in our case. In
electrophoresis at alkaline pH, Higgins et al.[5]
observed bands at the C [HbD Punjab/ HbQ India], S [HbD] and A [HbA]
positions, while the electrophoresis at acid pH showed two bands, at
the S position [HbD Punjab/HbQ India] and at the A position [HbA and
HbD Punjab]. Similar observations were reported in two Indian patients
double heterozygous for HbQInida/HbD Punjab by Mutreja et al. 2013.[6]
It
has been suggested that of the HbE and HbD mutations were originated in
India and dispersed to other parts of the world due to migration.[10] HbQ, a rare α chain variant, was first reported by Vella et al., in 1958 in a Chinese family.[11] Three variants of HbQ have been described, namely, HbQ-India, HbQ-Thailand [c.223G>C] and HbQ-Iran [c.226G>C].[2] The first case of HbQ India was reported by Sukumaran et al., in 1972 in a Sindhi family with associated β thalassaemia.[12]
The affected residue, α64 [E13], lies on the surface of the haemoglobin
tetramer and charge changes at this position do not alter the tertiary
structure of the haemoglobin molecule.[13] Hence, the presence of HbQ India does not impart any functional deficit and lacks any clinical manifestation.[14]
64 cases of HbQ India trait and its interaction with β thalassaemia have also been reported from India.[15] However, a few concerns have been raised for considering it as an entirely benign disorder; Panigrahi et al.,[16]
in 2005 reported mild anaemia and microcytosis in heterozygotes of HbQ
India and observed that apart from co-existent α globin/β globin
genotypes, iron deficiency also affects HbQ levels.
Das et al.,[6]
in 2013 studied the intra-ethnic heterogeneity of the
haemoglobinopathies in 1,498 Sindhis and observed the incidence of HbD
Punjab and HbQ-India to be 1.87% and 0.4% respectively. Giordano et al.[17]
reported in 2006 a β thalassemia heterozygote where the diagnosis of
high A2 β thalassemia was masked by the presence of an α chain variant,
HbJ Meerut. The presence of α chain variants leads to the generation of
split HbA2 peaks thereby reducing the HbA2 value by 20%. The generation of this variant might be the reason that HbA2 level in our case (2.2%) was lower than usual.
Rahimi et al.[18]
reported in 2007 an unusual case of an Iranian child with HbQ-Iran
[alpha75 (EF4) Asp→His] /- α 3.7 kb / βIVSII.1 G→A, who presented as a
minor beta-thalassemia with moderate anemia. In our case as the husband
of the propositus was a β thalassemia carrier, the couple was at risk
of having a child with double heterozygosity for HbD Punjab - β
thalassemia, or HbQ-India - β thalassemia or HbQ-India -Hb D
Punjab - β thalassemia. A combination of these hemoglobinopathies in
our experience does not lead to a severe disorder. Hence, the couple
was not advised to undergo prenatal diagnosis. However, when one of the
partners has a β thalassemia trait, it is always advisable to do
the full molecular work up of the spouse to ensure that they are not at
risk of having a child with a severe hemoglobin disorder.
Conclusion
Often unusual clinical presentations may be explained by the interaction of several Hb abnormalities and their identification may require further investigations. This is the third literature report of the compound heterozygosity of HbQ India/HbD Punjab. The couple was genetically counseled and was advised not to undergo prenatal diagnosis.
Acknowledgement
This work was funded by Indian Council of Medical Research (ICMR), New Delhi, India.Author’s contribution
All authors have contributed sufficiently to the project to be included as authors.References

[TOP]