Received: January 19, 2015
Accepted: February 10, 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015024, DOI 10.4084/MJHID.2015.024
This article is available on PDF format at:

Giovanni D’Arena1, Elvira Grandone2, Matteo Nicola Dario Di Minno3,4, Pellegrino Musto5 and Giovanni Di Minno3
1 Hematology and Stem Cell
Transplantation Unit, IRCCS Centro di Riferimento Oncologico della
Basilicata, Rionero in Vulture, Italy.
2 Hemostasis and Thrombosis Unit, IRCCS “Casa Sollievo della Sofferenza” Hospital, San Giovanni Rotondo, Italy.
3
Department of Clinical Medicine and Surgery, Regional reference Center
for Coagulation Disorders, Federico II University, Naples, Italy.
4 Unit of cell and molecular biology in cardiovascular diseases, Centro Cardiologico Monzino, IRCCS, Milan, Italy.
5 Scientific Direction, IRCCS Centro di Riferimento Oncologico della Basilicata, Rionero in Vulture, Italy.
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Acquired hemophilia A (AHA) is a rare bleeding disorder due to the development of specific autoantibodies against factor VIII. The anti-CD20 monoclonal antibody Rituximab has been proven to be effective in obtaining a long-term suppression of inhibitors of AHA, besides other immunosuppressive standard treatments. Here we describe a case of idiopathic AHA in a 60-year old man successfully treated with rituximab. He showed a complete clinical response with a normalization of clotting parameters after 5 weekly courses of rituximab given at a dose of 375 mg/sqm., but after stopping rituximab, an initial worsening of coagulation parameters induced the addition of 3 further courses. At present, the patient is in complete clinical and hematological remission after 200 days. This case confirms that Rituximab may be a safe and useful tool to treat AHA and, a prolonged administration can overcome the initial resistance. However, the precise position of this drug in the therapeutic strategy (first or second-line, alone or in combination with other drugs) remains to be established and warrants further investigation. |
Introduction
Rituximab is a chimeric human/murine monoclonal antibody targeting CD20 antigen on B-cell surface.[1]
It is extensively used to treat CD20 positive hematologic malignancies
and is now increasingly employed to treat several autoimmune
disorders. Acquired hemophilia A (AHA), is a rare bleeding
disorder caused by the development of specific autoantibodies, the
so-called inhibitors, against naturally occurring factor VIII (FVIII),
and has been treated with rituximab too.[2,3]
The treatment of such a disorder is aimed to control bleeding and to suppress inhibitors, as well.[4,5]
These results are usually obtained by using standard immunosuppressive
therapy (steroids, cyclophosphamide, azathioprine). Rituximab is
generally considered a second-line treatment option. About 171 patients
have been treated with this particular approach so far.
Here we
describe a paradigmatic patient with AHA, who experienced a complete
and long-lasting hematological response to Rituximab.
Case Report
A non-hemophilic 60-year old man was admitted to another Hospital
because of the sudden appearance of subcutaneous hemorrhage in his
upper right arm following minor trauma. Laboratory investigations
revealed a markedly prolonged activated partial thromboplastin time
(aPTT, 99 seconds,) (ratio 2.7), not corrected with normal plasma (1:1)
after a 2 hour incubation: (Prothrombin time (PT) was in normal range).
At the same time, heparin contamination, Lupus anticoagulants, and
other autoimmune diseases were excluded. Hemoglobin level was 11.2 g/dL
while platelets count was normal (252.000/μL). No o ther
biochemical or clotting system abnormalities were identified.
Antibodies against hepatitis B and C viruses and HIV were found
negative. Neoplastic biomarkers and a total body tomography
computerized scan were normal.
The clinical history was also negative for the use of drugs known to be associated with the development of AHA.
Consistent
with a diagnosis of AHA, the coagulation factor assay revealed that
FVIII levels were 2.6 %, with a titer of 4 Bethesda Units (BU), thus
confirming the presence of an acquired FVIII inhibitor.
The
patient initially received prednisone at a dose of 1 mg/Kg body weight
orally given and, due to acute bleeding stage, a treatment with rFVIIa
(Novoseven) was also started at a dose of 90 mcg/Kg every 2 hours
(4 doses) and then every 4 hours for 6 doses. Despite the low inhibitor
title at diagnosis, no response to corticosteroids was obtained. The
bleeding persisted notwithstanding the prolonged treatment with rFVIIa.
The patient was then sent to our Institution, where he initiated a
treatment with Rituximab at a dose of 375 mg/sqm weekly in combination
with prednisone. After the fifth dose, BU was undetectable and aPTT
normalized and bleding stopped. For these reasons, prednisone was then
slowly tapered, but at +57 days from the start of Rituximab therapy
aPTT was found again prolonged (40 s; ratio 1.4) and FVIII levels
reduced (27%) with 1.7 BU, in absence of any new hemorrhagic
manifestation. Prednisone was then reintroduced at the dose of 1 mg/Kg
and Rituximab given for 3 additional infusions, with normalization of
aPTT and disappearance of inhibitor since a week after the eighth
Rituximab infusion (Figure 1).
The patient stopped prednisone therapy at +150 days form start of
Rituximab without clinical signs of bleeding and normal clotting tests.
At the last follow-up (+200 days) the patients is still in clinical and
laboratory continue complete remission. Overall, Rituximab infusions
were well tolerated, without evidence of infusion and/or late
reactions. Finally, no infections have been reported so far.
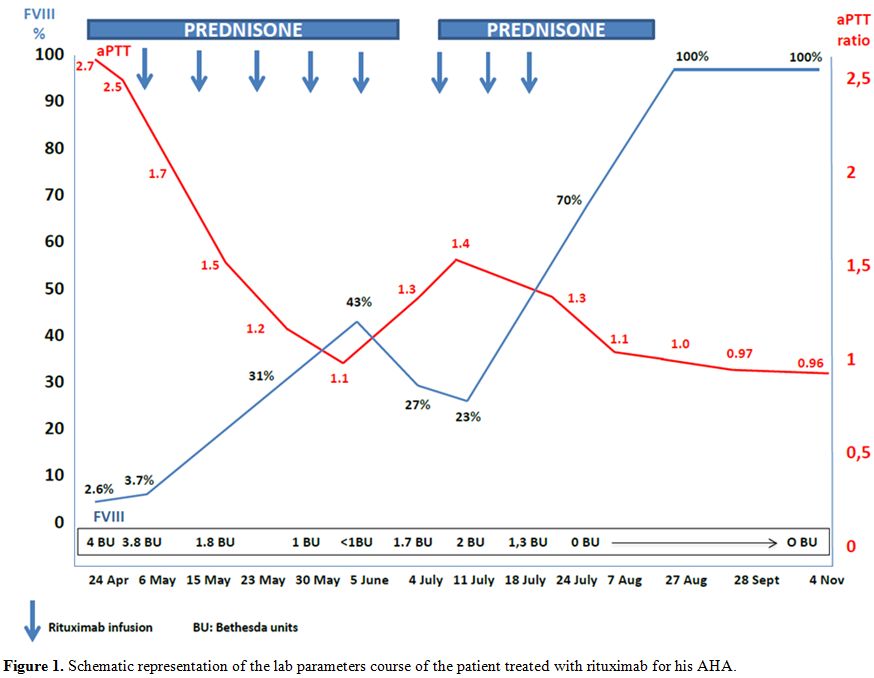 |
Figure 1. Schematic representation of the lab parameters course of the patient treated with rituximab for his AHA. |
Discussion
AHA is a rare bleeding disorder due to acquired autoantibodies
against FVIII. Its incidence has been estimated to be 0.2-1.0 case per
million population per year, but it is probably underestimated.[3]
Soft tissue bleeding manifestations are often severe and may occur
spontaneously or after minor trauma. Inhibitors are idiopathic in
nature in approximately half of patients. In the remaining cases,
various conditions are associated with FVIII inhibitors development,
such as connective tissue and inflammatory bowel diseases, puerperium,
malignancies, and dermatologic disorders.
An unexplained
prolongation of the aPTT, not corrected by the in vitro addition of
normal plasma (mixing test), is the typical laboratory feature of AHA.
FVIII level is reduced, and the presence of an inhibitor is revealed by
Bethesda assay. Treatment is aimed at the control of the acute bleeding
and the suppression of the inhibitor, as well. Acute bleeding is
managed through normalization of factor VIII level. Despite some
authors recommend choosing hemostatic treatment, such as recombinant
activated FVII (rFVII, Novoseven; Novo, Nordisk) and the activated
prothrombin complex concentrate (APCC, FEIBA; Baxter Healthcare),
according to the Bethesda assay (<5 BU or >5 BU, respectively),
some considerations need. As reviewed in detail by Tiede et al, in
contrast to congenital hemophilia, inhibitors in acquired hemophilia do
not follow a log-linear dose-response relationship, that is the basis
for quantification in Bethesda assay.[6] In addition,
an analysis of data from the EACH2 registry demonstrated that bypassing
agents are more effective than FVIII infusion.[5]
Human FVIII, porcine FVIII, and desmopressin are also used.
Immunosuppression is the mainstay to obtain inhibitor eradication.
Steroids alone or combined with cytotoxic agents (i.e.,
cyclophosphamide, azathioprine, vincristine, or combination therapy),
are also frequently used. The combined treatment of steroids with
cyclophosphamide is able to eradicate inhibitors in about 70% of
patients with AHA.
The monoclonal antibody Rituximab, is a chimeric antibody targeting CD20 antigen on B-cell surface.[1]
It has been reported to be effective in eradicating the inhibitors in
AHA and 172 patients with this condition have been treated so far,
including the present case. Rituximab has been used alone or in
combination with other immunosuppressive drugs, such as steroids and
cyclophosphamide, as salvage or first line-therapy. Overall, 157
patients (91%) showed a response, with 146 patients (85%) achieving
complete response and 20 patients (12%) partial response. Five patients
(3%) did not respond to rituximab therapy. The dose of Rituximab
infused in almost all cases was 375 mg/sqm, similar to that employed in
the lymphoma treatment. However, lower dose has been also used (100
mg). The number of rituximab infusions was very variable (from a single
low-dose to 12 standard doses). In addition, in the majority of cases 4
standard doses were the schedule used and the time to response was also
heterogeneous (from 1 week to more than one year).[7]
Of note, re-treatment with Rituximab was generally effective. Overall,
the administration of Rituximab in AHA was well tolerated with very few
infusional side effects, and no infectious complications have been
reported, so far. We choose to treat our patient with eight weekly
doses of rituximab according to the response to treatment (Figure 1).
The present case suggests that in certain circumstances a more
extensive course of rituximab infusions needs to reach a threshold of
circulating B-lymphocytes whose antibodies production is not able to
support the autoimmune disorder. In our patient, a B-cell number of
0.5/μL was achieved after the 8th doses, when a complete response was also seen (Table 1).
At variance of cases reported by Franchini,[7]
the rate of a stable remission is much lower in some published
reports: 71% by the European Acquired Haemophilia (EACH2) registry and
48% by the AHA Working group of the German, Austrian and Swiss
Thrombosis and Hemostasis society (GTH).[5,8] Finally, a treatment algorithm was proposed by Franchini and Mannucci very recently.[9]
Authors stated that first-line therapy for patients with AHA should be
the association of prednisone and cyclophosphamide while Rituximab ±
immunosuppressive therapy should be reserved for second-line treatment.
In
conclusion, the use of anti-CD20 monoclonal antibody Rituximab is
increasingly used to treat autoimmune disorders. In the case of AHA,
its definitive role in eradicating inhibitors (first or second-line,
high or low inhibitor titers, older and/or frail patients for whom
corticosteroid and cytotoxic therapy are unsuitable) requires further
evaluation. However, randomized-controlled trials are very hard to be
designed because of the rarity of AHA. Notwithstanding additional data
have to be acquired in order to optimize the use and to compare the
efficacy of this drug to that of other treatments, current evidences
confirm that Rituximab should be considered as part of the therapeutic
armamentarium of AHA.
 |
Table 1. Circulating lymphocytes and B-cell number before and after Rituximab infusions. |
References

[TOP]