Received: September 22, 2015
Accepted: January 11, 2016
Mediterr J Hematol Infect Dis 2016, 8(1): e2016015, DOI 10.4084/MJHID.2016.015
This article is available on PDF format at:

Abdel Badih El Ariss1, Mohamad Younes2, Jad Matar3 and Zeina Berjaoui4
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Objective: The objective of
this study was to assess the prevalence, gender differences, and time
trends of Sickle Cell Trait in the Southern Suburb of Beirut, Lebanon,
as well as to highlight the importance of screening for Sickle Cell
Trait carriers in this population. Another objective was to describe a
new screening technique for Sickle Cell Trait carriers. Methods: This was a retrospective cohort study carried out at a private laboratory in the Southern Suburb of Beirut, Lebanon between 2002 and 2014. The sickling test was carried out for each patient using two methods: the classical “sodium metabisulfite sickling test”, and the new “sickling test method” used in the private lab. As a confirmatory test, hemoglobin electrophoresis was run on a random sample of 223 cases which were found to be positive using the two sickling tests. Results: A total of 899 cases were found to be positive for the sickle cell trait out of 184,105 subjects screened during the 12-year period, prevalence = 0.49% (95% CI: 0.46 – 0.52). Among the total sample, females were found to have higher prevalence, where no time trend over the studied period was noted. The haemoglobin electrophoresis method confirmed the results of this new sickling test technique among the random sample of the 223 cases. Conclusion: We found that the prevalence of sickle cell trait is lower as compared to other Arab countries, higher in females, with no significant time trend. The sickle cell test was found to be an accurate, simple and cheap test that could be easily added as a requirement for the pre-marital testing to screen for Sickle Cell Trait carriers. |
Introduction
Sickle syndromes include several inherited diseases that cause red
blood cells to sickle in vivo, where the most recognized ones are
Sickle Cell Disease (SCD), Sickle Cell Trait (SCT), and Sickle
Cell/Thalassemia (S/β thalassemia)[1] Phenotypically,
only persons with double recessive genes of sickle cell homozygotes
(SS) do manifest disease, while the heterozygotes (AS) are being
referred to as Sickle Cell Trait carriers.[2] It is
estimated that each year more than 330,000 children are born worldwide
with haemoglobinopathies, 83% of which are sickle cell disorders.[3]
Sickle
Cell Disease is a disorder of haemoglobin synthesis that results from
the substitution of glutamic acid at the sixth position of its β-globin
chain by valine (HbS).[4] It is also known as
sickle-cell anemia (SCA) and is characterized by an abnormality in the
oxygen-carrying haemoglobin molecule in red blood cells.[5]
It is one of the most common haemoglobinopathies in Africa, the Middle
East and India. Sickle cell disease is now found throughout the world,
and its incidence has increased in Europe and North America because of
the high rate of migration from areas in which the disease is
prevalent.[3,6] In 2006, the World Health Organization (WHO) recognized sickle cell disease as a global public health problem.[7]
According to Diallo et al., Africa is the most highly affected
continent with 200,000 newborn affected by sickle cell disease per
year.[2] In the United States, sickle cell disease affects about 72,000 people.[8]
Moreover, Piel et al. expected that the annual global number of
newborns with sickle cell disease will increase from 305,800 patients
in 2010 to 404,200 patients in 2050 (about one-third increase).[9]
The clinical manifestations of the sickle cell disease are diverse
where any organ system may be affected. These signs are commonly
divided into vaso-occlusive (where the episodes of pain, acute chest
syndrome, splenic infarction, stroke, and avascular necrosis of joints
predominate) and hematologic (where severe anemia, leg ulcers, and
pulmonary hypertension predominate).[10,11] Early
diagnosis and treatment can ameliorate the course of these diseases and
improve survival. More specifically, prophylactic penicillin,
immunizations, comprehensive care, and parental education about serious
sickle cell disease complications has been shown to significantly give
a better survival rate and quality of life for sickle cell disease
patients.[12,13]
Sickle Cell Trait is present in
approximately 300 million people worldwide, with the highest prevalence
of approximately 30% to 40% in sub-Saharan Africa.[11]
Despite
the benign nature of the Sickle Cell Trait, several potentially serious
complications have been described with it including, gross hematuria,
increased urinary tract infection in women, complications of hyphema,
splenic infarction with altitude hypoxia or exercise, and
life-threatening complications of exercise.[14] Hematuria is the most common manifestation of sickle cell trait.[15]
Spontaneous sickling can occur in the renal papilla (normally under low
oxygen pressure) in patients with renal papillary necrosis; therefore,
5% of patients with sickle cell trait can suffer episodes of hematuria
at some point during their lives.[16,17] Studies in
America, England and Jamaica reported that the rates of urinary tract
infection are higher for women with sickle cell trait in comparison to
racially matched controls.[14] This finding is well
established for asymptomatic bacteriuria of pregnancy, in which the
rate is approximately doubled with sickle cell trait.[18]
People with sickle cell trait are more susceptible to complications
following treatment of hyphema. The slow flow of relatively hypoxic
fluid in the chamber of the eye out of the filtration apparatus is a
location in which both polymerization of hemoglobin S and obstruction
of flow by rigid erythrocytes is likely. All this can result in
glaucoma and secondary hemorrhage.[14] Splenic
Infarction from sickle cell trait is favoured by exercise at high
altitude but has occurred with altitude exposure at rest or during
exercise at sea level.[11] The spleen is susceptible
to vaso-occlusion related to hemoglobin S polymerization and red cell
deformation. When subjects with hemoglobin S are exposed acutely to
high altitude hypoxia, the spleen is the organ most consistently
injured by microvascular obstruction.[19] Moreover,
there has been growing concern about the possible association of Sickle
Cell Trait and exercise-related morbidity and mortality in healthy
young college athletes due to the effects of extreme physical exertion,
dehydration and relative hypoxia (typically at high altitudes).[1]
The risks of microvascular complications in Sickle Cell Trait carriers
in response to exercise are dependent on alterations in blood rheology
and vascular adhesion processes. These abnormalities include higher red
blood cell rigidity and higher blood viscosity in the sickle cell trait
carriers compared with the non-carriers, particularly 24 and 48 hours
after exercise.[20]
Pre-marital screening to
identify carriers of genetic disorders, especially those with
recessively inherited diseases, has been shown to decrease the
incidence of inherited Sickle Cell Disease.[21] This
approach is more needed for countries with high prevalence of carriers,
such as the Mediterranean region. Various diagnostic tests are
available to differentiate between blood samples from normal healthy
individuals, Sickle Cell Trait carriers and Sickle Cell Disease
patients. Examples of screening tests include sickling test, solubility
test, and alkaline haemoglobin electrophoresis on agarose gel.[22,23]
On the other hand, high specific diagnostic tests include isoelectric
focusing, citrate agar electrophoresis, and high-performance liquid
chromatography, as well as the capillary method.[22,24]
A
study carried out by Khoriaty et al. screened Lebanese neonates for
Sickle Cell Disease and other Hemoglobin variants in all public
hospitals in Lebanon, between 2010 and 2013. Among newborns, 0.1% were
found to have Sickle Cell Disease, and 2.1% were found to have an
abnormal Hb variant with HbS being the most common (84.4% corresponding
to 1.77% of the studied population) which were distributed in all the
regions. The carrier rate of Hb variant varried amongst regions,
between 5.7 and 50.2%, being the highest in Northern Lebanon, whereas
it is 13.2% (0.27% of the studied population) in Beirut.[25]
This
study was conducted to assess the prevalence of Sickle Cell Trait among
a sample from a private laboratory (Modern Medical Center / Modern
Laboratory) in the Southern Suburb of Beirut, as well as to assess any
time trends over a 12-year period, and any differences in gender.
Moreover, the importance of screening for Sickle Cell Trait carriers in
this population is also highlighted. Another objective was to
describe a new “sickling test method” for the screening of Sickle Cell
Trait carriers, as well as to assess the correlation of the results
with them of the “sodium metabisulfite sickling test”, and to confirm
findings with the hemoglobin electrophoresis technique (Sebia
Capillarys).
Methods
Study design and setting:
This research was a retrospective cohort study using data from a
private laboratory (Modern Medical Center / Modern Laboratory,
established in 1963) in the Southern Suburb of Beirut, Lebanon from
2002 till 2014. Population served by this laboratory includes
mainly subjects from a Southern suburb of Beirut, irrespective of their
socio-economic status. All patients coming for a complete blood count
with differential (CBCD) underwent a free-of-charge sickling test.
Inclusion and exclusion criteria:
Eligible for this study were all subjects referred to our laboratory
for CBCD testing, without any exclusion based on age, gender,
socioeconomic status or MCV findings. The sample included in our
analyses comprised all eligible subjects, where no sampling was
applied. Moreover, repetitive results for same patients were
excluded.
Blood testing method:
Blood samples (3 ml) were drawn from patients using EDTA
(Ethylenediaminetetraacetic acid) tubes. As a primary screening
procedure, the sickling test was carried out for each patient using two
methods. In the first method (the new “sickling test method”), one drop
of blood was placed on a slide and a cover slide which was sealed using
nitrocellulose dissolved in butyl acetate usually found in nail
varnish. The slides were incubated at 37°C for 24 hours. As for the
second method (“sodium metabisulfite sickling test”), one drop of 2%
Sodium metabisulfite was added to a drop of blood between a slide and a
cover slide, sealed using the same sealing method and incubated at 37°C
for 10 minutes or at room temperature for one hour. For both sets, the
slides were inspected under the microscope (Olympus, dry objective 40),
and the red blood cells found to be sickling in shape were indicative
of a positive sickling test (Figure 1-2).
Patients found to be positive were notified of their condition and
advised to confirm their results by haemoglobin electrophoresis.
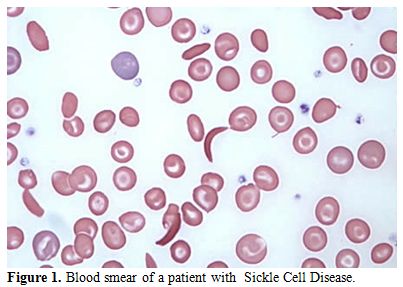 |
Figure 1. Blood smear of a patient with Sickle Cell Disease. |
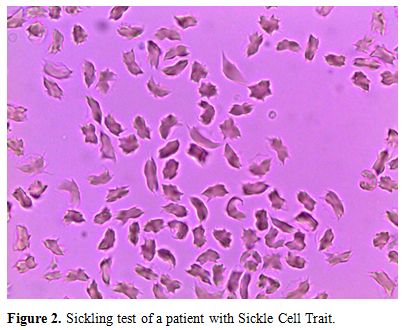 |
Figure 2. Sickling test of a patient with Sickle Cell Trait. |
As a confirmatory test, haemoglobin electrophoresis was run
on a random sample of 223 subjects who were found to be positive using
the sickling test. Another sample of whole blood was drawn from
these patients in EDTA tube, and then the red cells were separated from
plasma by centrifugation. The Capillarys, an automated capillary
electrophoresis by Sebia (Sebia, Surrey, UK), was used in the
separation of haemoglobins. The sample was placed in a precise
position required by the machine, as well as a red blood cell (RBC)
lysis solution at position 27 of the machine and control run
simultaneously. Capillarys uses buffer- filled, narrow bore capillaries
and detects haemoglobin fractions using UV-visible at 200 nm.
Separation occurs according to the electrolyte pH and electroosmotic
flow. The test can detect abnormal levels of HbS, the form associated
with Sickle Cell Trait and Sickle Cell Disease, as well as other
abnormal hemoglobin-related blood disorders.
Data Collection:
Data were collected by extracting the information from the electronic
and paper files. For the analysis of the prevalence, information
available were a date, age, gender, and MCV. On the other hand, for the
confirmatory analysis, information collected included age, gender, and
the hematologic parameters such as RBC, Hb (haemoglobin), Hct
(hematocrit), MCV (mean corpuscular volume), MCH (mean cell
hemoglobin), and MCHC (mean corpuscular hemoglobin concentration).
Moreover, information about different types of haemoglobin: HbA1
(haemoglobin, alpha 1), HbA2 (hemoglobin, alpha 2), HbF (fetal
hemoglobin) and HbS (hemoglobin S) were included.
Ethical Considerations:
Patients enrolled in our study provided oral consent for the tests to
be performed, as well as to be included in the study. The sickling
tests were carried out free of charge. Moreover, institutional
review board (IRB) of the American University of Science and Technology
(AUST) in Lebanon was obtained. Data collected were kept
confidential at the principal investigator’s office.
Statistical analyses:
Data entered into a Microsoft Excel spreadsheet and then transferred
into the Statistical Package for Social Sciences (SPSS) version 21,
which was used for data cleaning, management and analyses. Descriptive
analyses were carried out by presenting the number and percent for
categorical variables, whereas mean and standard deviation were
performed for continuous ones. The prevalence of the sickle cell
trait was calculated along with the 95% confidence interval (95%
CI). The difference in the prevalence of sickle cell trait, as
well as the other hematological parameters between males and females,
were assessed by using the students t-test. Statistical
significance was identified at 0.05 level.
Results
During the 12-year period between January 2002 and December 2014, a total of 184,105 subjects were screened for sickle cell trait. Over this period, 899 patients were found to be positive for the sickle cell trait, which yielded an overall 12-year sickle cell prevalence of 0.49% (95% CI: 0.46–0.52). There was a decrease in prevalence between 2002 from 0.75% (95% CI: 0.61- 0.90) to 0.37% (95% CI: 0.28-0.47) in 2009 and then an increase was found from 0.40% (95% CI: 0.30-0.51) in 2010 to 0.56% (95% CI: 0.43-0.70) in 2014 (Table 1 and Figure 3). Moreover, it was found that 325 males were positive for the sickle cell trait, out of 92,053 subjects yielding a prevalence of 0.35% (95% CI: 0.32-0.39), whereas, 574 out of 92,053 were females yielding a prevalence of 0.62% (95% CI: 0.57-0.67).Table 2 presents the distribution of sickle cell trait by gender and year. Among males, there was a decrease in prevalence of sickle cell trait between 2002 (0.54%, 95% CI: 0.37-0.72) and 2009 (0.15%, 95% CI: 0.07-0.24). On the other hand, the sickle cell trait prevalence increased during the period between 2010 and 2014, where the highest prevalence was found to be in 2012 (0.55%, 95% CI: 0.38 - 0.72). As for females, there was a decrease in prevalence of sickle cell trait from 0.96% in 2002 (95% CI: 0.73-1.12) to 0.46% in 2004 (95% CI: 0.30-0.61). We did not find any changes in the trends of sickle cell trait prevalence between the years 2005 and 2014. The highest prevalence during this period was found to be in 2012 (0.76%, 95% CI: 0.55-0.96), and the lowest was found to be in 2008 (0.45%, 95% CI: 0.30-0.61). Worth noting is the finding that the prevalence of sickle cell in each year was lower among males compared to that among females. In some years this difference was statistically significant: p-values of 0.004, 0.01, 0.004, 0.0002, <0.0001, 0.04, 0.009, and 0.04 for the years 2002, 2003, 2005, 2007, 2009, 2011, 2013, and 2014, respectively. These trends are presented in figure 4.
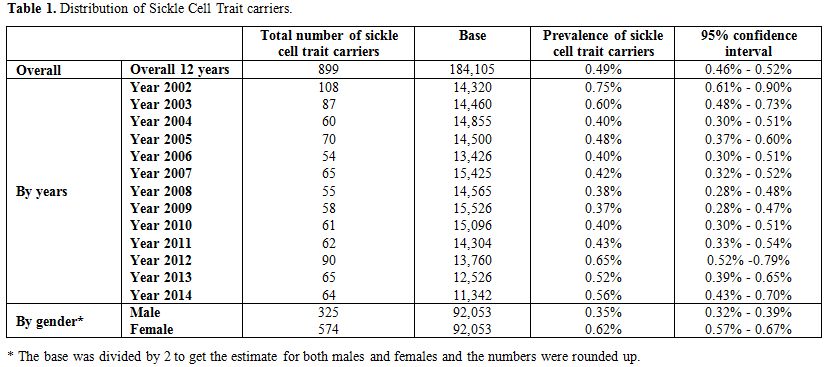 |
Table 1. Distribution of Sickle Cell Trait carriers. |
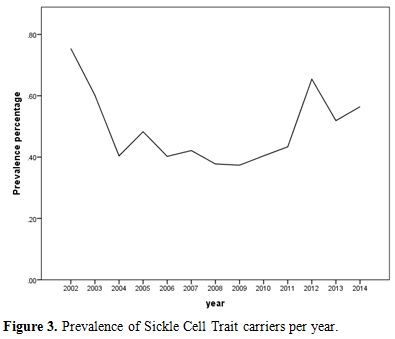 |
Figure 3. Prevalence of Sickle Cell Trait carriers per year. |
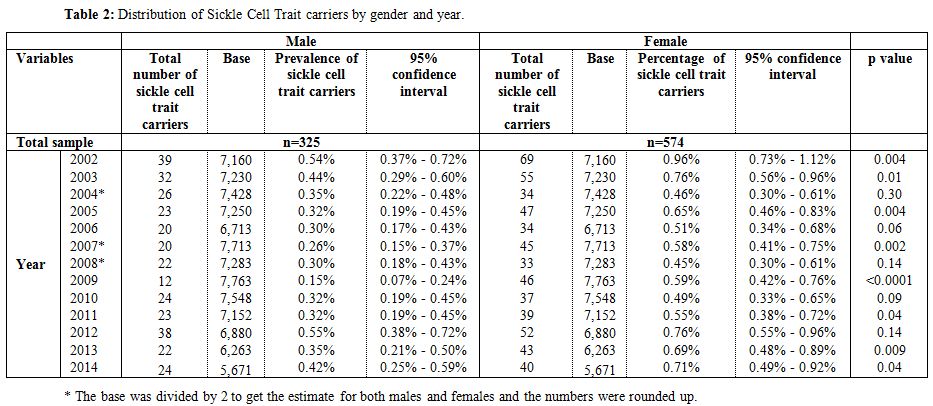 |
Table 2. Distribution of Sickle Cell Trait carriers by gender and year. |
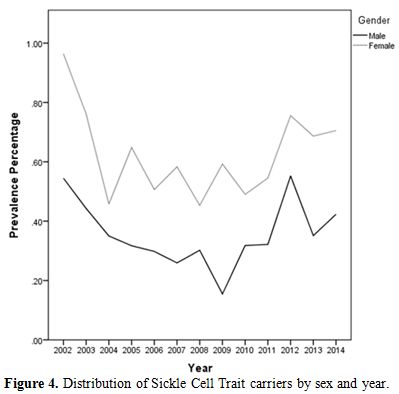 |
Figure
4. Distribution of Sickle Cell Trait carriers by sex and year. |
Table 3 presents the
average MCV by year and gender. The overall average MCV was found to be
84.3 (±7.6). Stratified by gender, the average MCV for males and
females were found to be 84.7(±7.4) and 84.1 (±7.8), respectively, for
which the difference was not statistically significant
(p-value=0.24). The average MCV for the different years ranged
between 81.8 (±8.3) in 2006 and 86.9 (±9.2) in 2011. The
differences in MCV between the genders in the years2002 and 2004 were
found to be statistically significant with p-values of 0.01 and 0.02,
respectively.
The demographic and hematological characteristics
of the 223 cases of positive sickle cell trait which were confirmed by
the Hb electrophoresis are summarized in Table 4.
The average age of patients was found to be 24.9 years (±15.6) with
40.8% being males. The values of the laboratory results were as
follows: RBC=4.7 million/ul (±0.6), Hb=12.9 g/dl (±7.6), Hct=38.1%
(±5.1), MCV=80.6 fL (±9.1), MCH=26.3 pg (±3.1), MCHC=32.5 g/dl (±1.1),
HbA1=57.3% (±7.6), HbA2=2.8% (±0.5), HbF=0.6% (±1.8), and HbS=38.8%
(±5.3).
Finally, Table 5
presents the differences in age and the different hematological
parameters between males and females, for the 223 cases with positive
HbS screening. Females were older than males with the average age being
26.9 years (±14.3) and 22.1 years (±17.0) for females and males,
respectively, p-value=0.03. A statistically significant difference
between males and females was found in the RBC, Hct, and HbA2, the
males having highest levels. The p-values were <0.0001 for each of
the parameters.
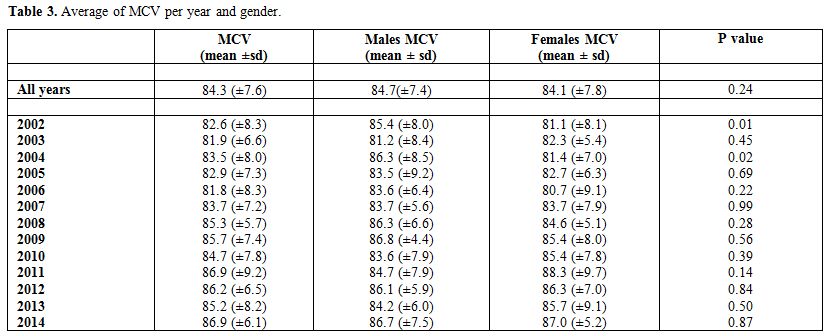 |
Table 3. Average of MCV per year and gender. |
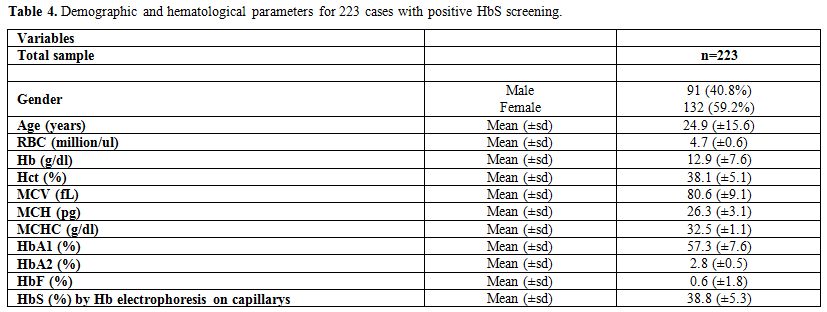 |
Table 4. Demographic and hematological parameters for 223 cases with positive HbS screening. |
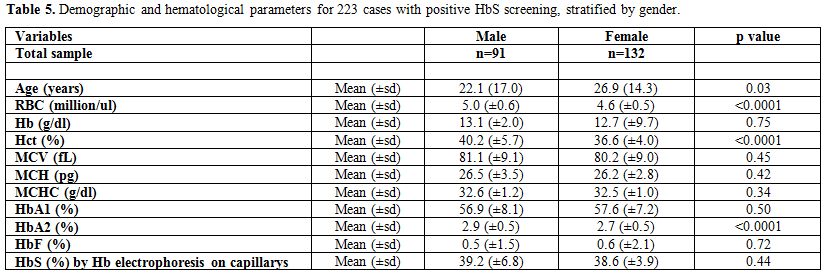 |
Table 5. Demographic and hematological parameters for 223 cases with positive HbS screening, stratified by gender. |
Discussion
In
this retrospective cohort study, that was carried out in a private
laboratory in the Sowthern Suburb of Beirut between 2002 and 2014, we
assessed the prevalence of Sickle Cell Trait, and time trends over a
12-year period, and any differences in prevalence between the two
genders. Moreover, we described a new technique for screening of sickle
cell trait carriers, assessing the correlation of the results with
those of the known sickling test, and confirmed the findings with the
haemoglobin electrophoresis technique (Capillarys). We found the
prevalence of Sickle Cell Trait to be 0.49% (95% CI: 0.46-0.52). No
major differences in trends were found over the 12-year period. We also
found that the prevalence of Sickle Cell Trait was higher in females as
compared to males, consistently over the years. The average MCV value
among carriers of the sickle cell haemoglobin. Were not diferent from
nomal controls. Therefore, the sickling test was found to be a good
diagnostic test for Sickle Cell Trait. The prevalence of Sickle Cell
Trait in our study was found to be 0.49% (95% CI: 0.46- 0.52). Our
results were higher than that reported in Khoriaty et al. study in
Beirut (0.27% of all Hb variants) but lower than that in all regions of
Lebanon (2.1% for all Hb variants and 1.77% for HbS). This
difference could be due to the change in the demographics of the
Lebanese population, mainly due to emigration and
immigration. Another factor that could have affected our results
could be due to the change in the ethnic background, as reported by
Khoriaty et al..[25] Also our results were lower
than some of the reported prevalence of Sickle Cell Trait
internationally. In a study conducted by Ojudu et al., it was
reported that in 44 states of the United States for which data were
available, of all infants screened for the Sickle Cell Trait in 2010,
1.5% tested positive. These states represent approximately 88% of the
U.S. population.[26] Moreover, a systematic review
carried out by Lervolino et al. reported a prevalence ranging
from 1.1 % to 9.8% for the Sickle Cell Trait in different
Brazilian regions,[27] and Ballardini et al. reported a 0.7% prevalence of Sickle Cell Trait in a single center in Italy.[28] On
the other hand, Guler et al. assessed the prevalence of sickle cell
trait in Konya, the urban area of Turkey, in a study carried out in
2007, where they reported a prevalence of 0.05%, which is lower than
that reported in our study.[29] Compared to other
Arab countries, Jastaniah et al. reported in a survey carried out
in 2011 that the prevalence of sickle-cell trait in Saudi Arabia
varies significantly in different parts of the country and ranges from
2% to 27%.[30] This rate is higher than that shown in
our study. Similarly, some surveys peformed in Middle Eastern
countries show that our carrier rate is lower than that reported in
many other Arab countries like Bahrain (11.2–16.4%),[31] Iraq (6.5%),[32] Jordan (4% among females and 6% among males),[33] Libya (4.5%),[34] Tunisia (1.9%),[35] UAE (1.1%)[36] and Yemen (2.2%).[37]
This difference could be explained in part by the lower rate of
consanguineous marriages among the Lebanese population compared to
other Arab countries.
The
results of the present study showed some gender differences, where the
prevalence of Sickle Cell Trait in females (0.62%, 95% CI: 0.57-0.67)
was higher than that of males (0.35%, 95% CI: 0.32-0.39), consistently
over the years. Similarly, significant gender differences have been
also reported in adults with Sickle Cell Trait carriers. For instance,
In a study carried out by Kamble et al. in India, it was reported that
male: female ratio was 1.71:1 in HbAS cases.[38]
We
did not find any significant differences in the trends of the Sickle
Cell Trait over the 12-year period. No studies have been found in the
literature reporting on the trends of the Sickle Cell Trait prevalence
over time. Nevertheless, our findings indicate a relatively stable
prevalence of Sickle Cell Trait over time probably because our studied
subjects were born in Lebanon and of Lebanese ancestry. Accordingly, no
crucial demographic changes or marital habits (consanguineous) were
observed in the past 12 years.
In
our study, the overall average MCV was found to be 84.3 fl (±7.6).
Similarly, a comparable standard MCV value has been
reported among sickle cell trait carriers in France by Tripette
et al. in 2009.[39]
On
the other hand, in a study done in India, Dangi, et al. in
2010 reported an MCV value of 73.4 fl among the sickle cell trait
carriers, which was lower than that reported in our study.[40]
However, this study was performed in an anemic population, and most of
these subjects were affected by sickle anemia with beta thalassaemia
trait,[40] and also the iron deficiency cannot be
excluded in this setting of patients. We found that the MCV level of
the 223 cases of positive sickle cell trait, which were confirmed by
the Hb electrophoresis, to be 80.6 (±9.1). MCV levels among the whole
sample, as well as the 223 cases, that were tested carrying the Sickle
Cell Trait, were within the normal range. Thus, MCV is not useful
for screening sickle cell trait, and the microcytosis does not exclude
the presence of Sickle Cell Trait as reported by Dangi,[40]
and an accurate screening test, like the sickling test, should also be
made in patients with microcytosis (microdrepanocytosis according to
Silvestroni).[41] Of course, the results should be confirmed by Hb electrophoresis (100% accuracy).
The
present study had several limitations. First, it was an observational
study without a control group. Another limitation is that the sample
was collected from a single center that might not be representative of
the whole Lebanese population. As such, the results of the study should
be interpreted with these limitations in mind. On the other hand, the
study had several strengths, such as the large sample size, the
extended period, the prospective data collection, as well as being
among the few studies carried out in Lebanon and the region assessing
the prevalence of Sickle Cell Trait.
Conclusion
In this study, we found that the prevalence of Sickle Cell Trait is lower as compared to other Arab countries, higher in females, and with no significant trend over the 12-year period. Moreover, we showed that the MCV, though was not a screening test for Sickle Cell Trait carriers, was useful. In fact, subjects with microcytosis should not be excluded from the selection and could have a contemporaneously thalassemic trait, or sideropenia. On the other hand, the sickle cell test was found to be an accurate, simple and cheap test (It costs approximately two US dollars for both sickling tests) which could be easily added as a requirement for the pre-marital testing to screen for Sickle Cell Trait carriers.
References

[TOP]