Antonio Marzollo1, Elisabetta Calore1, Manuela Tumino1, Marta Pillon1, Maria Vittoria Gazzola1, Roberta Destro1, Raffaella Colombatti1, Piero Marson2, Tiziana Tison2, Anna Colpo2, Chiara Mainardi1, Maria Gabelli1, Maria Paola Boaro1, Sara Rossin1, Aurora Strano1, Nadia Quaglia1, Federica Menzato1, Giuseppe Basso1, Laura Sainati1 and Chiara Messina1
1 Pediatric Hematology-Oncology Unit, Department of Women’s and Children’s Health, University of Padova, Italy
2 Department of Transfusion Medicine, Azienda Ospedaliera Padova
Corresponding
author: Antonio Marzollo. Pediatric Hematology-Oncology Unit,
Department of Women’s and Children’s Health, University of Padova, Via
Giustiniani 3, Padova 35128, Italy. Tel: +39-049 8213579; Fax: +39-049
8213510. E-mail:
antonio.marzollo@unipd.it
Published: February 15, 2017
Received: October 10, 2016
Accepted: January 12, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017014 DOI
10.4084/MJHID.2017.014
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background and objectives: Lack
of suitable donors and regimen related toxicity are major barriers for
hematopoietic stem cell transplantation (HSCT) in patients with sickle
cell disease (SCD). The aim of the study is the assessment of efficacy
and toxicity of Treosulfan-based conditioning regimen for SCD also when
alternative donors such as mismatched unrelated donor and
haploidentical donor are employed.
Methods:
We report our single-center experience: 11 patients with SCD received
HSCT with a Treosulfan/Thiotepa/Fludarabine/Anti-thymoglobulin
conditioning regimen between 2010 and 2015. The donor was a matched
sibling donor (n= 7), a haploidentical parent (n= 2), a matched
unrelated donor (n= 1) or a mismatched unrelated donor (n=1). The
haploidentical and mismatched unrelated donor grafts were manipulated
by removing TCRαβ and CD19 positive cells.
Results:
All patients survived the procedure and achieved stable engraftment.
Stable mixed chimerism was observed in 5/11 patients. Grade III-IV
regimen related toxicity was limited to mucositis and no grade III-IV
graft-versus-host disease (GvHD) occurred. No SCD manifestation was
observed post-transplant and cerebral vasculopathy improved in 3/5
evaluable patients. Organ function evaluation showed no pulmonary,
cardiac or renal toxicity but gonadal failure occurred in 1/4 evaluable
patients.
Conclusion:
Our data suggest that Treosulfan is associated with low toxicity and
may be employed also for unrelated and haploidentical donor HSCT.
|
Introduction
Sickle
cell disease (SCD) is the most frequent haemoglobinopathy worldwide. A
point mutation in the beta-globulin gene alters the hemoglobin
structure and results in chronic hemolytic anemia and increased blood
viscosity. This leads to an heterogeneous phenotypic spectrum:
increased morbidity and mortality is due to a higher susceptibility to
infections, intermittent vaso-occlusive events and ischemic tissue
injury with progressive organ dysfunction.[1] Hematopoietic stem cell
transplantation (HSCT) is the most consolidated curative treatment.[2]
When considering HSCT for SCD patients, expected SCD morbidity should
be balanced against the risk of transplant related mortality and
morbidity, keeping in mind that SCD is a disease with a life expectancy
of over 50 years with contemporary treatment.[3]
More than 200
matched sibling donor (MSD) transplants after a myeloablative
conditioning regimen based on Busulfan and Cyclophosphamide were
reported. The limitations associated with this strategy are the
significant regimen related toxicity and the risk of graft failure. The
transplant associated mortality is around 2-8% and graft failure is
observed in around 10-15% of patients.[4-11] Although better outcomes
have been recently reported with the use of targeted Busulfan therapy,
the use of Busulfan-based conditioning regimen is established only in
the setting of MSD transplant and only few SCD patients in need of a
HSCT have a matched sibling available.[12-15] The use of alternative
donors such as matched unrelated donor (MUD), mismatched unrelated
donor (MMUD), unrelated umbilical cord blood (UCB) and haploidentical
family members is associated with higher mortality and morbidity, due
to pre-existing organ dysfunction, alloimmunisation and risk of
GvHD.[2,16] Moreover, suitable matched unrelated donors are difficult
to find and experience is limited for umbilical cord blood or
haploidentical donor HSCT.[17-21] In order to overcome the limitations
of Busulfan-based conditioning regimen and allow the use of alternative
donors, different conditioning strategies have been proposed.[22,23]
Recently, Treosulfan became attractive to substitute Busulfan due to
its lower toxicity and good immune suppressive and myeloablative
potential.[24-28] This led to the use of Treosulfan in patients with
pre-existent morbidity (mainly primary immune deficiency or
β-thalassemia) or receiving a second transplant for malignant
disorders.[25,29,26,30-35] For these reasons, since 2010, at our
institution Busulfan was substituted with Treosulfan as standard
conditioning regimen for SCD patients.
We present our
single-centre experience of HSCT performed for SCD patients employing
Treosulfan-based conditioning regimen also in haploidentical and MUD
HSCT.
Methods
From
April 2010 to December 2015, all SCD patients undergoing HSCT at the
Pediatric Hematology-Oncology Unit of the University of Padova received
a Treosulfan-based conditioning regimen and are described in this
retrospective cohort study. Eligibility for HSCT was determined on the
basis of published guidelines and criteria included cerebral
vasculopathy, recurrent episodes of acute chest syndrome (ACS) or
vaso-occlusive crises despite hydroxycarbamide treatment.[36] Donors
were chosen on the basis of availability and considered in the
following order: MSD, MUD, Haploidentical donor and MMUD. The preferred
stem cell source was bone marrow or umbilical stem cells for MSD, bone
marrow for MUD and T-depleted peripheral blood stem cells for
haploidentical donors and MMUD. The use of a combination of umbilical
cord blood and bone marrow was employed in MSD HSCT if the cellularity
of the umbilical cord graft was low.[20] Before T-cell depleted or MUD
HSCT, autologous bone marrow stem cells were harvested and
cryopreserved in order to be re-infused in case of graft rejection, due
to the higher risk of this event after T-cell depletion.[37] All
patients received either red cell exchange transfusion or simple
transfusion the day before the start of conditioning regimen in order
to obtain a proportion of HbS < 30% and an Hb level ≥ 100g/L.
The
haploidentical donors underwent PBSC collection after mobilization with
subcutaneous Filgrastim 10 µg/Kg twice daily from day -5 to day -1 and
once on the day of collection. PBSC were collected using a COBE®
Spectra Apheresis System (BCT Terumo, Lakewood, CO). T-cell depletion
was performed by removing TCRαβ positive and CD19 positive cells
through immuno-magnetic selection (CliniMACS; Miltenyi Biotec, Bergisch
Gladbach, Germany).
All patients received Thiotepa (8 mg/kg or 10 mg/kg in 2 doses on day -7), Treosulfan (14 g/m2/day for 3 days from day -6 to day -4 ) and Fludarabine (40 mg/m2/day
for 4 days from day -6 to day-3).[29] Fresenius® anti-thymocyte
globulins (ATG) at the dose of 20 mg/kg/day were administered for 3
days in MSD and MUD transplants and 5 mg/kg/day for 4 days in
T-depleted transplants. Patient 8 and patient 11 received Rituximab
(200 mg/m2)
at day -1 to reduce the risk of EBV reactivation and GvHD after
HSCT.[38] Graft-vs-Host Disease (GvHD) prophylaxis for T-replete
transplants consisted in short term Methotrexate (10mg/kg for 4 doses),
Cyclosporine 1 mg/kg/day on days -7 to -2, and Cyclosporine aiming at a
pre-dose level of 100-200 µg/L for 6 months post HSCT. No GvHD
prophylaxis was given for T-deplete transplants. The supportive
measures, diagnosis and treatment strategy for acute GvHD employed at
our institution were recently described.[39]
Engraftment and
chimerism were serially tracked after HSCT. Samples were obtained every
2-3 weeks up to day +100 and monthly thereafter up to 18 months
post-transplant in patient with complete donor chimerism. Follow up was
longer for patients with mixed chimerism. Chimerism analysis was
performed by PCR testing for informative short tandem repeats. Adverse
events were graded according to common terminology criteria for adverse
events (CTCAE) v4. Neutrophil and platelet recovery were defined as a
neutrophil count ≥ 0.5×109/L for 3 consecutive days and as a platelet count ≥ 50×109/L independently of platelet transfusions for 7 consecutive days.
Organ
function was assessed pre-transplant and every year post-transplant by
pulmonary function testing, echocardiography, growth and puberty
evaluation and hormonal dosage (estradiol/testosterone, FSH, LH, T4 and
TSH levels). Transcranial Doppler Ultrasonography (TCD) was performed
for all patients prior to the initiation of chronic transfusion and
pre-HSCT; data were evaluated according to the criteria defined by the
STOP trial.[40] Brain magnetic resonance imaging (MRI) and magnetic
resonance angiography (MRA) were performed pre-transplant, and repeated
one year post transplant and every two years thereafter if lesions were
detected. MRI and MRA were evaluated according to a standardized
scoring system.[41,42] Cerebral vasculopathy was defined as abnormal
Transcranial Doppler velocity associated with cerebral artery stenosis.
Immune reconstitution was evaluated by total lymphocyte count, CD4+
cell count and immunoglobulin dosage. Data were recorded at day +30,
+90, +180 and +365.
Results
Patients:
Eleven consecutive children affected by SCD (7 females, 4 males) were
transplanted at the Pediatric Hematology-Oncology Unit of the
University of Padova. The origin of patients was African (n=6),
Caucasian (n=4) or Caribbean (n=1). Patient and transplant
characteristics are summarized in table 1.
Seven patients were diagnosed at birth due to family history, the
remaining at their first disease manifestation, between 7 month and 5
years of age. The Hb genotype was HbSS (n=10) or HbSβ0 (n=1, P7).
Before transplant patients were treated with chronic transfusion (n=7),
monthly red cell exchange transfusion (n=2) and/or hydroxycarbamide
(n=5). Patients were eligible for HSCT due to cerebral vasculopathy
(n=7), recurrent episodes of acute chest syndrome (ACS) or
vaso-occlusive crises despite hydroxycarbamide treatment (n=4). Patient
7 suffered also from recurrent splenic sequestration.[36] The median
age at HSCT was 6.5 years (range: 4 – 16.3 years). At the time of HSCT,
only two patients had significant comorbidities: relapsing autoimmune
hepatitis (P8) and an association of Chiari I malformation and
syringomyelia (P11). Donor source was an HbS/A MSD (n = 5), an HbA/A
MSD (n=2), a haploidentical HbS/A parent (mother, n=1 and father, n=1),
HbA/A MUD (n=1) or HbA/A MMUD (n=1, 8/12 HLA loci donor/recipient
matching). Stem cell source was bone marrow (n=6, median total
nucleated cells, TNC=4,9x10^8/kg), combined bone marrow and umbilical
cord blood (n=2, median TNC for bone marrow = 2,67x108/kg; median TNC for cord blood = 2,44x107/kg) or peripheral blood stem cells (PBSC) (n=3, two haploidentical grafts and one MUD graft, median CD34+ cells = 14,3x106/kg). One apheresis was sufficient to reach the target dose of CD34+cell (10x106/kg recipient) in both haploidentical donors. Both donors complained only grade I-II myalgia and fatigue.
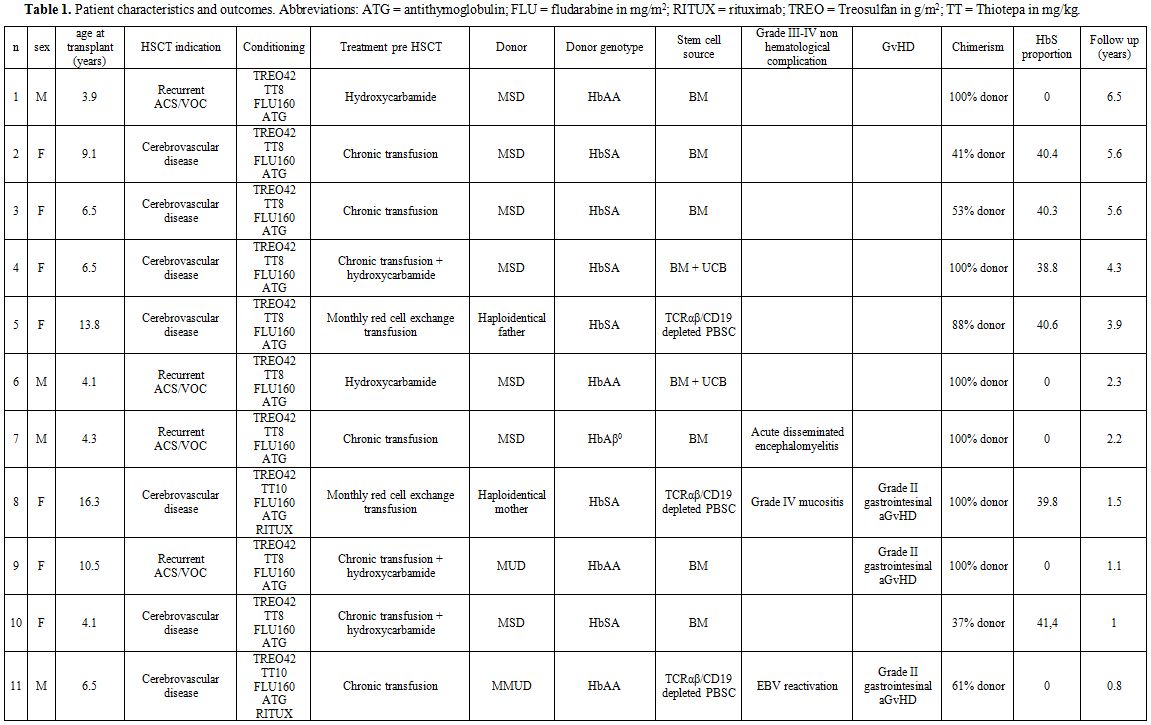 |
Table
1. Patient characteristics and outcomes. Abbreviations: ATG = antithymoglobulin; FLU = fludarabine in mg/m2; RITUX = rituximab; TREO = Treosulfan in g/m2; TT = Thiotepa in mg/kg. |
Transplant-related outcomes:
All patients achieved neutrophil and platelets engraftment at a median
of 20 days (range: 15-34 days) and 22 days (range: 12-31 days) from HSC
infusion, respectively. No patient experienced primary graft failure.
All patients experienced Grade III anemia, grade IV thrombocytopenia
and grade IV neutropenia. Grade III-IV non hematological toxicity
occurred in 2 patients and consisted in grade IV stomatitis in one
patient and acute disseminated encephalomyelitis in one patient. All
toxicity resolved completely. Grade I-II gastrointestinal acute GvHD
was diagnosed in 3 patients (haploidentical transplant, n=1; MUD
transplant, n=1, MMUD transplant, n=1). These patients were
successfully treated with calcineurin inhibitors (n=3), steroid (n=1)
and extracorporeal photochemotherapy (n=3) as per institutional
protocol.[39] No grade III-IV acute GvHD or chronic GvHD were observed.
No secondary graft failure was observed.
Full donor chimerism
was demonstrated in 6/11 patients and stable mixed chimerism was
observed in 5/11 patients (45%). Donor hematopoiesis ranged from 37% to
90%, but did not affect the HbS proportion: HbS was absent after a
transplant from HbA/A MUD (n=1) or compatible with HbS carrier (median
40.5%, range 40.3-41,4%) in patients transplanted from a HbS/A MSD
(n=4).
The lymphocyte count reached normal values for age at day
+180 in all patients except P11. Median time to reach a CD3+CD4+ cell
count higher than 400/µL was 148 days for T-replete grafts (range
57-203 days) and 245 days (range 237-253 days) for T-deplete grafts.
Immunoglobulin replacement was necessary only in one patient (P11) who
experienced EBV reactivation and received one dose of rituximab.
Despite serial monitoring no viral reactivation was documented in any
other patient.
Organ damage and SCD-related outcomes:
The median follow-up was 2.35 years (range: 0.8-6.5 years). All
patients are alive and well. No episode compatible with acute chest
syndrome, stroke or other sickle cell disease manifestation occurred.
No patient experienced renal or hepatic dysfunction following transplantation.
TCD was normal for patients without cerebral vasculopathy. Data for patients with cerebral vasculopathy are reported in Table 2.
Brain MRI and MRA data are available for 10 patients. Three patients
had normal pre-transplant brain imaging. Two patients had cerebral
vasculopathy before transplantation, but no post-transplant imaging.
Five patients had alteration on the pre transplant MRI and evaluable
data on follow-up (Table 3).
Resolution or improvement of the vascular stenosis was detected in 3/5
patients. Last post-transplant evaluation was performed after a median
of 1315 days (range: 268-1417).
Pre-transplant organ function
was within normal limits for all patients. Post-transplant lung
function evaluation was performed in 7 patients (P1-3 and P5-8) and was
normal for all of them after a median of 1104 days from transplant
(range 369-2304 days). Hormonal function was evaluated in 7 patients
(P1-3 and P5-8) after a median of 804 days from transplant (range
205-1518 days). Height, weight and thyroid function were normal for all
explored patients. Three patients were pre-pubertal at last assessment.
Puberty was evaluable in 4 patients (P1, P2, P5 and P8: 1 male and 3
females): 3 had normal pubertal development and 1 patient (P8)
experienced secondary gonadal failure. Follow-up echocardiography (data
available for 7 patients) and eye examination (data available for 5
patients) were normal.
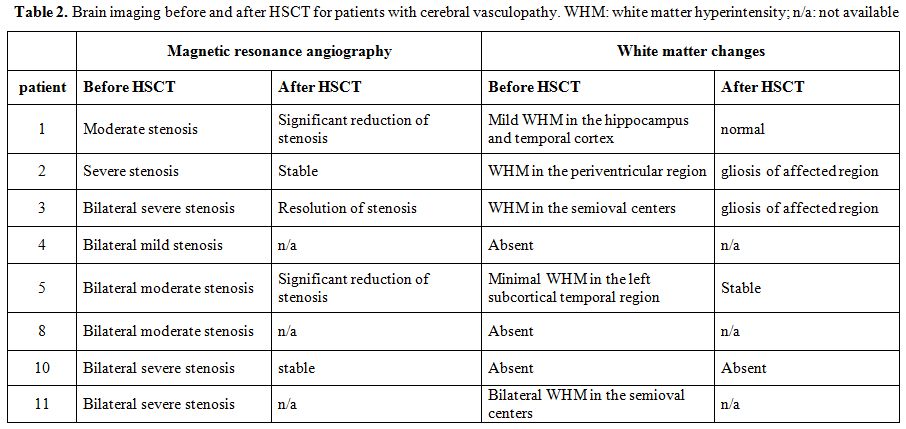 |
Table 2. Brain imaging before and
after HSCT for patients with cerebral vasculopathy. WHM: white matter
hyperintensity; n/a: not available |
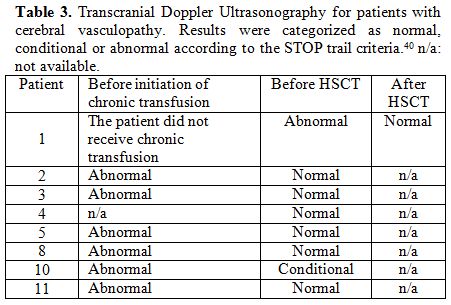 |
Table 3. Transcranial Doppler
Ultrasonography for patients with cerebral vasculopathy. Results were
categorized as normal, conditional or abnormal according to the STOP
trail criteria.40 n/a: not available. |
Discussion
We
report a retrospective case series of 11 SCD patients who received HSCT
after a Treosulfan-based conditioning regimen. Sustained engraftment
was observed in all patients. Stable mixed chimerism was detected in a
significant proportion of patients (45%), did not change after the
discontinuation of immunosuppressive treatment and resulted in a cure
of SCD for all patients. Previous experiences have demonstrated that
full donor chimerism is not needed to cure SCD due to the survival
advantage for donor red cell in peripheral blood: pulmonary, gonadal
and central nervous system status can be significantly ameliorated also
when stable mixed chimerism is obtained.[43-47] Indeed, no clinical
manifestation correlated with SCD occurred after HSCT in our cohort.
Since cerebral vasculopathy was the cause for transplant in 7 patients,
we focused our attention on the evaluation of TCD and brain MRI and
MRA. Chronic transfusions resulted in normalization of pre-transplant
TCD in all patients receiving this treatment. However, cerebral artery
stenosis persisted on pre-HSCT MRA for all patients. Post-HSCT MRA
data, evaluated with a standardized scoring system, were available for
5 patients and showed either a stabilization of the stenosis or
amelioration. Although improvement in vascular stenosis has been
previously described in patients treated with chronic transfusion,
hydroxycarbamide or HSCT, the rate of improving patients in our cohort
compares favorably with previous reports.[37,48-51] These satisfactory
outcomes could be possibly due to the screening program for cerebral
vasculopathy performed at our center that led to the fact that all
patients were transplanted before any clinically evident stroke.[42]
The
safety profile of Treosulfan conditioning regimen was excellent and
incidence of adverse events was comparable to previous reports: no
transplant-related mortality was observed and grade III-IV non
hematological toxicity was limited to mucositis which resolved
completely without sequelae.[25,26] The neurological event in our case
series cannot be attributed with certainty to the Treosulfan
conditioning. This toxicity profile is similar to results obtained in
adult patients transplanted after a non-myeloablative
conditioning.[52-55] Grade I-II acute GvHD was observed in 3/11
patients in our cohort (27%) with no grade III-IV acute GvHD or chronic
GvHD. The GvHD cases were all among patients receiving an alternative
donor transplant, no GvHD was observed among the 7 patients receiving a
MSD HSCT and all the patients experiencing GvHD responded rapidly to
first line treatment. To the best of our knowledge, data regarding
organ damage related to HSCT has not been previously reported for SCD
patients undergoing HSCT after a Treosulfan-based conditioning regimen.
In our cohort, the decline in pulmonary and renal function observed
after Busulfan-based conditioning regimen was not present and growth,
thyroid and cardiac function were preserved after HSCT.[37,43] Although
3 patients had normal pubertal development, 1 patient that had reached
puberty before HSCT and for whom pre-transplant ovarian
cryopreservation was performed, experienced secondary gonadal failure.
This event highlights the opportunity to attentively evaluate possible
long term effects on reproductive health and propose mitigating
strategies before HSCT.[56]
Current knowledge about outcomes of
Treosulfan based conditioning regimen in SCD is limited to a
single-center experience reporting 15 patients who received a MSD or
MUD HSCT.[26] We have nearly doubled the number of reported patients
and we have described the use of Treosulfan-based conditioning regimen
for MMUD or haploidentical donor HSCT, which are considered
investigational approaches in SCD.[16,37,57] To mitigate the risk of
rejection and GvHD, TCRαβ+ and CD19+ cell depletion was performed.[38]
In the MMUD setting, this approach was reported as safe and efficacious
for patients with acute myeloid leukemia or Hurler syndrome but no SCD
patient has been described yet.[58,59] If further investigations will
confirm its feasibility and efficacy, haploidentical or MMUD HSCT in
SCD may open the possibility of cure for many patients without a MSD or
MUD donor available.[17-19] When employing alternative donors, a higher
risk of GvHD and delayed immune reconstitution should be taken into
account; however, in our experience, these drawbacks can be managed by
supportive therapy and are outweighed by the satisfactory outcomes.
Although PBSC mobilization with G-CSF in HbS heterozygous parents is
often perceived as risky, no significant adverse events were reported
and, in our experience, both the haploidentical donors underwent PBSC
mobilization and collection safely.[60,61]
The main limitation of
our study is its retrospective nature and the report of a single center
experience; sample size was also limited and warrants further
confirmation. Moreover, in order to perform the TCRαβ and CD19
depletion, a facility with experience in stem cell manipulation is
needed.
Conclusions
Our
data show that HSCT after Treosulfan based conditioning regimen for SCD
patients is effective and associated with low toxicity. End organ
damage may be halted or even ameliorated as shown by the regression of
cerebral vessel stenosis and white matter changes. This strategy is
suitable also for alternative donor transplants and, if our data are
confirmed in larger cohorts, could pave the way for expanding the
access to HSCT also to SCD patients lacking a matched sibling or
matched unrelated donor.
Author Contributions
C.M.
and A.M. wrote the manuscript. E.C., M.P., R.C. and L.S. critically
reviewed the manuscript. E.C., M.T., M.P., R.C., P.M., T.T., A.C.,
M.C., M.G., M.P.B., S.R., N.Q., F.M., G.B. and L.S. were involved in
the clinical management of patients. M.V.G., R.D. and A.S. were
responsible for stem cell product manipulation. All authors contributed
to the intellectual content of this paper and approved the final
manuscript.
References
- Rees DC, Williams TN, Gladwin MT. Sickle-cell
disease. Lancet. 2010 Dec;376(9757):2018-31.
https://doi.org/10.1016/S0140-6736(10)61029-X

- Talano
J-A, Cairo MS. Smoothing the crescent curve: sickle cell disease.
Hematology. 2014 Dec 1;2014(1):468-74.
https://doi.org/10.1182/asheducation-2014.1.468 PMid:25696896
- Quinn
CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children
and adolescents with sickle cell disease. Blood. 2010 Apr
29;115(17):3447-52. https://doi.org/10.1182/blood-2009-07-233700
PMid:20194891 PMCid:PMC2867259
- Walters
MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, Davies
SC, Ohene-Frempong K, Bernaudin F, Matthews DC, Storb R, Sullivan KM.
Bone Marrow Transplantation for Sickle Cell Disease. N Engl J Med. 1996
Aug 8;335(6):369-76. https://doi.org/10.1056/NEJM199608083350601
PMid:8663884
- Vermylen
C, Cornu G, Ferster A, Brichard B, Ninane J, Ferrant A, Zenebergh A,
Maes P, Dhooge C, Benoit Y, Beguin Y, Dresse MF, Sariban E.
Haematopoietic stem cell transplantation for sickle cell anaemia: the
first 50 patients transplanted in Belgium. Bone Marrow Transplant. 1998
Jul;22(1):1-6. https://doi.org/10.1038/sj.bmt.1701291 PMid:9678788
- Walters
MC, Storb R, Patience M, Leisenring W, Taylor T, Sanders JE, Buchanan
GE, Rogers ZR, Dinndorf P, Davies SC, Roberts IA, Dickerhoff R, Yeager
AM, Hsu L, Kurtzberg J, Ohene-Frempong K, Bunin N, Bernaudin F,
Wong WY, Scott JP, Margolis D, Vichinsky E, Wall DA, Wayne AS, Pegelow
C, Redding-Lallinger R, Wiley J, Klemperer M, Mentzer WC, Smith FO,
Sullivan KM. Impact of bone marrow transplantation for symptomatic
sickle cell disease: an interim report. Multicenter investigation of
bone marrow transplantation for sickle cell disease. Blood. 2000 Mar
15;95(6):1918-24. PMid:10706855
- Bernaudin
F, Socie G, Kuentz M, Chevret S, Duval M, Bertrand Y, Vannier J-P,
Yakouben K, Thuret I, Bordigoni P, Fischer A, Lutz P, Stephan J-L,
Dhedin N, Plouvier E, Margueritte G, Bories D, Verlhac S, Esperou H,
Coic L, Vernant J-P, Gluckman E. Long-term results of related
myeloablative stem-cell transplantation to cure sickle cell disease.
Blood. 2007 Oct 1;110(7):2749-56.
https://doi.org/10.1182/blood-2007-03-079665 PMid:17606762
- Panepinto
JA, Walters MC, Carreras J, Marsh J, Bredeson CN, Gale RP, Hale GA,
Horan J, Hows JM, Klein JP, Pasquini R, Roberts I, Sullivan K, Eapen M,
Ferster A. Matched-related donor transplantation for sickle cell
disease: Report from the Center for International Blood and Transplant
Research. Br J Haematol. 2007;137(5):479-85.
https://doi.org/10.1111/j.1365-2141.2007.06592.x PMid:17459050
- McPherson
ME, Hutcherson D, Olson E, Haight A, Horan J, Chiang K-Y. Safety and
efficacy of targeted busulfan therapy in children undergoing
myeloablative matched sibling donor BMT for sickle cell disease. Bone
Marrow Transplant. 2011 Jan 22;46(1):27-33.
https://doi.org/10.1038/bmt.2010.60 PMid:20305698
- Dedeken
L, Lê PQ, Azzi N, Brachet C, Heijmans C, Huybrechts S, Devalck C, Rozen
L, Ngalula M, Ferster A. Haematopoietic stem cell transplantation for
severe sickle cell disease in childhood: a single centre experience of
50 patients. Br J Haematol. 2014 May;165(3):402-8.
https://doi.org/10.1111/bjh.12737 PMid:24433465
- Locatelli
F, Pagliara D. Allogeneic hematopoietic stem cell transplantation in
children with sickle cell disease. Pediatr Blood Cancer. 2012
Aug;59(2):372-6. https://doi.org/10.1002/pbc.24177
PMid:22544533
- Walters
MC, Patience M, Leisenring W, Eckman JR, Buchanan GR, Rogers ZR,
Olivieri NE, Vichinsky E, Davies SC, Mentzer WC, Powars D, Scott JP,
Bernaudin F, Ohene-Frempong K, Darbyshire PJ, Wayne A, Roberts IA,
Dinndorf P, Brandalise S, Sanders JE, Matthews DC, Appelbaum FR, Storb
R, Sullivan KM. Barriers to bone marrow transplantation for sickle cell
anemia. Biol Blood Marrow Transplant. 1996 May;2(2):100-4.
PMid:9118298
- Gaziev
J, Isgrò A, Mozzi AF, Petain A, Nguyen L, Ialongo C, Dinallo V, Sodani
P, Marziali M, Andreani M, Testi M, Paciaroni K, Gallucci C, De Angelis
G, Alfieri C, Ribersani M, Lucarelli G. New insights into the
pharmacokinetics of intravenous busulfan in children with sickle cell
anemia undergoing bone marrow transplantation. Pediatr Blood Cancer.
2015 Apr;62(4):680-6. https://doi.org/10.1002/pbc.25376
PMid:25557687
- Maheshwari
S, Kassim A, Yeh RF, Domm J, Calder C, Evans M, Manes B, Bruce K, Brown
V, Ho R, Frangoul H, Yang E. Targeted Busulfan therapy with a
steady-state concentration of 600-700 ng/mL in patients with sickle
cell disease receiving HLA-identical sibling bone marrow transplant.
Bone Marrow Transplant. 2014;49(3):366-9.
https://doi.org/10.1038/bmt.2013.188 PMid:24317124
- Lucarelli
G, Isgrò A, Sodani P, Marziali M, Gaziev J, Paciaroni K, Gallucci C,
Cardarelli L, Ribersani M, Alfieri C, De Angelis G, Armiento D,
Andreani M, Testi M, Amato A, Akinyanju OO, Wakama TT. Hematopoietic
SCT for the Black African and non-Black African variants of sickle cell
anemia. Bone Marrow Transplant. 2014;49(11):1376-81.
https://doi.org/10.1038/bmt.2014.167 PMid:25068420
- Gluckman
E. Allogeneic transplantation strategies including haploidentical
transplantation in sickle cell disease. Hematology. 2013 Dec
1;2013(1):370-6. https://doi.org/10.1182/asheducation-2013.1.370
PMid:24319206
- Krishnamurti
L, Abel S, Maiers M, Flesch S. Availability of unrelated donors for
hematopoietic stem cell transplantation for hemoglobinopathies. Bone
Marrow Transplant. 2003 Apr;31(7):547-50.
https://doi.org/10.1038/sj.bmt.1703887 PMid:12692619
- Justus
D, Perez-Albuerne E, Dioguardi J, Jacobsohn D, Abraham A. Allogeneic
donor availability for hematopoietic stem cell transplantation in
children with sickle cell disease. Pediatr Blood Cancer. 2015
Jul;62(7):1285-7. https://doi.org/10.1002/pbc.25439 PMid:25663074
- Gragert
L, Eapen M, Williams E, Freeman J, Spellman S, Baitty R, Hartzman R,
Rizzo JD, Horowitz M, Confer D, Maiers M. HLA Match Likelihoods for
Hematopoietic Stem-Cell Grafts in the U.S. Registry. N Engl J Med. 2014
Jul 24;371(4):339-48. https://doi.org/10.1056/NEJMsa1311707
PMid:25054717
- Ruggeri
A, Eapen M, Scaravadou A, Cairo MS, Bhatia M, Kurtzberg J, Wingard JR,
Fasth A, Lo Nigro L, Ayas M, Purtill D, Boudjedir K, Chaves W, Walters
MC, Wagner J, Gluckman E, Rocha V. Umbilical Cord Blood Transplantation
for Children with Thalassemia and Sickle Cell Disease. Biol Blood
Marrow Transplant. 2011 Sep;17(9):1375-82.
https://doi.org/10.1016/j.bbmt.2011.01.012 PMid:21277376
PMCid:PMC3395002
- Locatelli
F, Kabbara N, Ruggeri A, Ghavamzadeh A, Roberts I, Li CK, Bernaudin F,
Vermylen C, Dalle J-H, Stein J, Wynn R, Cordonnier C, Pinto F,
Angelucci E, Socie G, Gluckman E, Walters MC, Rocha V. Outcome of
patients with hemoglobinopathies given either cord blood or bone marrow
transplantation from an HLA-identical sibling. Blood. 2013 Aug
8;122(6):1072-8. https://doi.org/10.1182/blood-2013-03-489112
PMid:23692854
- King
AA, Kamani N, Bunin N, Sahdev I, Brochstein J, Hayashi RJ, Grimley M,
Abraham A, Dioguardi J, Wah Chan K, Douglas D, Adams R, Andreansky M,
Anderson E, Gilman A, Chaudhury S, Yu L, Dalal J, Hale G, Cuvelier G,
Jain A, Krajewski J, Gillio A, Kasow KA, Delgado D, Hanson E, Murray L,
Shenoy S. Successful matched sibling donor marrow transplantation
following reduced intensity conditioning in children with
hemoglobinopathies. Am J Hematol. 2015 Dec;90(12):1093-8.
https://doi.org/10.1002/ajh.24183 PMid:26348869
- Matthes-Martin
S, Lawitschka A, Fritsch G, Lion T, Grimm B, Breuer S, Boztug H,
Karlhuber S, Holter W, Peters C, Minkov M. Stem cell transplantation
after reduced-intensity conditioning for sickle cell disease. Eur J
Haematol. 2013 Apr;90(4):308-12. https://doi.org/10.1111/ejh.12082
PMid:23369103
- Burroughs
LM, Nemecek ER, Torgerson TR, Storer BE, Talano J-A, Domm J, Giller RH,
Shimamura A, Delaney C, Skoda-Smith S, Thakar MS, Baker KS, Rawlings
DJ, Englund J a., Flowers MED, Deeg HJ, Storb R, Woolfrey AE.
Treosulfan-Based Conditioning and Hematopoietic Cell Transplantation
for Nonmalignant Diseases: A Prospective Multi-Center Trial. Biol Blood
Marrow Transplant. 2014 Sep; https://doi.org/10.1016/j.bbmt.2014.08.020
PMid:25196857 PMCid:PMC4324724
- Slatter
MA, Boztug H, Pötschger U, Sykora K-W, Lankester A, Yaniv I, Sedlacek
P, Glogova E, Veys P, Gennery AR, Peters C. Treosulfan-based
conditioning regimens for allogeneic haematopoietic stem cell
transplantation in children with non-malignant diseases. Bone Marrow
Transplant. 2015 Dec 10;50(12):1536-41.
https://doi.org/10.1038/bmt.2015.171 PMid:26259076
- Strocchio
L, Zecca M, Comoli P, Mina T, Giorgiani G, Giraldi E, Vinti L, Merli P,
Regazzi M, Locatelli F. Treosulfan-based conditioning regimen for
allogeneic haematopoietic stem cell transplantation in children with
sickle cell disease. Br J Haematol. 2015 Jun;169(5):726-36.
https://doi.org/10.1111/bjh.13352 PMid:25818248
- Feit
PW, Rastrup-Andersen N, Matagne R. Studies on epoxide formation from
(2S,3S)-threitol 1,4-bismethanesulfonate. The preparation and
biological activity of (2S,3S)-1,2-epoxy-3,4-butanediol
4-methanesulfonate. J Med Chem. 1970 Nov;13(6):1173-5.
https://doi.org/10.1021/jm00300a034 PMid:5479859
- Sjöö
F, Hassan Z, Abedi-Valugerdi M, Griskevicius L, Nilsson C, Remberger M,
Aschan J, Concha H, Gaughan U, Hassan M. Myeloablative and
immunosuppressive properties of treosulfan in mice. Exp Hematol. 2006
Jan;34(1):115-21. https://doi.org/10.1016/j.exphem.2005.09.015
PMid:16413398
- Bernardo
ME, Piras E, Vacca A, Giorgiani G, Zecca M, Bertaina A, Pagliara D,
Contoli B, Pinto RM, Caocci G, Mastronuzzi A, La Nasa G, Locatelli F.
Allogeneic hematopoietic stem cell transplantation in thalassemia
major: results of a reduced-toxicity conditioning regimen based on the
use of treosulfan. Blood. 2012 Jul 12;120(2):473-6.
https://doi.org/10.1182/blood-2012-04-423822 PMid:22645178
- Slatter
MA, Rao K, Amrolia P, Flood T, Abinun M, Hambleton S, Nademi Z, Goulden
N, Davies G, Qasim W, Gaspar HB, Cant A, Gennery AR, Veys P.
Treosulfan-based conditioning regimens for hematopoietic stem cell
transplantation in children with primary immunodeficiency: United
Kingdom experience. Blood. 2011 Apr 21;117(16):4367-75.
https://doi.org/10.1182/blood-2010-10-312082 PMid:21325599
- Beier
R, Schulz A, Hönig M, Eyrich M, Schlegel P-G, Holter W, Stachel KD,
Ehlert K, Greil J, Nürnberger W, Wößmann W, Bader P, Urban C, Müller I,
Suttorp M, Sauer M, Gruhn B, Meisel R, Zimmermann M, Sykora K-W.
Long-term follow-up of children conditioned with Treosulfan: German and
Austrian experience. Bone Marrow Transplant. 2013 Apr 22;48(4):491-501.
https://doi.org/10.1038/bmt.2012.188 PMid:23085832
- Lehmberg
K, Albert MH, Beier R, Beutel K, Gruhn B, Kroger N, Meisel R, Schulz A,
Stachel D, Woessmann W, Janka G, Muller I. Treosulfan-based
conditioning regimen for children and adolescents with hemophagocytic
lymphohistiocytosis. Haematologica. 2014 Jan 1;99(1):180-4.
https://doi.org/10.3324/haematol.2013.094730 PMid:24162790
PMCid:PMC4007927
- Dinur-Schejter
Y, Krauss AC, Erlich O, Gorelik N, Yahel A, Porat I, Weintraub M, Stein
J, Zaidman I, Stepensky P. Bone marrow transplantation for
non-malignant diseases using treosulfan-based conditioning. Pediatr
Blood Cancer. 2015 Oct;62(2):299-304. PMid:25284797
- Boztug
H, Zecca M, Sykora K-W, Veys P, Lankester A, Slatter M, Skinner R,
Wachowiak J, Pötschger U, Glogova E, Peters C. Treosulfan-based
conditioning regimens for allogeneic HSCT in children with acute
lymphoblastic leukaemia. Ann Hematol. 2015 Feb 19;94(2):297-306.
https://doi.org/10.1007/s00277-014-2196-8 PMid:25231927
- Morillo-Gutierrez
B, Beier R, Rao K, Burroughs L, Schulz A, Ewins A-M, Gibson B, Sedlacek
P, Krol L, Strahm B, Zaidman I, Kalwak K, Talano J-A, Woolfrey A,
Fraser C, Meyts I, Muller I, Wachowiak J, Bernardo ME, Veys P, Sykora
K-W, Gennery AR, Slatter M. Treosulfan based conditioning for
allogeneic HSCT in children with chronic granulomatous disease: a
multicentre experience. Blood. 2016 May 23;
https://doi.org/10.1182/blood-2016-03-704015 PMid:27216217
- Angelucci
E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini
MD, Dalle J-H, Di Bartolomeo P, de Heredia CD, Dickerhoff R, Giardini
C, Gluckman E, Hussein AA, Kamani N, Minkov M, Locatelli F, Rocha V,
Sedlacek P, Smiers F, Thuret I, Yaniv I, Cavazzana M, Peters C.
Hematopoietic stem cell transplantation in thalassemia major and sickle
cell disease: indications and management recommendations from an
international expert panel. Haematologica. 2014 May 1;99(5):811-20.
https://doi.org/10.3324/haematol.2013.099747 PMid:24790059
PMCid:PMC4008115
- Dallas
MH, Triplett B, Shook DR, Hartford C, Srinivasan A, Laver J, Ware R,
Leung W. Long-Term Outcome and Evaluation of Organ Function in
Pediatric Patients Undergoing Haploidentical and Matched Related
Hematopoietic Cell Transplantation for Sickle Cell Disease. Biol Blood
Marrow Transplant. 2013 May;19(5):820-30.
https://doi.org/10.1016/j.bbmt.2013.02.010 PMid:23416852
PMCid:PMC4712646
- Bertaina
A, Merli P, Rutella S, Pagliara D, Bernardo ME, Masetti R, Pende D,
Falco M, Handgretinger R, Moretta F, Lucarelli B, Brescia LP, Li Pira
G, Testi M, Cancrini C, Kabbara N, Carsetti R, Finocchi A, Moretta A,
Moretta L, Locatelli F. HLA-haploidentical stem cell transplantation
after removal of aß+ T and B cells in children with nonmalignant
disorders. Blood. 2014 Jul 31;124(5):822-6.
https://doi.org/10.1182/blood-2014-03-563817 PMid:24869942
- Calore
E, Marson P, Pillon M, Tumino M, Tison T, Mainardi C, De Silvestro G,
Rossin S, Franceschetto G, Carraro E, Pescarin M, Varotto S, Destro R,
Gazzola MV, Basso G, Messina C. Treatment of Acute Gvhd in Childhood
with Extracorporeal Photochemotherapy/Phofotopheresis: the Padova
Experience. Biol Blood Marrow Transplant. 2015;1-10.
- Adams
RJ, McKie VC, Hsu L, Files B, Vichinsky E, Pegelow C, Abboud M,
Gallagher D, Kutlar A, Nichols FT, Bonds DR, Brambilla D. Prevention of
a first stroke by transfusions in children with sickle cell anemia and
abnormal results on transcranial Doppler ultrasonography. N Engl J Med.
1998 Jul 2;339(1):5-11. https://doi.org/10.1056/NEJM199807023390102
PMid:9647873
- Montanaro
M, Colombatti R, Pugliese M, Migliozzi C, Zani F, Guerzoni ME, Manoli
S, Manara R, Meneghetti G, Rampazzo P, Cavalleri F, Giordan M, Paolucci
P, Basso G, Palazzi G, Sainati L. Intellectual function evaluation of
first generation immigrant children with sickle cell disease: the role
of language and sociodemographic factors. Ital J Pediatr. 2013 Jun
4;39:36. https://doi.org/10.1186/1824-7288-39-36 PMid:23735165
PMCid:PMC3704731
- Manara
R, Talenti G, Rampazzo P, Ermani M, Montanaro M, Baracchini C, Teso S,
Basso G, Sainati L, Colombatti R. Longitudinal evaluation of cerebral
white matter hyperintensities lesion volume in children with sickle
cell disease. Br J Haematol. 2016 Mar 27;i.
- Walters
MC, Hardy K, Edwards S, Adamkiewicz T, Barkovich J, Bernaudin F,
Buchanan GR, Bunin N, Dickerhoff R, Giller R, Haut PR, Horan J, Hsu LL,
Kamani N, Levine JE, Margolis D, Ohene-Frempong K, Patience M,
Redding-Lallinger R, Roberts IAG, Rogers ZR, Sanders JE, Scott JP,
Sullivan KM. Pulmonary, Gonadal, and Central Nervous System Status
after Bone Marrow Transplantation for Sickle Cell Disease. Biol Blood
Marrow Transplant. 2010;16(2):263-72.
https://doi.org/10.1016/j.bbmt.2009.10.005 PMid:19822218
PMCid:PMC2919571
- Walters
M., Patience M, Leisenring W, Rogers Z., Aquino V., Buchanan G.,
Roberts IA., Yeager A., Hsu L, Adamkiewicz T, Kurtzberg J, Vichinsky E,
Storer B, Storb R, Sullivan K. Stable mixed hematopoietic chimerism
after bone marrow transplantation for sickle cell anemia. Biol Blood
Marrow Transplant. 2001 Dec;7(12):665-73.
https://doi.org/10.1053/bbmt.2001.v7.pm11787529
PMid:11787529
- Krishnamurti
L, Kharbanda S, Biernacki MA, Zhang W, Baker KS, Wagner JE, Wu CJ.
Stable Long-Term Donor Engraftment following Reduced-Intensity
Hematopoietic Cell Transplantation for Sickle Cell Disease. Biol Blood
Marrow Transplant. 2008;14(11):1270-8.
https://doi.org/10.1016/j.bbmt.2008.08.016 PMid:18940682
- Kean
LS, Manci EA, Perry J, Balkan C, Coley S, Holtzclaw D, Adams AB, Larsen
CP, Hsu LL, Archer DR. Chimerism and cure: Hematologic and pathologic
correction of murine sickle cell disease. Blood. 2003;102(13):4582-93.
https://doi.org/10.1182/blood-2003-03-0712 PMid:12933586
- Andreani
M, Testi M, Gaziev J, Condello R, Bontadini A, Tazzari PL, Ricci F, De
Felice L, Agostini F, Fraboni D, Ferrari G, Battarra M, Troiano M,
Sodani P, Lucarelli G. Quantitatively different red cell/nucleated cell
chimerism in patients with long-term, persistent hematopoietic mixed
chimerism after bone marrow transplantation for thalassemia major or
sickle cell disease. Haematologica. 2011 Jan 1;96(1):128-33.
https://doi.org/10.3324/haematol.2010.031013 PMid:20935000
PMCid:PMC3012776
- Lefèvre
N, Dufour D, Gulbis B, Lê P-Q, Heijmans C, Ferster A. Use of
hydroxyurea in prevention of stroke in children with sickle cell
disease. Blood. 2008 Jan 15;111(2):963-4; author reply 964.
https://doi.org/10.1182/blood-2007-08-102244 PMid:18182580
- Grace
RF, Su H, Sena L, Poussaint TY, Heeney MM, Gutierrez A. Resolution of
cerebral artery stenosis in a child with sickle cell anemia treated
with hydroxyurea. Am J Hematol. 2009;85(2):NA-NA.
- Abboud
MR, Cure J, Granger S, Gallagher D, Hsu L, Wang W, Woods G, Berman B,
Brambilla D, Pegelow C, Lewin J, Zimmermann RA, Adams RJ, STOP study.
Magnetic resonance angiography in children with sickle cell disease and
abnormal transcranial Doppler ultrasonography findings enrolled in the
STOP study. Blood. 2004 Apr 1;103(7):2822-6.
https://doi.org/10.1182/blood-2003-06-1972 PMid:14684415
- Bernaudin
F, Verlhac S, Arnaud C, Kamdem A, Hau I, Leveille E, Vasile M, Kasbi F,
Madhi F, Fourmaux C, Biscardi S, Gluckman E, Socie G, Dalle J-H, Epaud
R, Pondarre C. Long-term treatment follow-up of children with sickle
cell disease monitored with abnormal transcranial Doppler velocities.
Blood. 2016 Apr 7;127(14):1814-22.
https://doi.org/10.1182/blood-2015-10-675231 PMid:26851292
- Iannone
R, Casella JF, Fuchs EJ, Chen AR, Jones RJ, Woolfrey A, Amylon M,
Sullivan KM, Storb RF, Walters MC. Results of minimally toxic
nonmyeloablative transplantation in patients with sickle cell anemia
and ß-thalassemia. Biol Blood Marrow Transplant. 2003 Aug;9(8):519-28. https://doi.org/10.1016/S1083-8791(03)00192-7
- Horan
JT, Liesveld JL, Fenton P, Blumberg N, Walters MC. Hematopoietic stem
cell transplantation for multiply transfused patients with sickle cell
disease and thalassemia after low-dose total body irradiation,
fludarabine, and rabbit anti-thymocyte globulin. Bone Marrow
Transplant. 2005 Jan 8;35(2):171-7. https://doi.org/10.1038/sj.bmt.1704745 PMid:15531901
- Hsieh
MM, Kang EM, Fitzhugh CD, Link MB, Bolan CD, Kurlander R, Childs RW,
Rodgers GP, Powell JD, Tisdale JF. Allogeneic Hematopoietic Stem-Cell
Transplantation for Sickle Cell Disease. N Engl J Med. 2009 Dec
10;361(24):2309-17. https://doi.org/10.1056/NEJMoa0904971 PMid:20007560 PMCid:PMC3627532
- Saraf
SL, Oh AL, Patel PR, Jalundhwala Y, Sweiss K, Koshy M, Campbell-Lee S,
Gowhari M, Hassan J, Peace D, Quigley JG, Khan I, Molokie RE, Hsu LL,
Mahmud N, Levinson DJ, Pickard a S, Garcia JG, Gordeuk VR, Rondelli D.
Nonmyeloablative Stem Cell Transplantation with Alemtuzumab/Low-Dose
Irradiation to Cure and Improve the Quality of Life of Adults with
Sickle Cell Disease. Biol Blood Marrow Transplant. 2016
Mar;22(3):441-8. https://doi.org/10.1016/j.bbmt.2015.08.036 PMid:26348889
- Schattman GL. Cryopreservation of Oocytes. Solomon CG, editor. N Engl J Med. 2015 Oct 29;373(18):1755-60. https://doi.org/10.1056/nejmcp1307341
- Bola-os-Meade
J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, Brodsky R a.
HLA-haploidentical bone marrow transplantation with posttransplant
cyclophosphamide expands the donor pool for patients with sickle cell
disease. Blood. 2012;120(22):4285-91. https://doi.org/10.1182/blood-2012-07-438408 PMid:22955919 PMCid:PMC3507140
- Maschan
M, Shelikhova L, Ilushina M, Kurnikova E, Boyakova E, Balashov D,
Persiantseva M, Skvortsova Y, Laberko A, Muzalevskii Y, Kazachenok A,
Glushkova S, Bobrynina V, Kalinina V, Olshanskaya Y, Baidildina D,
Novichkova G, Maschan A. TCR-alpha/beta and CD19 depletion and
treosulfan-based conditioning regimen in unrelated and haploidentical
transplantation in children with acute myeloid leukemia. Bone Marrow
Transplant. 2016 May 25;51(5):668-74. https://doi.org/10.1038/bmt.2015.343 PMid:26808573
- Mainardi
C, Tumino M, Gazzola M V, Rampazzo A, Scarpa M, Messina C. TCRaß CD19
depletion in allogeneic haematopoietic stem cell transplantation
performed for Hurler syndrome. Bone Marrow Transplant. 2016 Mar
9;51(3):438-9. https://doi.org/10.1038/bmt.2015.258 PMid:26551775
- Kang
EM. Mobilization, collection, and processing of peripheral blood stem
cells in individuals with sickle cell trait. Blood. 2002 Feb
1;99(3):850-5. https://doi.org/10.1182/blood.V99.3.850 PMid:11806986
- Fitzhugh
CD, Hsieh MM, Bolan CD, Saenz C, Tisdale JF. Granulocyte
colony-stimulating factor (G-CSF) administration in individuals with
sickle cell disease: time for a moratorium? Cytotherapy. 2009
Jan;11(4):464-71. https://doi.org/10.1080/14653240902849788 PMid:19513902 PMCid:PMC2747259
[TOP]