Vincenzo De Sanctis1, Christos Kattamis2, Duran Canatan3, Ashraf T. Soliman4 , Heba Elsedfy5, Mehran Karimi6, Shahina Daar7, Yasser Wali8, Mohamed Yassin9, Nada Soliman10, Praveen Sobti11, Soad Al Jaouni12, Mohamed El Kholy5, Bernadette Fiscina13 and Michael Angastiniotis14
1 Pediatric and Adolescent Outpatient Clinic, Quisisana Hospital, Ferrara, Italy.
2 First Department of Paediatrics, University of Athens, Athens, Greece.
3 Director of Thalassemia Diagnosis Center of Mediterranean Blood Diseases Foundation, Antalya, Turkey.
4 Department of Pediatrics, Division of Endocrinology, Alexandria University Children's Hospital, Alexandria, Egypt.
5 Department of Pediatrics, Ain Shams University, Cairo, Egypt.
6 Hematology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran.
7 Department of Haematology, College of Medicine and Health Sciences, Sultan Qaboos University, Sultanate of Oman
8
Pediatric Hematology Unit, Child Health Department, Sultan Qaboos
University Hospital, Muscat, Oman and Department of Pediatrics,
Alexandria University Children's Hospital, Egypt.
9 National Center for Cancer Care and Research, Medical Oncology Hematology Section HMC, Doha, Qatar.
10 Primary Health Care, Ministry of Health, Alexandria, Egypt.
11 Professor, Pediatric Hemato-Oncology, Christian Medical College and Hospital, Ludhiana, Punjab, India.
12 Head,
Division of Pediatric Hematology Oncology, Deputy Chair of Hematology
and Head, Section of Hematology Research Lab, King Fahd Medical
Research Center, Department of Hematology Faculty of Medicine, King
Abdulaziz University, Jeddah, Kingdom of Saudi Arabia.
13 Department of Pediatrics, NYU School of Medicine, New York, USA.
14 Medical Advisor, Thalassemia International Federation (TIF), Nicosia, Cyprus.
Corresponding
author: Vincenzo De Sanctis MD,
Pediatric and Adolescent Outpatient Clinic, Quisisana Hospital, 44100
Ferrara, Italy; Tel.: +39 0532 770243; E-mail:
vdesanctis@libero.it
Published: February 20, 2017
Received: November 20, 2016
Accepted: February 5, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017018 DOI
10.4084/MJHID.2017.018
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Haemoglobinopathies constitute the commonest recessive monogenic
disorders worldwide, and the treatment of affected individuals presents
a substantial global disease burden. β -thalassaemia is characterised
by the reduced synthesis (β+) or absence (βo) of the β-globin chains in
the HbA molecule, resulting in accumulation of excess unbound
α-globin chains that precipitate in erythroid precursors in the bone
marrow and in the mature erythrocytes, leading to ineffective
erythropoiesis and peripheral haemolysis. Approximately 1.5% of the
global population are heterozygotes (carriers) of the β-thalassemias;
there is a high incidence in populations from the Mediterranean basin,
throughout the Middle East, the Indian subcontinent, Southeast Asia,
and Melanesia to the Pacific Islands.
Aim:
The principal aim of this paper is to review, from a historical
standpoint, our knowledge about an ancient disease, the β-thalassemias,
and in particular, when, how and in what way β-thalassemia spread
worldwide to reach such high incidences in certain populations.
Results:
Mutations involving the ß-globin gene are the most common cause of
genetic disorders in humans. To date, more than 350 β-thalassaemia
mutations have been reported. Considering the current distribution of
β- thalassemia, the wide diversity of mutations and the small number of
specific mutations in individual populations, it seems unlikely that
β-thalassemia originated in a single place and time.
Conclusions:
Various processes are known to determine the frequency of genetic
disease in human populations. However, it is almost impossible to
decide to what extent each process is responsible for the presence of a
particular genetic disease. The wide spectrum of β-thalassemia
mutations could well be explained by looking at their geographical
distribution, the history of malaria, wars, invasions, mass migrations,
consanguinity, and settlements. An analysis of the distribution of the
molecular spectrum of haemoglobinopathies allows for the development
and improvement of diagnostic tests and management of these disorders.
|
Introduction
Haemoglobinopathies
constitute the commonest recessive monogenic disorders worldwide. They
fall into two main groups: the thalassemia syndromes and the structural
hemoglobin variants (abnormal hemoglobins). α, β, and δβ thalassemias
are the main types of thalassemia with clinical importance; the most
frequent and clinically important structural hemoglobin variants are
HbS, HbE, HbC and HbD. The treatment of patients presents a substantial
global disease burden.
Variants of thalassaemias and main
abnormal haemoglobins interact to produce a wide range of clinical
disorders of varying severity.[1-3] Homozygotes for
β-thalassemia may develop either thalassemia major or thalassemia
intermedia. Individuals with thalassemia major are usually diagnosed
within the first 2 years of life and require regular blood transfusions
to survive.[3] Patients with thalassemia intermedia
are diagnosed later and as a rule, have milder anemia and do not depend
on transfusion for survival.
The clinical manifestations of
β-thalassemias are highly variable. They range from asymptomatic cases
with mild (silent) mutations, to those with mild hypochromic anemia, to
others with moderate and severe lifelong transfusion-dependent anemia
and multi organ involvement.[1]
β-thalassemia is
mainly caused by mutations resulting in a single nucleotide
substitution, small deletions or insertions within the β-globin gene or
its immediate flanking sequence and, rarely, by gross deletions.[2,3] To date, more than 350 β-thalassaemia mutations have been reported in the IthaGenes database.[4]
Nearly
all β-thalassemia variants are inherited in a Mendelian recessive
manner, but there is a small subgroup of β-thalassemia alleles that
behave in a dominant fashion.[1]
The principal
aim of this paper is to review, from a historical standpoint, our
knowledge of an ancient disease, the β-thalassemias, and to discuss
when, how and in what way β-thalassemia has spread in the old
world to reach such high incidences in certain populations. To this
end, knowledge of the molecular defects that prevail in each country
provides valuable information that can be used for population screening
in prevention programmes and facilitates effective prenatal diagnosis.[2,3] Epidemiology and Global Burden of the Thalassemia Disorders
The
thalassemias have a high incidence in a broad area extending from the
Mediterranean basin and parts of Africa, throughout the Middle East,
the Indian subcontinent, Southeast Asia, and Melanesia in to the
Pacific Islands.[4,5]
The carrier frequencies for
β-thalassemia in these areas range from 1 to 20%, and rarely may be
higher. The frequencies for the milder forms of β-thalassemia
are much greater, varying from 10 to 20% in parts of sub- Saharan
Africa, to 40% or more in some Middle Eastern and Indian populations,
to as high as 80% in northern Papua New Guinea and some isolated groups
in the northeast of India.[6]
Globally, it is
estimated that there are 270 million carriers with abnormal
haemoglobins and thalassemias, of which 80 million are carriers of
β-thalassemia. Recent surveys suggest that between 300,000 and 400,000
babies are born with a serious hemoglobin disorder each year (23,000
with β-thalassemia major) and that up to 90% of these births occur in
low- or middle-income countries.[1-6]
Origin, Spread and Evolutionary History of Beta-Globin Gene Mutations
In
the major hemoglobinopathies, adult haemoglobin A, composed of two
α-globin and two β-globin chains, is altered by genetic variants that
encode single amino acid substitutions in β-globin (as in HbS, HbC, HbD
and HbE) or reduced production of β-globin chains as in β-thalassaemia.[1-3] Rarely do β-thalassemias result from gross β- gene deletion.[3]
β-thalassaemia is characterised by the partial reduction of synthesis (mild=β++, severe=β+) or total reduction (βo),
of the β-globin chains in the HbA molecule. This results in
accumulation of excess, unbound α-globin chains that precipitate in
erythroid precursors in the bone marrow and in the mature erythrocytes,
leading to ineffective erythropoiesis and peripheral haemolysis.[1-3]
While α-
and β- thalassemias are caused by more than 450 mutations, almost 1200
mutations have been reported that change the structure of the α- and β-globin genes to create abnormal hemoglobin tetramers of HbA.[1-3] Among this broad spectrum of mutations, some are very frequent and population specific.
DNA
haplotypes in the β-globin gene can help to determine the date of
origin, track gene flow of a particular β-globin gene mutation and
detect major migrations.
The β0-39 nonsense (C→T) and the β+ IVS-1-110 (G>A) mutations are largely prevalent in the Mediterranean basin.1-3 β+ IVS-1-110 is found in the eastern part of the Mediterranean area, including Turkey,[7] Lebanon,[8] and Egypt.[9] The β+ IVS-1-110 seems to be older than the β0-39 mutation because of its nearly exclusive association with ancestral haplotype I.[10] In addition, β+
IVS-1-110, which is believed to have arisen in the Middle Orient and
reaches its highest frequencies in the Eastern Mediterranean
populations, may have been introduced to the other countries by a
variety of settlers from the East. Its diffusion to the occident
probably happened at the time of the expansion of Greek civilization,
starting from the eighth century B.C.
Most Common β-Thalassemia Variants in the Mediterranean Belt and Arabic Countries
1. Italy: In the Po Delta area (northern Italy) β+ thalassemia represents approximately 50% of the total thalassemia major population. The production of β globin in a β+ homozygote and in a β+, β °39 (nonsense mutation at codon 39) double heterozygote is approximately 20% and 10%, respectively, of total non-α globin synthesis. This suggests that in the Po River Delta region the most common thalassemic genes are β °39 and β+
IVS-1-110 (G>A) mutation. It is possible that IVS-1-110 (G>A)
mutation came from Greece to this area where Greek influence has been
reported in the past.[11]
On the other hand, the β0-39 (C>T) mutation is found almost exclusively in Sardinia.[3]
This dominating mutation seems to have undergone a significant founder
effect influence on the island. It is also highly prevalent in
continental Italy[12] and Spain[13] and is frequently encountered in Portugal[14] and Tunisia.[15]
The exclusive presence of β0-thalassemia
type in southern Sardinia, which most likely arose from a single
mutational event, is not surprising since Sardinia has been free from
external colonisation for a long period of time. In fact, Greek
colonisation was localised to the north-east of the island while Romans
and Carthaginians settled only along the southern coast.[16]
Moreover, other populations, such as Vandals, Goths, Saracens, Pisans,
and Spanish did not stay on the island long enough or in a sufficient
number to modify the genetic structure of the autochthonous population
significantly. The relatively high prevalence of β+- thalassemia in the northern part of the island was probably due to the Greek colonisation.[16,17]
More heterogeneity is detected in Sicily, where there are a variety of mutations and where the frequency of the β0-39 mutation is only 37%.[17]
2. Cyprus: In Cyprus, the estimated β-thalassaemia carrier rate is around 12–15% of the population.[18]
The most common β-globin gene mutation is IVS-1-110 (G> A), with a
percentage of 74-80%, followed by three other alleles, specifically
IVS-2-745 (C> G), IVS-1-6 (T> C), IVS-1-1 (G> A), with
frequencies of 5–8%.[19] The presence of various
haemoglobin variants in low prevalence, such as Hb S, Hb D, and Hb
Lepore, is likely to be directly linked to the history of Cyprus, as
archeological monuments have been found throughout the island
signifying the presence for many years of the Greeks, Syrians,
Persians, Arabs, Byzantines, Franks, Venetians, and Turks.[20]
3. Greece: Greece is a country of approximately 11 million people with a mean frequency of β-thalassaemia carriers of 7.4%.[21]
Extensive
studies have disclosed that the distribution of β-thalassemia carriers
is extremely uneven. The low fertile areas of Thessaly, Western
Peloponnesus, and Western Epirus, as well as the islands of Rhodes,
Lesbos, Corfu and other areas, displayed frequencies of up to 15%. In
contrast, in the high altitude areas (that were free of malaria) and
Macedonia, the prevalence of heterozygotes was significantly lower.[22]
The
molecular basis of β-thalassaemia is very heterogeneous in Greece, and
up to 30 β-thalassaemia mutations have been observed. The molecular
characterization of β- thalassaemia was evaluated in three studies on
857 thalassemia patients (106 with thalassaemia intermedia (TI)
covering more than 25% of the 3,241 patients with TM and TI registered
in 2010.[23]
In the three studies, 27 β-thalassemia mutations were observed. Of interest are:
i)
the rates of the prevalent β-thalassaemia mutations. In order of
frequency they were: IVS-1-110(G>A) with an incidence of 43.8%;
Codon 39(C>T), 18%; IVS-1-1(G>A), 12.7%; IVS-1-6(T>C), 6.9%;
IVS-2-745(G>A), 4.7% and IVS- 2-1(G>A), 3%. The five mutations
cover 89.1 % and the six 91.4% of the β-thal genes.
(ii) the
three very mild mutations (+1480 ( C>T), -101 (C>T) and + 33
C>G ) that were found only in TI patients covering 22% of
TI β-thalassaemia mutations.[21-23]
4. Turkey:
The overall incidence of β-thalassemia trait in Turkey is 2.1% (range:
0.6-13%). At present, the Turkish population is about 80 million, with
1.6 million people with thalassemia trait and about 5500 patients with
homozygous thalassemia and other hemoglobinopathies.[24]
By
April 1999, the total number of β-thalassemia alleles described in the
Turkish population was ~40, and this number can be considered as a
testimony of past settlements in Asia Minor.[23] The
most frequent thalassemia allele in the Turkish population is IVS-I-110
(G>A) mutation (40%), being the most common thalassemia mutation in
the majority of the high risk regions of the Mediterranean area. The
other common mutations in Turkey are: IVS-1-6(T>C), FSC-8(-AA),
IVS-1-1(G>A), IVS-2-745(G>A), Cd39 (C>T), -30 (T>A) and
FSC-5 (-CT). Malarial selection for the oldest β-thalassemia allele in
Anatolia (i.e., IVS-1-110 G>A) may have occurred between 6,500 and
2,000 BC. Since then, Arab, Byzantine, Sumerian, Hittite and Turkish
societies have played an important role in the history of the country,
leading to a complex ethnic structure.
Currently, the minority
of the population living in Adana and Mersin, in southern Turkey, is of
Arab and African origin, and are known as “Eti-Turks”, whose ancestors
immigrated from Syria and Egypt centuries ago. From that date on, most
of the common β-thalassemia mutations in Turkey were established, and
by the 13th century A.D., most of them were present at frequencies close to those 7% observed today.[25,26]
Turkey's large molecular heterogeneity can be explained by its unique
geographical position and rich history, an important crossroad between
cultures, civilizations, and continents for several centuries.
The three most prevalent mutations include: IVS-1-110 (G>A): 39.5%, IVS -1-6 (T>C):10.1% and FSC -8 (-AA): 5.5%.
5. Arab populations.
The standard definition of the Arab world includes the 22 states and
territories from the Atlantic Ocean in the west to the Arabian Sea in
the east, and from the Mediterranean Sea in the north to the Horn of
Africa and the Indian Ocean in the southeast. It has a combined
population of around 350 million people, one third of who are under 15
years of age. β-thalassemia is encountered in polymorphic frequencies
in almost all Arab countries with carrier rates ranging from 1 to 11%.[27]
Various papers reporting the spectrum of β-thalassemia mutations in different Arab countries have been published.[28-46] However, data for some countries is still lacking or only preliminary.
The spectrum of β-thalassemia mutations in the Arab populations has been extensively reported by Zahed in 2001,[41] Tadmouri in 2003[42] Haj Khelil in 2010[40] and Hamamy in 2013.[27]
The
number of mutations detected in each population varies depending on its
origin and interaction with other populations as well as the methods
used for characterisation.[27-43]
Among Arabs,
the heterogeneity of these mutations varies from 44 different
mutations in UAE to 10 in Eastern Saudi Arabia. The most widespread and
common mutation among Arabs is IVS-1-110 (G>A). The latter mutation
has its highest prevalence in Cyprus and Greece suggesting that it may
be of Greek origin. In the Eastern Arabian Peninsula, the Asian Indian
mutations (IVS-1-5 (G>C), codons 8/9 (+G) and IVS-1 (−25 bp del) are
more common.[27]
Although β-thalassemia
mutations reported are mainly of Mediterranean and Asian origin, some
countries have unique mutations, e.g. Hb Dhofar in Oman.[29]
Codon 39 (C>T) has been found in all Arab countries without
exception. IVSI-110 (G>A), the most common mutation, has rates of
12%–38% in Arab countries of the eastern Mediterranean but reaches
lower frequencies in countries around the Gulf (0%–2%). IVS-2-1
(G>A) has been detected in all countries except Tunisia and Algeria.
The highest frequency has been reported in North Jordan (20%) (IVS-2-1)[43] and in Kuwait.[44]
The
most widespread and common mutations are presumably the oldest. Codon
39 (C>T) is believed to be of Roman origin, and IVS-1-110 is
believed to have arisen in Turkey. It reaches its highest frequency in
the Eastern Mediterranean Arab countries and may have been introduced
to other countries by a variety of settlers from the East, including
Turks, Greeks, or Phoenicians.[41]
a. Egypt:
Egypt is divided into three main geographical regions: the Nile Valley,
the Eastern desert and the Western desert. The Nile Valley represents
4% of the area of Egypt and is divided into Upper Egypt, Lower Egypt,
Suez Canal, and Northern coast lakes regions.
Cairo’s population has risen to more than 18 million (the highest population density in Egypt).
Egyptians
are mainly descended from ancient Egyptians (94%). Ethnic minorities in
Egypt include Nubians, Berbers, Bedouin Arabs, Beja and Dome (4%) and
others (2%). The position of Egypt in the center of the Middle East,
contiguous with the Mediterranean countries, has facilitated genetic
admixture of Egyptians with several populations of diverse geographic
and ethnic origins.[35]
El-Hashemite et al.
reported that of 1.5 million annual live births, approximately 1000
babies are born with β-thalassemia major. The most common mutations in
Egyptian children with β-thalassemia are IVS-1-110(G>A) 48%,
IVS-1-6 (T>C) 40%, IVS-1-1(G>A) 24%, IVS-1-5(G>C) 10%,
IVS-2-848 (C>A) 9%, IVS-2-745(C>G) 8%, IVS-2-1(G>A) 7%.[35]
b. Oman:
The nature of the β-thalassaemia mutations that have been determined in
the Omani population suggests that the majority have been introduced by
gene flow. There was active commerce between Oman and the area around
the Indus Valley for centuries. Burial sites and pottery dating to
around 3,000 B.C. have also been unearthed in the surrounding Gulf
countries. Using archaeological data from all these various sites it
has been possible to establish that there was trade between Mesopotamia
(present day Iraq), the lands bordering the Arabian Gulf and the Indus
Valley peoples. Historically, Oman was a major maritime nation with
links to the Far East and India on the one hand and East Africa and
Egypt on the other, as well as trade in the Arabian Gulf. IVS-1-5
(G>C), a severe β+ allele, is the
most prevalent of the mutations thus far described in Oman. A detailed
analysis of the distribution of β-thalassaemia mutations in Pakistan
shows that the IVS I-5 (G>C) mutation is widespread in that country,
reaching a peak incidence in the Baluchis (76.2%) and having a high
prevalence among the Sindhis (43.9%). It is also found at a high
frequency in India.[27,29]
Other
common β-thalassemia alleles common in Oman are the codon 44 (-C)
(frequent in Iraq),37 IVS-1-3’ –25bp (epicentre Bahrain),[34] IVS-2-1 (G>A) (Indian subcontinent)[47] mutations, and the 619bp deletion at the 3’ end of the beta globin gene (Indian subcontinent),[47]
all of which almost certainly were introduced to Oman through gene
flow. The only mutation unique to Oman is Hb Dhofar, which has
originated in the southern region of the Sultanate and thus far has
only been described in Omanis.[29]
c. Qatar:
Qatar is a peninsula bordering the Arabian Gulf and Saudi Arabia. The
country’s population has been roughly split with 20% native Qatari,
largely tribal, and 25% other Arabs from Egypt, Syria, Iraq, Lebanon,
Yemen, Palestine, and Jordan. The rest of the population (55%) consists
of expatriate workers from the East and the West.
Qatar’s
history is very rich indicating the various phases that led to the
ultimate development of the present State. The first trace of human
settlements was found in the Qatar peninsula around 4,000 B.C. At the
beginning of the 16th century, Qatar
fell under the control of the Portuguese who were successful in
establishing their control in many parts of the Arabian Peninsula.
Later, the Ottomans ruled Qatar for four centuries.
The
frequency of β- thalassemia heterozygotes is estimated to be 2-3%.
Al-Obaidli et al. analyzed the molecular basis of β-thalassemia in
Qatar. They found the most common mutant alleles were IVS-1-5 (G>C)
and codon 8/9 (+G), representing 35.4% and 26.1% of the total,
respectively. Most of these two mutations are homozygous, probably
because of the high rate of consanguinity. The frequencies of three
other common mutant alleles, IVS-2-1 (G>A), 25bp deletion and
IVS-1-110 (G>A) are higher in surrounding locations, such as
Southern Iran, Kuwait, eastern Saudi Arabia and Bahrain.[38]
Recently,
a novel β-thalassemia deletional variant allele in an ethnic Qatari
patient was reported. The deletion spans exon 1, the entire intron 1
and the first two bases of exon 2 causing a frameshift and the
premature appearance of a stop codon. The presence of this novel
deletional allele in a compound heterozygous state with a
non-deletional allele is alarming in a diagnostic setting, especially
in the absence of family studies.[39]
6. Iran:
Iran, centrally located in the Asian continent, has served for
centuries as a gateway for movement of human populations across diverse
spheres of Asia and Europe. The Iranian population consists of diverse
ethnic and linguistic groups namely Arabs, Armenians, Assyrians,
Azeris, Baluchis, Gilaks, Mazandarani, Kurds, Lurs, Persian, Turkmen,
and Zoroastrians.
In the eastern Mediterranean region, Iran is one
of the major centers for the prevalence of β-thalassemia. Due to the
high consanguinity in the population, it is estimated that there are
more than three million β-thalassemia carriers (4-8%) and 20,000
patients.
The gene frequency of β-thalassemia is high and varies
considerably between areas, with double the country average rate in
Mazandaran, Sistan and Baluchistan, Fars, Hormozgan, and Kerman
provinces and half the average country rate in provinces of Tehran,
East Azerbaijan, Khorasan, Hamedan, Yazd and West Azerbaijan. There are
more than 47 different β-globin gene mutations. Among them, the most
common is IVS-2-1 (G > A; beta0) mutation, followed in decreasing order of frequency by IVS-1-5 (G > C, beta+), codons 8/9 (+G, beta0), IVS-1-110 (G > A, beta+), IVS-1-1(G > A, beta0), 25 bp deletion (beta0), IVS-I-6 (T > C, beta+), codon 5 (-CT, beta0), and codon 39 (C >T, beta0) mutations.[46]
It seems that codons 8/9 (+G) might reflect two historical
events: one connected to the secular trade along the great Silk
Road which extends from Xian in China through the Indian subcontinent
to Iran and the Eastern Mediterranean and the other to the invasion of
the Mongols (1,220 A.D.) and the Tatars (1,380–87 A.D.). The prevalence
of codon 8/9 (+G) in the Northeast of Iran, and toward the center and
northwest, may be related to the Oghuz Turks who migrated there from
central Asia.[48]
From extensive studies
on the geographical distribution of the molecular basis of
β-thalassemia, it has been shown that, despite the wide spectrum of
mutations, the number of prevalent molecular defects for each
population is limited.
The information provided in Table 1
is derived from published reports. The reported data often do not
represent the entire population. Therefore, each single publication
should be judged and used according to its merits, including sample
size, the population group studied, geographical location within a
country, and methodology used. A complete updated list of β-thalassemia
variants is available through the Globin Gene Server Web Site (http://www.globin.cse.psu.edu).
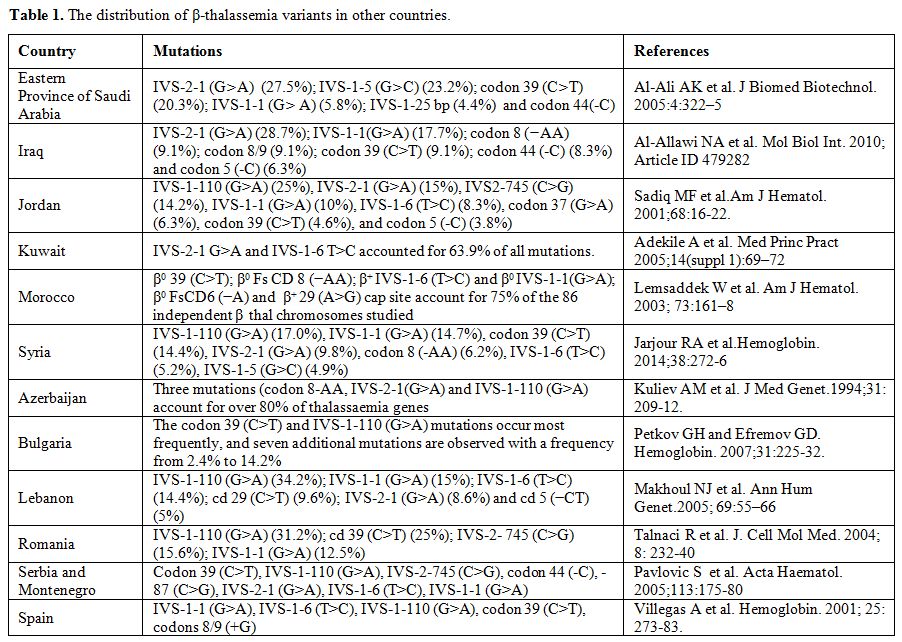 |
Table 1. The distribution of β-thalassemia variants in other countries. |
The Phylogeography and Phytogeographic Brief History of People in Ancient Times
Genetic
data on living human populations have been used to reconstruct the
evolutionary history of the human species by considering how global
patterns of human variation could be produced, given different
evolutionary scenarios. Various processes (selection, mutation,
migration and genetic drift) are known to determine the frequency of
genetic diseases in human populations, but so far it has proved almost
impossible to identify to what extent each process is responsible for
the presence of a particular genetic disease.[49-54]
The
most controversial issue at present is exactly when and how these
anatomically and genetically modern populations first spread from
Africa to other parts of Asia and Europe. There are two main
possibilities. The first is that the initial expansion occurred via
North Africa and the Nile valley, with subsequent dispersals to the
west into Europe and to the east into Asia.[50-54]
The second is that the initial dispersal was from Ethiopia, across the
mouth of the Red Sea, and then either northward through Arabia or
eastward along the south Asian coastline to Australasia.[55,56] The strongest evidence at present for the second pothesis is provided by the mtDNA lineage- analysis patterns.[50,51]
This model would mean that modern populations in southwest Asia and
Europe must have reached these areas substantially later, via western
or central Asia.[51]
Why did modern human populations disperse from Africa ca. 60,000 years ago?
Childe[57]
suggested that, after the glaciations, North Africa and South-west Asia
became drier and humans began to aggregate in areas where water was
available.
The hypothesis of Childe on the role of the Younger
Dryas (a geological period from c. 12,900 to c. 11,700 calendar years
ago, characterized by a cool and dry climate) which was contemporary
with the beginning of cultivation (c. 10,000 B.C.) in the region, is
supported by recent studies from ancient sites of the Middle Euphrates
region.[58-61]
These suggest that the world’s
first civilization emerged in the late fourth millennium B.C. in
southern Iraq, developing urban centers situated along the banks of the
Tigris and Euphrates Rivers (Mesopotamia). This area, a small region of
south-east Turkey and north-east Syria around the Middle Euphrates,
might be the cradle of wheat agricultural innovation.[62-66]
After domestication, free-threshing wheat spread west through the Mediterranean basin, reaching its western edge by 5,000 B.C.[67,68].
Between 2,000 B.C. and 1,100 A.D., Anatolia served as the ground for
several civilizations (Hittite, Persian, Greek, Roman, Byzantine, and
Seljuk), resulting in an intensive flow of mutations into and out of
present-day Turkey. Before reaching the Western Europe, agriculture
spread in Greece during 4th millennium B.C.; it is possible that during that period Greece was one of the most malarious regions of the Mediterranean.[69,70]
During the 8th
century B.C. the Greeks arrived in Italy. They came from Euboea,
Argolis, Locris, Crete and the Aegean islands, settling on the southern
coasts (from Campania to Apulia) and eastern and southern Sicily. They
founded many prosperous colonies whose economy was generally based on
agriculture and commerce.
It is possible that thalassemia
existed among Sicily's native populations (Lymians, Sicanians, and
Sicels) before that time, as it is postulated that some of these
earlier inhabitants migrated to Sicily from the eastern Mediterranean,
albeit probably before 4,000 B.C. A further historical influx of the
thalassemia genes may be associated with mass immigration of the
Saracens (Arabs) in the ninth and tenth centuries, as this population
had a strong Middle-eastern Semitic component.
Silvestroni and
Bianco recorded a very high percentage of β-thalassemia trait in people
originating from the area of the Po River’s delta (Upper Adriatic): 10%
in Ferrara, 16% in Codigoro and 12% in Pomposa. These towns in the past
were in direct connection to the important Greek emporium of Spina
through an ancient branch of the Po River.[71] Spina was a commercial port founded at the beginning of the 6th
century B.C. by Etruscans. Because of its location, along the Po River
delta, the port became an ideal community to facilitate trade with
the Greeks. The port was abandoned towards the end of the 3rd century B.C.[71]
The expansion of the Ottoman Empire (15th and 16th
centuries) towards Eastern Europe, Northern Africa, and Central Asia
permitted further spread of β-thalassemia mutations in and out of
Anatolia making it a melting-pot of a large number of alleles. The
migration of many Muslim groups living in former Turkish territories in
Southeastern Europe during the decline of the Ottoman Empire (starting
from 1914 AD) contributed more to the racial mixture (i.e., the
introduction of IVS-2-745, -87, IVS-2-654) .[24-26].
Factors Influencing the Global Distribution of Thalassemias
Several other factors have contributed to the global distribution of thalassemias. These factors include:
1.The resistance to malaria:
World-wide, the distribution of malaria and the common
haemoglobinopathies have largely confirmed the close relationship
between malaria and high prevalence of hemoglobinopathy traits (HbS and
β-thalassemia) and G-6PD deficiency in populations living in highly
malarious areas. Micro-population epidemiological surveys have
established this association. [72,73]
In 1948,
Haldane suggested that the heterozygote state in β-thalassaemia
provided a selective advantage against attack by the malarial parasite.[74]
This theory was based on the fact that homozygous beta thalassaemia
results in a lethal condition, whereas the heterozygote only has a mild
microcytic anaemia. If the carrier had no selective advantage over the
normal or was less fit, then a very high mutation rate for beta
thalassaemia would be required to secure equilibrium. Haldane felt
that the heterozygote was fitter than normal, and he suggested that
this might be due to the resistance of the microcytic erythrocytes to
attack by the malaria parasite.[74,75]
It is not
possible at this point to say exactly when malarial parasites evolved.
Although there is no direct evidence as to the antiquity of malaria,
there are many historical references to intermittent fevers and
splenomegaly. Most authors believe that malaria was established in
pre-historic times; it may have originated in tropical areas of the Old
World, but the Pleistocene glaciations (130,000- 10,000 years ago)
delayed its spread in the Northern Hemisphere.[76,77]
Until
about 3,000 - 4,000 years ago population groups were small and isolated
and infection could not spread easily. In addition, the vector for
malaria, Anopheles gambiae, breeds in stagnant water that does not
generally occur in the unbroken tropical rain forest. It is only when
forest is cut down to make way for agriculture that these stagnant
pools are found. The spread of malaria probably evolved with the
development of tools during the Neolithic period (~6,000 B.C.–1,000
B.C.), which has traditionally been associated with the origins of
farming and more settled human groups. It is possible that the
infection then spread to most of the tropical and much of the temperate
regions of the world.
Genetic variants that affect the structure
and production of the β- or α-chains of haemoglobin are variously
associated with protection from a range of clinical manifestations of P. falciparum
infection. The degree of protection varies between haemoglobinopathies,
but in general is greatest against severe malaria, moderate against
uncomplicated malaria, and absent against asymptomatic P. falciparum parasitaemia.[78-83]
HbS and to a lesser extent HbC protect from malaria but not from
parasitaemia, suggesting that these haemoglobin variants prevent the
transition from asymptomatic parasitaemia to malaria. This protective
effect may derive from the abnormal display of parasite virulence
factors on the surface of parasitized HbC and HbS erythrocytes,[78,79]
possibly owing to the disruption of the parasite’s remodeling of
erythrocyte’s intracellular trafficking network by HbS and HbC.[80] Additionally, the age-dependent nature of malaria protection due to HbAS[81,82] and α-thalassaemia[83] among children in recent reports support a protective mechanism based upon an enhanced acquisition of malaria immunity.
Additional
possible mechanisms for protection due to haemoglobinopathies include
an enhanced clearance of parasitized erythrocytes,[84] impaired parasite growth,[85] or the induction of protective immunomodulatory mechanisms by parasitized erythrocytes.[86]
2. Consanguinity:
The roots of Western attitudes toward consanguinity extend back over
1,500 years. Consanguineous marriage is especially common throughout
the Eastern Mediterranean, Anatolia, North Africa and the Indian
sub-continent, where 25-70% of unions involve related family members.
Religious, cultural and economic factors are commonly perceived reasons
for such marriage. As a consequence, at least 8.4% of the world’s
children have related parents. The practice is also accepted in South
America and parts of sub-Saharan Africa. The highest overall prevalence
of consanguineous unions is in poor rural communities, which are
typified by low levels of maternal education, early age at marriage and
first birth, short birth intervals, and longer reproductive spans.[87-89]
3. Nutrition and infections:
Another important factor is the diseases’ epidemiological transition.
Because social and public health standards have gradually improved in
developing countries, nutritional and infectious diseases are less
frequent causes of mortality compared to the past.[1,90,91]
Furthermore,
despite premarital screening and prenatal diagnosis, sociocultural
challenges (traditional customs, religious beliefs or superstition)
especially in countries with high thalassemia trait populations, still
impact on β-thalassaemia major birth rates and global distribution of
thalassemia.[91]
4. Migration: The consistent multi-ethnic migrations of the last decades have considerably changed the epidemiology of hemoglobinopathies (Figure 1).
Healthy carriers of these conditions are living today in many
nonendemic countries, and severely affected children are now born in
areas where these diseases were previously rare or unknown. In view of
population migration, the distribution of the thalassemia gene appears
to be worldwide affecting all ethnic groups except those originating
from western, central and north Europe. The number of patients with
haemoglobinopathies has been estimated to be almost 44,000 in the 10 EU
countries, varying from 150 cases in Sweden to 10,500 in France.[92]
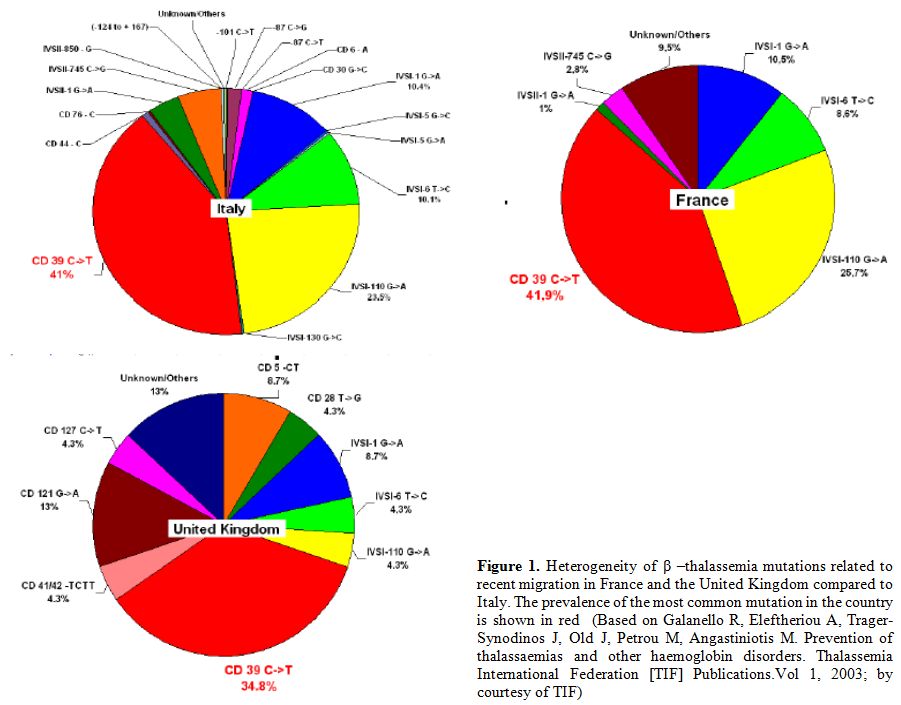 |
Figure 1. Heterogeneity of β–thalassemia
mutations related to recent migration in France and the United Kingdom
compared to Italy. The prevalence of the most common mutation in the
country is shown in red (Based on Galanello R, Eleftheriou A,
Trager-Synodinos J, Old J, Petrou M, Angastiniotis M. Prevention of
thalassaemias and other haemoglobin disorders. Thalassemia
International Federation [TIF] Publications. Vol 1, 2003; by courtesy
of TIF) |
Since β-thalassemia
major has increased markedly in some European countries due to
immigration from Africa and Asia, therefore national programmes of care
and prevention are needed. Furthermore, major health organizations and
funding agencies must support these initiatives.
5. Prevention:
In the late 1970s, pilot programs directed to prevent β-thalassemia
major started in several at-risk populations in the Mediterranean area.
At present, several countries have set up comprehensive national
prevention programs, which include public awareness and education,
carrier screening, and counseling, as well as information on prenatal
diagnosis and preimplantation diagnosis.[3] The
efficacy of the prevention programmes in Italy, Cyprus, and Greece is
reflected by the fact that the number of babies with thalassemia major
has decreased substantially in the last two decades. In Sardinia,
for instance, the number of thalassemia major children born per year
predicted on the basis of the carrier rate, assuming random mating,
shows a reduction from 1:250 live births to 1:1660 in 2009 with an
effective prevention of 85% of the cases.[3] In
Greece, the expected number of 160 annual births of
affected newborns per 100,000 live birth decreased to 8 during 2005 to
2009.[5]
Archeological Remains Suggestive of Hereditary Anemias
Besides
malaria, the evolution of β-thalassemia has been studied from two other
perspectives: macroscopic examination of archeological skeletal remains
on the basis of bone pathology, and analysis of distribution of
mutations in various living populations.
Angel analysed 2334
skeletal specimens coming from several archaeological sites in the
Eastern Mediterranean (in particular from Turkey, Greece, Cyprus, and
Morocco), belonging to the Upper Paleolithic period to Middle Ages. He
used bone modifications, in particular, the porotic hyperostosis of the
skull, as a marker of thalassemia trait and on this basis, started
researching the appearance and diffusion of malaria and thalassemia.
Porotic hyperostosis was classified as a thickening of the cranial
vault caused by expansion of the diploë due to anaemia usually
accompanied by porosity on the external cranial vault (cribra cranii)
and often on the orbital roof (cribra orbitalia).[93]
Changes can be observed in both adults and older children and may occur
in an active or healed condition. Healed and activecribra orbitalia
differ by size of the affected bone tissue, size of the perforating
lesions and thickness of the porous bone.
An infant skull (2-4
months old) with porotic hyperostosis was found in a cemetery in the
old city of Nicosia between two churches of the late Byzantine and
early Frankish period (around 1,200 A.D.) (Angastiniotis et al.,
unpublished data). The researchers tried to isolate DNA and to
identify mutations but even though fragmented DNA was isolated no
mutations were found.
Several other documented skeletal remains
with bone pathology suggestive of β-thalassemia were discovered in
various prehistoric sites of the world in areas such as Greece,
Albania, Australia,[94] the Middle East[95] and Southeast Asia.[96]
It
is now it is known that porotic hyperostosis is not a characteristic
mark of β-thalassemia, as Angel thought, but is an unspecific indicator
of several diseases such as megaloblastic anae¬mia, deficiency of
vitamin C and B12 and chronic iron-deficiency anemia. These
difficulties are characteristic of all morphological skeletal diagnosis
of ancient diseases.[97-99]
Rabino Massa et al.
examined samples from the Marro Collection, belonging to the
Anthropological and Ethnographic Museum of Turin, to determine the
presence of malaria antigens. They assessed the specimens belonging to
predynastic mummies (3,200 B.C.) from Gebelen excluding only the poorly
conserved mummies. They analyzed about fifty of the 85 individuals of
this collection. In these Egyptian mummies, the authors detected the
presence of malaria.[100]
The existence of a
hemolytic disorder was observed in histological preparations of one
individual, indicating pathological variations of hemoglobin.
Radiographic examination confirmed the existence of hemoglobinopathy:
in some mummified heads, there was the classical presence of "brush
skulls".[101]
Although skeletal changes are
important additional findings for some clinical assessments, they
cannot be used alone for diagnosis. Many other criteria have to be used
for accurate diagnosis. Most recently, a variety of chemical methods
has been used to study human bones at the molecular level including DNA
analysis.
In 1995, Filon et al. presented the first direct proof
of the occurrence of an inherited anemia in the archeological remains
of a child, eight years old, with severe bone pathology consistent with
β-thalassemia major. The remains came from an Israeli archaeological
site, Akhziv, thought to date to the Ottoman period, sometime between
the 16th and the 19th
centuries. DNA analysis has shown that the child was homozygous for the
frameshift mutation at codon 8 (-AA) as well as the variant (C-T) in
the second codon of the β-globin gene.[102]
Unfortunately,
quantitation of aDNA templates was not carried out in this analysis.
Quantitation of aDNA should be carried out in aDNA studies to estimate
the authenticity of aDNA sequences and the risk of contamination with
modern DNA.[103,104] If amounts of the aDNA
templates are insufficient, then it is possible that PCR amplifications
may have preferentially amplified one specific allele (mutant codon 8)
over another (normal codon 8), resulting in homozygous sequences.
Conclusions
Ancient
literature alludes to conditions which appear to have been caused by
the thalassemias, but infant mortality was so high (from this and other
causes), and so little was known about hemolytic disorders that the
true causes of the thalassemic syndromes were only determined in the
second half of the 20th century, with a better knowledge of genetics.
Before
the advent of agriculture, roughly 5,000 years ago, it is unlikely that
humans were exposed to large malaria outbreaks and that this stage was
only reached whenever Neolithic (early) farmers settled in
mosquito-infested soft and marshy soil near standing water. In these
surroundings young carriers of the thalassemias (α
and β) had better chances than normal to survive malaria during
infancy, and were able to reproduce and pass the trait to the next
generation, thus increasing the gene frequency. The higher and varying
distribution of certain hemoglobin disorders in different populations
may also reflect the high frequency of consanguineous marriages,[105]
mass migration, improvement of nutritional status, prevention and
treatment of infectious diseases, settlements and the strong
constitutions of inhabitants.[96-98]
Considering
the current distribution of β-thalassemia and the diversity of mutation
patterns, it is unlikely that β-thalassemia originated from a single
place and time and then spread throughout the rest of the malaria belt.
In fact, the contiguous distribution of β-thalassemia, as well as other
β-globin gene mutations in localized areas of the world, had led to the
conclusion that these mutations had emerged even before malaria became
an important selective factor.
Because hemoglobinopathies are
endemic or have expanded following mobility flows, they are present in
all European countries creating an important impact on health services[99]
that has not yet been effectively addressed by Member States Health
authorities. Recent surveys suggest that between 300,000 and 400,000
babies are born with a serious hemoglobin disorder each year (23,000
babies with β-thalassemia major) and that up to 90% of these births
occur in low- or middle-income countries.[1-6]
The
global distribution of the various globin gene mutations poses a number
of interesting and important conundrums. Nowadays, a suitable strategy
for identifying the β-globin mutations calls for the knowledge of the
frequency of mutations, common or rare, in the ethnic group of the
individuals.
The chronicity of the disease and the high cost of
life-long treatment make prevention strategies crucial in the
management of this disease. This requires intervention in high
risk/targeted populations. Thanks to the implementation of massive
prevention programs, mainly for thalassemia, in several countries the
incidence of the disease has been reduced substantially, and in some
areas, the annual rate of affected births has decreased by more than
90%.[105-107]
References
-
Weatherall DJ, Clegg
JB. 2001. The thalassaemia syndromes. 4th Edition. Oxford: Blackwell Science
Ltd 2001 https://doi.org/10.1002/9780470696705

- Thein SL. The
molecular basis of ß-thalassemia. Cold Spring Harb Perspect Med. 2013 May
1;3(5):a011700. doi: 10.1101/cshperspect.a011700. https://doi.org/10.1101/cshperspect.a011700
-
Cao A, Kan YW. The
prevention of thalassemia. Cold Spring Harb Perspect Med. 2013 Feb
1;3(2):a011775. doi: 10.1101/cshperspect.a011775 https://doi.org/10.1101/cshperspect.a011775
-
Kountouris P, Lederer
CW, Fanis P, Feleki X, Old J, Kleanthous M. IthaGenes: an interactive database
for haemoglobin variations and epidemiology. PLoS One. 2014 Jul
24;9(7):e103020. doi: 10.1371/journal.pone.0103020. eCollection 2014 https://doi.org/10.1371/journal.pone.0103020
-
Ladis V.Kaagiorga–Langana M, Tsiarta I Chouliaras. Thirty-year experience in preventing hemoglobinopathies in Greece:
achievements and potentials for optimisations. Eur J Haematol .2013; 90:
313-322 https://doi.org/10.1111/ejh.12076 PMid:23331260
-
Williams TN,
Weatherall DJ. World distribution, population genetics, and health burden of
the hemoglobinopathies. Cold Spring Harb Perspect Med. 2012 Sep 1;2(9):a011692 https://doi.org/10.1101/cshperspect.a011692 PMid:22951448 PMCid:PMC3426822
- Basak AN. The
molecular pathology of beta-thalassemia in Turkey: the Bogaziçi University
experience. Hemoglobin. 2007;31:233-241 https://doi.org/10.1080/03630260701296735 PMid:17486506
- Chehab FF, Der Kaloustian V, Khouri FP, Deeb SS, Kan
YW. The molecular basis of beta-thalassemia in Lebanon: application to prenatal
diagnosis. Blood 1987;69:1141–1145
PMid:3828533
-
El-Hazmi MA,
al-Swailem AR, Warsy AS. Molecular defects in beta-thalassaemias in the
population of Saudi Arabia. Hum Hered.1995;45:278–285 https://doi.org/10.1159/000154314 PMid:7590760
- Zhong Chen L, Easteal S, Board PG. Evolution of the
ß-globin haplotypes in human populations. Mol Biol Evol. 1990;7: 423–437
-
Del Senno L, Pirastu M, Barbieri R, Bernardi F, Buzzoni
D, Marchetti G, Perrotta C, Vullo C, Kan YW, Conconi F. beta (+)-Thalassaemia
in the Po River Delta region (northern Italy): genotype and beta globin
synthesis. J Med Genet. 1985;22:54-58 https://doi.org/10.1136/jmg.22.1.54 PMid:2580095 PMCid:PMC1049378
- Pirastu M, Saglio G,
Camaschella C, Loi A, Serra A, Bertero T, Gabutti W, Cao A. Delineation of
specific beta-thalassemia mutations in high-risk areas of Italy: a prerequisite
for prenatal diagnosis. Blood 1987;71:983–988
-
Amselem S, Nunes V,
Vidaud M, Estivill X, Wong C, d'Auriol L, Vidaud D, Galibert F, Baiget M, Goossens
M. Determination of the spectrum of beta-thalassemia genes in Spain by use of
dot-blot analysis of amplified beta-globin DNA. Am J Hum Genet. 1988;
43:95–100 PMid:2897787 PMCid:PMC1715291
- Faustino P, Pacheco
P, Loureiro P, Nogueira PJ, Lavinha J. The geographic pattern of
beta-thalassaemia mutations in the Portuguese population. Br J Haematol.
1999;107:903–904 https://doi.org/10.1046/j.1365-2141.1999.01821.x PMid:10671033
- Fattoum S, Guemira F,
Oner C, Li HW, Kutlar F, Huisman TH. Beta-thalassemia, HB S-beta-thalassemia
and sickle cell anemia among Tunisians. Hemoglobin. 1991;15:11–21 https://doi.org/10.3109/03630269109072481 PMid:1917531
- Falchi A, Giovannoni L, Vacca L, Latini V, Vona G, Varesi
L.ß-globin gene cluster haplotypes associated with beta-thalassemia on Corsica
island. Am J Hematol. 2005;78:27-32 https://doi.org/10.1002/ajh.20199 PMid:15609277
- Maggio A, Giambona A, Cai SP, Wall J, Kan YW, Chehab FF. Rapid and simultaneous typing of hemoglobin S, hemoglobin C, and seven
Mediterranean beta-thalassemia mutations by covalent reverse dot-blot analysis:
application to prenatal diagnosis in Sicily. Blood.1993;81:239–242 PMid:8417793
- Kountouris P,
Kousiappa I, Papasavva T, Christopoulos G, Pavlou E, Petrou M, Feleki X,
Karitzie E, Phylactides M, Fanis P, Lederer CW, Kyrri AR, Kalogerou E, Makariou
C, Ioannou C, Kythreotis L, Hadjilambi G, Andreou N, Pangalou E, Savvidou I,
Angastiniotis M, Hadjigavriel M, Sitarou M, Kolnagou A, Kleanthous M, Christou
S. The molecular spectrum and distribution of haemoglobinopathies in Cyprus: a
20-year retrospective study. Sci Rep. 2016 May 20;6:26371. doi:
10.1038/srep26371 https://doi.org/10.1038/srep26371
- Baysal E, Indrak K,
Bozkurt G, Berkalp A, Aritkan E, Old J, Ioannou P, Angastiniotis M, Droushiotou
A, Yüregir GT. The beta thalassaemia mutations in the population of Cyprus. Br
J Haematol. 1992; 81: 607-609 https://doi.org/10.1111/j.1365-2141.1992.tb03000.x PMid:1390250
- Kyrri AR, Felekis X,
Kalogerou E, Wild BJ, Kythreotis L, Phylactides M, Kleanthous M. Hemoglobin
variants in Cyprus. Hemoglobin 2009;33:81-94 https://doi.org/10.1080/03630260902813502 PMid:19373583
- Malamos B, Fessas Ph,
Stamatoyiannopoulos G. Types of thalassemia trait carriers, as revealed by a
study of their incidence in Greece. Br J Haemat. 1962;8:5-14 https://doi.org/10.1111/j.1365-2141.1962.tb06489.x
- Loukopoulos D.
Current status of thalassemia and sickle cell syndromes in Greece. Sem
Hematol.1996;33:76-86 PMid:8714587
- Kattamis C, Hu H,
Cheng G, Reese J, Gonzalez-Redondo M Kutlar A, Kutlar F, Huisman THJ. Molecular
characterization of ß–thalassemia in 174 Greek patients with thalassemia major.
Br.J Haem.1990;74:342-346 https://doi.org/10.1111/j.1365-2141.1990.tb02593.x
- Canatan D.
Thalassemias and hemoglobinopathies in Turkey. Hemoglobin 2014;38:305-307 https://doi.org/10.3109/03630269.2014.938163 PMid:25030194
- Tadmouri GO, Garguier
N, Demont J, Perrin P, Basak AN. History and origin of beta-thalassemia in
Turkey: sequence haplotype diversity of beta-globin genes. Hum Biol.
2001;73:661-674 https://doi.org/10.1353/hub.2001.0075
PMid:11758688
-
Aksoy M, Ikin EW,
Mourant AE Lehmann H. Blood groups, haemoglobins, and thalassaemia in Turks in
southern Turkey and Eti-Turks. Br Med J. 1958;2:937-939 https://doi.org/10.1136/bmj.2.5102.937 PMid:13584804 PMCid:PMC2026570
- Hamamy HA, Al-Allawi NA. Epidemiological
profile of common haemoglobinopathies in Arab countries. J Community Genet.
2013;4:147-167 https://doi.org/10.1007/s12687-012-0127-8 PMid:23224852 PMCid:PMC3666833
- Agouti I, Badens C,
Abouyoub A, Levy N, Bennani M. Molecular basis of beta-thalassemia in Morocco:
possible origins of the molecular heterogeneity. Genet Test. 2008;12:563-568 https://doi.org/10.1089/gte.2008.0058 PMid:18976160
- Daar S, Gravell D,
Hussein HM, Pathare AV, Wali Y, Krishnamoorthy R. Haematological and clinical
features of beta-thalassaemia associated with Hb Dhofar. Eur J Haematol.
2008;80:67-70 https://doi.org/10.1111/j.1600-0609.2007.00989.x PMid:18173741
- De Leo R, Deidda G,
Novelletto A, El-Kalla S, Mathews AR, Felicetti L. Analysis of ß-thalassemia
mutations in the United Arab Emirates provides evidence for recurrent origin of
the IVSI nt 5 (G-C) mutation. Hum Mutat. 1995;5:327–328 https://doi.org/10.1002/humu.1380050409 PMid:7627187
- Kyriacou K,
Al-Quobaili F, Pavlou E, Christopoulos G, Ioannou P, Kleanthous M. Molecular
characterization of ß-thalassemia in Syria. Hemoglobin 2000;24:1–13 https://doi.org/10.3109/03630260009002268 PMid:10722110
- Zahed L, Qatanani M,
Nabulsi M, Taher A. ß- thalassemia mutations and haplotype analysis in Lebanon.
Hemoglobin 2000;24:269–276 https://doi.org/10.3109/03630260008993133 PMid:11186256
- Filon D, Oron V,
Shawa R, Shawa R, Elborno E, Najjar K, Tulchinsky T, Rachmilewitz E, Rund D,
Oppenheim A. Spectrum of ß- thalassemia mutations in the Gaza area. Hum Mutat.
1995;5:351–353 https://doi.org/10.1002/humu.1380050416 PMid:7627194
- Jassim N, Merghoub T,
Pascaud O, al Mukharraq H, Ducrocq R, Labie D, Elion J, Krishnamoorthy R,
Arrayed SA. Molecular basis of ß-thalassemia in Bahrain. Ann NY Acad Sci.
1998;850:407–409 https://doi.org/10.1111/j.1749-6632.1998.tb10505.x PMid:9668568
- El-Hashemite N,
Petrou M, Khalifa AS, Heshmat NM, Rady MS, Delhanty JD. Identification of novel
Asian Indian and Japanese mutations causing ß-thalassemia in Egyptian
population. Hum Genet 1997;99:271-274 https://doi.org/10.1007/s004390050352 PMid:9048934
- El-Shanshory M, Hagag
A, Shebl S, Badria I, Abd Elhameed A, Abd El-Bar E, Al-Tonbary Y, Mansour A,
Hassab H, Hamdy M, Alfy M, Sherief L, Sharaf E. Spectrum of beta globin gene
mutations in Egyptian children with ß-Thalassemia. Mediterr J Hematol Infect
Dis. 2014 Nov 1;6(1):e2014071 https://doi.org/10.4084/mjhid.2014.071 PMid:25408857 PMCid:PMC4235483
- Al-Allawi NA,
Al-Mousawi BM, Badi AI, Jalal SD.The spectrum of ß-thalassemia mutations in
Baghdad, Central Iraq. Hemoglobin 2013;37:444-453 https://doi.org/10.3109/03630269.2013.810641 PMid:23826747
- Al-Obaidli A, Hamodat
M, Fawzi Z, Abu-Laban M, Gerard N, Krishnamoorthy R. Molecular basis of
thalassemia in Qatar. Hemoglobin 2007;31:121-127 https://doi.org/10.1080/03630260701288815 PMid:17486492
- Al-Obaidli A, Gerard
N, Al Zadjali S, Fawzi Z, Pravin S, Pathare A, Krishnamoorthy R. A novel
deletional betathalassemic variant in an ethnic Qatari patient. Hemoglobin.
2009; 33:214-219 https://doi.org/10.1080/03630260903081398 PMid:19657835
- Haj Khelil A, Denden
S, Leban N, Daimi H, Lakhdhar R, Lefranc G, Ben Chibani J, Perrin P.
Hemoglobinopathies in North Africa: a review. Hemoglobin 2010;34:1-23 https://doi.org/10.3109/03630260903571286 PMid:20113284
- Zahed L. The spectrum
of beta-thalassemia mutations in the Arab populations. J Biomed Biotechnol.
2001;1:129-132 https://doi.org/10.1155/S1110724301000298 PMid:12488606 PMCid:PMC129059
- Tadmouri GO, Gulen
RI. Deniz: the electronic database for beta-thalassemia mutations in the Arab
world. Saudi Med J. 2003;24:1192-1198 PMid:14647552
- Sadiq MFG, Huisman
THJ. Molecular characterization of ß-thalassemia in North Jordan.
Hemoglobin1994;18:325–332 https://doi.org/10.3109/03630269408996198 PMid:7852087
- Adekile AD, Gu LH,
Baysal E, Haider MZ, al-Fuzae L, Aboobacker KC, al-Rashied A, Huisman TH.
Molecular characterization of alpha thalassemia determinants, beta thalassemia
alleles and beta S haplotypes among Kuwaiti Arabs. Acta Haematol.
1994;92:176–181 https://doi.org/10.1159/000204216 PMid:7701914
- Fattoum S, Guemira F,
Oner C, Li HW, Kutlar F, Huisman TH.. Beta-thalassemia, HbS-beta-thalassemia
and sickle cell anemia among Tunisians. Hemoglobin. 1991;15:11–21 https://doi.org/10.3109/03630269109072481 PMid:1917531
- Maryami F, Azarkeivan
A, Fallah MS, Zeinali S. A large cohort study of genotype and phenotype
correlations of beta-thalassemia in Iranian population. Int J Hematol Oncol
Stem Cell Res. 2015;9:198-202 PMid:26865931
PMCid:PMC4748687
- Varawalla NY, Fitches
AC, Old JM. Analysis of beta-globin gene haplotypes in Asian Indians: origin
and spread of beta-thalassaemia on the Indian subcontinent. Hum Genet.
1992;90:443-449 https://doi.org/10.1007/BF00220475 PMid:1362388
- Rezaee AR, Banoei MM,
Khalili E, Houshmand M. Beta-Thalassemia in Iran: new insight into the role of
genetic admixture and migration. Sc World J. 2012; 2012:635183. doi:
10.1100/2012/635183. Epub 2012 Dec 18 https://doi.org/10.1100/2012/635183
- Harcourt AH. Human
phylogeography and diversity. Proc Natl Acad Sci USA. 2016;113:8072- 8078 https://doi.org/10.1073/pnas.1601068113 PMid:27432967 PMCid:PMC4961125
- Lahr MM, Foley RA.
Towards a theory of modern human origins: geography, demography, and diversity
in recent human evolution. Am J Phys Anthropol. 1998;Suppl 27:137-176 https://doi.org/10.1002/(SICI)1096-8644(1998)107:27+<137::AID-AJPA6>3.0.CO;2-Q
- Liu H, Prugnolle F,
Manica A, Balloux F. A geographically explicit genetic model of worldwide human-settlement
history. Am J Hum Genet. 2006;79:230-237 https://doi.org/10.1086/505436 PMid:16826514 PMCid:PMC1559480
- Mellars P. Why did
modern human populations disperse from Africa ca. 60,000 years ago? A new
model. Proc. Natl. Acad. Sci. USA. 2006; 103:9381– 9386 https://doi.org/10.1073/pnas.0510792103 PMid:16772383 PMCid:PMC1480416
- O'Connell J. Allen J.
Dating the colonization of the Sahul (Pleistocene Australia-New Guinea): A
review of recent research. J Archaeol Sc. USA. 2004; 31:835– 853
- Armitage SJ, Jasim
SA, Marks AE, Parker AG, Usik VI, Uerpmann HP. The southern route "out of
Africa": evidence for an early expansion of modern humans into Arabia.
Science 2011;331:453-456 https://doi.org/10.1126/science.1199113 PMid:21273486
- Forster P, Matsumura
S. Evolution. Did early humans go north or south? Science 2005; 308: 965-966 https://doi.org/10.1126/science.1113261 PMid:15890867
- Stringer C.
Palaeoanthropology. Coasting out of Africa. Nature 2000;405:24-25 https://doi.org/10.1038/35011166 PMid:10811201
- Childe VG. New light
on the most ancient east. London: Routledge and Paul, 4th ed.1952 PMid:14946582
- Bar-Yosef O. On the
nature of transitions: the middle to upper Palaeolithic and the Neolithic
revolution. Cambridge Archaeol J. 1998;8:141–163
https://doi.org/10.1017/S0959774300001815
- Bar-Yosef O, Kislev
M. Early farming communities in the Jordan Valley. In: Harris DR, Hillman GC,
eds. Foraging and farming. The evolution of plant exploitation. London: Unwin
Hyman, 1989; pp. 633–642

- Zohary D, Hopf M,
Weiss R. Domestication of plants in the old world, 4th ed. New York: Oxford
University Press. 2012; pp.1-264 https://doi.org/10.1093/acprof:osobl/9780199549061.003.0001
- Araus JL, Ferrio JP,
Buxó R, Voltas J. The historical perspective of dryland agriculture: lessons
learned from 10,000 years of wheat cultivation. J Exp Bot. 2007;58:131-145 https://doi.org/10.1093/jxb/erl133 PMid:17050642
- Salamini F, Ozkan H,
Brandolini A, Schäfer-Pregl R, Martin W. Genetics and geography of wild cereal
domestication in the near east. Nat Rev Genet. 2002;3:429-441 PMid:12042770
- Kenoyer JM, Price TD,
Burton JH.A new approach to tracking connections between the Indus Valley and
Mesopotamia: initial results of strontium isotope analyses from Harappa and Ur.
J Archaeol Sci.2013;40: 2286–2297 https://doi.org/10.1016/j.jas.2012.12.040
- Crawford HEW. Sumer
and the Sumerians: Cambridge University Press, 2004; pp.1-11
- Wiercinski A. Anthropology of ancient Mesopotamia. In:
Mesopotamia, Ed. Braun, PWN - National Scientific Publishing. 1997; pp
42–45 PMid:9562802
- Araus JL, Buxo´ R.
Changes in carbon isotope discrimination in grain cereals from the
north-western Mediterranean Basin during the past seven millenia. Aust J Plant
Physiol.1993;20:117–128 https://doi.org/10.1071/PP9930117
- Buxo´ R. Arqueologi’a de las plantas: la explotacion
economica de las semillas y los frutos en el marco mediterraneo de la Peninsula
Iberica. Barcelona: Critica, Grijalbo eds, 1998; pp-
1-367
- Feldman M. Origin of
cultivated wheat. In: Bonjean AP, Angus WJ, eds. The world wheat book. a
history of wheat breeding. Paris: Lavoisier Publishing, 2001; pp.3–57
- Iandola F. La thalassemia dall'alba della civiltà
occidentale ad oggi. Tesi di Laurea, In: Fondazione De Marchi, www.demarchi.org/thalassemia.htm
- Grmek MD. Malaria in
the Eastern Mediterranean in prehistory and antiquity. Parassitologia,
1994;36:1–6 PMid:7898948
- Silvestroni E, Bianco I, Alfieri N. Sulle origini della
microcitemia in Italia e nelle altre regioni della terra. Medicina 1952;2:187–216
- Choremis C, Fessas
Ph, Kattamis C, Stamatoyiannopoulos G, Zannos-Mariolea L, Karaklis A, Belios G.
Three inherited red-cell abnormalities in a district of Greece. Thalassemia, sickling
and glucose-6-phosphate-dehydrogenase deficiency. Lancet 1963;1;907-909 https://doi.org/10.1016/S0140-6736(63)91688-X
- Flint J, Harding RM,
Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies.
Baillieres Clin Haematol. 1998;11:1-51 https://doi.org/10.1016/S0950-3536(98)80069-3
- Haldane JBS: Disease
and evolution. La Ricerca Scientifica 1949, 19:68–76
- Canali S. Researches
on thalassemia and malaria in Italy and the origins of the "Haldane
hypothesis". Med Secoli. 2008;20:827-846
PMid:19848219
- Ratcliffe AW. The
historical background of malaria; a reconsideration. J Indiana State Med Assoc.
1946;39:339-347 PMid:20988762
- Bruce-Chwatt LJ.
Aleogenesis and paleo-epidemiology of primate malaria. Bull World Health
Organ.1965;32:363-387 PMid:14315710 PMCid:PMC2555232
- Fairhurst RM, Baruch
DI, Brittain NJ, Ostera GR, Wallach JS, Hoang HL, Hayton K, Guindo A, Makobongo
MO, Schwartz OM, Tounkara A, Doumbo OK, Diallo DA, Fujioka H, Ho M, Wellems TE.
Abnormal display of PfEMP-1 on erythrocytes carrying haemoglobin C may protect
against malaria. Nature 2005;435:1117–1121 https://doi.org/10.1038/nature03631 PMid:15973412
- Cholera R, Brittain
NJ, Gillrie MR, Lopera-Mesa TM, Diakité SA, Arie T, Krause MA, Guindo A, Tubman
A, Fujioka H, Diallo DA, Doumbo OK, Ho M, Wellems TE, Fairhurst RM. Impaired
cytoadherence of Plasmodium falciparum-infected erythrocytes containing sickle
hemoglobin. Proc Natl Acad Sci U S A. 2008;105:991–996 https://doi.org/10.1073/pnas.0711401105 PMid:18192399 PMCid:PMC2242681
- Cyrklaff M, Sanchez
CP, Kilian N, Bisseye C, Simpore J, Frischknecht F, Lanzer M. Hemoglobins S and
C Interfere with Actin Remodeling in Plasmodium falciparum Infected
Erythrocytes. Science 2011;334:1283-1286 https://doi.org/10.1126/science.1213775 PMid:22075726
- Williams TN, Mwangi
TW, Roberts DJ, Alexander ND, Weatherall DJ, Wambua S, Kortok M, Snow RW, Marsh
K. An immune basis for malaria protection by the sickle cell trait. PLoS Med.
2005 May;2(5):e128 https://doi.org/10.1371/journal.pmed.0020128 PMid:15916466 PMCid:PMC1140945
- Gong L,
Maiteki-Sebuguzi C, Rosenthal PJ, Hubbard AE, Drakeley CJ, Dorsey G, Greenhouse
B. Evidence for both innate and acquired mechanisms of protection from
Plasmodium falciparum in children with sickle cell trait. Blood 2012 Apr
19;119(16):3808-14. doi: 10.1182/blood-2011-08-371062 https://doi.org/10.1182/blood-2011-08-371062
- Veenemans J, Jansen
EJ, Baidjoe AY, Mbugi EV, Demir AY, Kraaijenhagen RJ, Savelkoul HF, Verhoef H.
Effect of alpha (+) -thalassaemia on episodes of fever due to malaria and other
causes: a community-based cohort study in Tanzania. Malar J. 2011 Sep
22;10:280. doi: 10.1186/1475-2875-10-280 https://doi.org/10.1186/1475-2875-10-280
- Tan X, Traore B,
Kayentao K, Ongoiba A, Doumbo S, Waisberg M, Doumbo OK, Felgner PL, Fairhurst
RM, Crompton PD. Hemoglobin S and C heterozygosity enhances neither the
magnitude nor breadth of antibody responses to a diverse array of Plasmodium
falciparum antigens. J Infect Dis. 2011;204:1750–1761 https://doi.org/10.1093/infdis/jir638 PMid:21998476 PMCid:PMC3203232
- Cabrera G, Cot M,
Migot-Nabias F, Kremsner PG, Deloron P, Luty AJ. The sickle cell trait is
associated with enhanced immunoglobulin G antibody responses to Plasmodium falciparum
variant surface antigens. J Infect Dis. 2005;191:1631–1638 https://doi.org/10.1086/429832 PMid:15838789
- Ayi K, Turrini F,
Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes:
a common mechanism that may explain protection against falciparum malaria in
sickle trait and beta-thalassemia trait. Blood. 2004;104:3364–3371 https://doi.org/10.1182/blood-2003-11-3820 PMid:15280204
- Christianson A,
Howson CP, Modell B. March of Dimes global report on birth defects. March of
Dimes Birth Defects Foundation, New York, 2009; pp.21-23
- Rudra S, Chakrabarty P, Hossain MA, Ripon MJ, Rudra M,
Mirza TT. Awareness among parents of
ß-Thalassemia major patients regarding prenatal diagnosis and premarital
screening in day care centre of Transfusion Medicine Department. Mymensingh Med
J. 2016;25:12-17 PMid:26931242
- Canatan D, Karadogan
C, Oguz N, Balta N, Cosan R: The frequency of consanguineous marriages in
patients with hereditary blood disorders in South of Turkey. Community Genet.
2003.6:58
- Moghadam M. Karimi M,
Dehghani SJ, Dehbozorgian J, Montazeri S, Javanmardi E, Asadzade R, Amiri A,
Saghatoleslam Z, Sotodegan F, Morshedi N, Imanifard J, Afrasiabi A.
Effectiveness of ß-thalassemia prenatal diagnosis in Southern Iran: a cohort
study. Prenat Diagn. 2015; 35:1238-1242 https://doi.org/10.1002/pd.4684 PMid:26296249
- Weatherall DJ.
Thalassemia as a global health problem: Recent progress towards its control in
the developing countries. Ann NY Acad Sci. 2010;1202:17–23 https://doi.org/10.1111/j.1749-6632.2010.05546.x PMid:20712767
- Weatherall, D.J. The
inherited diseases of hemoglobin are an emerging global health burden. Blood
2010;115:4331–4336 https://doi.org/10.1182/blood-2010-01-251348 PMid:20233970 PMCid:PMC2881491
- Angel JL. Porotic
hyperostosis, anemias, malarias, and marshes in the prehistoric eastern
Mediterranean. Science 1966;153:760-763 https://doi.org/10.1126/science.153.3737.760 PMid:5328679
- Webb S. Cranial
thickening in an Australian hominid as a possible palaeoepidemiological
indicator. Am J Phys Anthropol.1990;82:403-411 https://doi.org/10.1002/ajpa.1330820402 PMid:2205102
- Hershkovitz I, Ring
B, Speirs M, Galili E, Kislev M, Edelson G, Hershkovitz A. Possible congenital
hemolytic anemia in prehistoric coastal inhabitants of Israel. Am J Phys
Anthropol. 1991;85:7-13 https://doi.org/10.1002/ajpa.1330850103 PMid:1853944
- Tayles N. Anemia,
genetic diseases, and malaria in prehistoric mainland Southeast Asia. Am J Phys
Anthropol. 1996;101:11-27 https://doi.org/10.1002/(SICI)1096-8644(199609)101:1<11::AID-AJPA2>3.0.CO;2-G
- Aufderheide AC.
Progress in paleopathology. Biomedical studies of human mummies. Minn
Med.1998;81:28-31 PMid:9842233
- Stuart-Macadam P.
Porotic hyperostosis: new evidence to support the anemia theory. Am J Phys
Anthropol. 1987;74:521-526 https://doi.org/10.1002/ajpa.1330740410 PMid:3327384
- Schultz M.
Paleohistopathology of bone: a new approach to the study of ancient diseases.
Am J Phys Anthropol. 2001;Suppl 33:106-147 https://doi.org/10.1002/ajpa.10024 PMid:11786993
- Rabino Massa E. Conservazione dei Globuli in Tessuti di
Mummie Egiziane. Archivio per L'Antropologia e L'Etnologia. La Nuova
Italia1967;3:181-182
- Rabino Massa E.
Presence of Thalassemia in Egyptian Mummies. J.
Hum. Evol. 1977;6:223-225
- Filon D, Faerman M,
Smith P, Oppenheim A. Sequence analysis reveals a beta-thalassaemia mutation in
the DNA of skeletal remains from the archaeological site of Akhziv, Israel. Nat
Genet. 1995;9:365-368 https://doi.org/10.1038/ng0495-365 PMid:7795641
- Handt O, Krings M, Ward
RH, Pääbo S. The retrieval of ancient human DNA sequences. Am J Hum Genet.
1996;59:368-376 PMid:8755923 PMCid:PMC1914746
- Paabo S. 1989.
Ancient DNA: extraction, characterization, molecular cloning, and enzymatic
amplification. Proc Natl Acad Sci USA.1989;86:1939-1943 https://doi.org/10.1073/pnas.86.6.1939 PMid:2928314 PMCid:PMC286820
- Christianson A,
Howson CP, Modell B. March of Dimes global report on birth defects. March of
Dimes Birth Defects Foundation, New York, 2006; pp.18-23
- Aguilar Martinez P,
Angastiniotis M, Eleftheriou A, Gulbis B, Ma-ú Pereira Mdel M, Petrova-Benedict
R, Corrons JL. Haemoglobinopathies in Europe: health & migration policy
perspectives. Orphanet J Rare Dis. 2014 Jul 1;9:97. doi: 10.1186/1750-1172-9-97
https://doi.org/10.1186/1750-1172-9-97
- Giordano PC,
Harteveld CL, Bakker E. Genetic epidemiology and preventive healthcare in
multiethnic societies: the hemoglobinopathies. Int J Environ Res Public Health.
2014;11:6136-6146 https://doi.org/10.3390/ijerph110606136 PMid:24921462 PMCid:PMC4078570
[TOP]