Hina Qamar1, Adrienne Lee2, Karen Valentine2, Leslie Skeith2 and Victor H Jimenez-Zepeda3
1 Department of Medicine, University of Alberta, Edmonton, AB, Canada.
2 Southern Alberta Rare Blood & Bleeding Disorders Comprehensive Care Program, Department of Medicine, Calgary, AB, Canada.
3 Tom Baker Cancer Center, Department of Medical Oncology and Hematology, Calgary, AB, Canada.
Published: May 1, 2017
Received: February 15, 2017
Accepted: April 10, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017034 DOI
10.4084/MJHID.2017.034
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Acquired
von Willebrand syndrome (AVWS) is a rare hemorrhagic disorder that
occurs in patients with no prior personal or family history of
bleeding. Here, we describe a case of AVWS occurring after autologous
stem cell transplantation (ASCT). Interestingly, AVWS developed after
bortezomib-based induction and conditioning regimens. Recent evidence
suggests that the proximity of the bortezomib therapy to the collection
of stem cells with consequent depletion of regulatory T cells after the
conditioning regimen could explain some of the unusual autoimmune
complications reported in patients receiving bortezomib prior to ASCT.
In addition, this patient developed a secondary MGUS post-ASCT, which
may have also contributed to the AVWS. To the best of our knowledge,
this is the first case of post-ASCT AVWS reported. Prospective data is
needed to better elucidate the mechanisms by which these unusual
complications occur in patients receiving bortezomib prior to ASCT.
|
Introduction
Immunoglobulin light-chain Amyloidosis (AL) is the most common type of systemic amyloidosis.[1]
The use of high-dose chemotherapy followed by Autologous Stem Cell
Transplantation (ASCT) has been recognized as an important therapy that
has dramatically changed the perspective of AL amyloidosis care.[2] Since a plasma cell dyscrasia is the underlying cause of AL amyloidosis,[3]
ASCT has also been adopted for the treatment of this entity, aiming to
induce a hematological and organ response by providing time for
the involved organs to slowly clear out some of the amyloid
protein that translates into survival prolongation.[4]
Because the depth of response is a critical determinant of treatment
outcome, different strategies have been employed, hoping to increase
the proportion of patients who ultimately achieve a complete
hematological response after ASCT. These strategies have included the
use of induction chemotherapy before ASCT, especially for those
patients with >10% bone marrow plasma cells.[5] A
recent, single-arm, prospective clinical trial investigated the role of
bortezomib and dexamethasone induction followed by ASCT for newly
diagnosed AL amyloidosis.[6] The study was successful
to show unprecedentedly high hematologic responses. A hematologic very
good partial response or better was seen in 77% of 35 patients at 6
months but was reported in 100% for the 27 patients who completed all
planned therapy. While exciting results were noted, also new emerging
complications after ASCT were seen (GVHD-like syndrome). The mechanisms
by which this complication occurred remain uncertain. Here, we describe
the first case of acquired von Willebrand syndrome (AVWS) in a patient
with AL amyloidosis who received cyclophosphamide, bortezomib and
dexamethasone induction followed by ASCT. Interestingly, this patient
also developed a GVHD-like process in the skin that was successfully
treated with steroids and a secondary IgM Monoclonal Gammopathy.
Case Presentation
A
previously healthy, 56-year/old male presented with asymptomatic
hypoalbuminemia detected on routine screening 2-years prior and
subsequently developed asymmetric inflammatory polyarthritis (MCP’s,
wrists and shoulders) and proteinuria. Further investigations led to
the diagnosis of AL Amyloidosis (Stage I) with a concurrent low-grade
B-cell neoplasm on bone marrow and no evidence of lymphadenopathy. The
patient presented mainly with kidney and soft tissue involvement.
Pre-treatment studies revealed lambda light chain of 3030 mg/L (normal
range 5.71-26.3), free kappa 59.3 mg/L (normal range 3.3-19.4) and
ratio of 0.02 (normal range 0.26-1.65), 24 hr urine collection showed
9.86 g/day of proteinuria with 0.16 g/day of monoclonal IgG lambda and
free lambda light chain. NT-pro-BNP and high-sensitive troponin-T were
normal, serum LDH was normal, CRP was elevated (23.6 mg/L), creatinine
was 108 µmol/L,
ALP was normal and hemostasis study was normal (PT, INR, PTT and factor
X activity were all normal). Cardiac MRI and echocardiogram did not
suggest amyloid involvement. Treatment with cyclophosphamide,
bortezomib and dexamethasone (CyBorD) was initiated, achieving Very
Good Partial Response (VGPR) after 3 cycles of therapy. Stem cell
mobilization was successfully performed followed by autologous stem
cell transplantation with bortezomib/melphalan conditioning. At-2
months post-ASCT the patient developed an erythematous rash affecting
more than 50% of the body surface area. Skin biopsy was consistent with
superficial perivascular lymphocytic infiltrate. In addition, a new
onset of moderate thrombocytopenia (platelet count of 45 10e9/L)
was noted and a bone marrow biopsy was performed. BM biopsy showed
normal cellularity and adequate megakaryocytes, no evidence of plasma
or lymphoproliferative disorders were seen. Treatment with prednisone
at 1mg/kg was initiated leading to complete resolution of these
symptoms. At day-100, response assessment was consistent with complete
hematologic response. Serum protein electrophoresis, however, showed
the presence of a secondary IgM lambda Monoclonal Gammopathy. Four
months post-ASCT patient presented to ER with prolonged epistaxis.
Prior to this time, he has had no history of bleeding. Epistaxis was
severe and required packing and cautery with mild improvement only.
Laboratory investigations revealed prolonged PTT (45.5 s), and decrease
von Willebrand factor activity (VWF:Act), von Willebrand factor antigen
(VWF:Ag), and factor VIII activity (FVIII:C) levels. VWF:Act (GP1b) 12
IU/dL (normal range: 41-144), VWF:Ag 31 IU/dL (normal range: 40-185),
FVIII:C 14 IU/dL (normal range: 54-147). Factor IX and XI levels were
normal, FXII was slightly decreased at 30 IU/dL. Mixing studies for
VWF:Act and FVIII:C showed no inhibitory effect on normal plasma
consistent with a non-neutralizing VWF antibody against non-functional
VWF domains. VWF propeptide to VWF antigen ratio (VWFpp:Ag) was
increased at diagnosis (7.7) suggesting increased VWF clearance (VWFpp
measured by VWFpp-specific monoclonal antibody enzyme-linked
immunosorbent assay (ELISA) using the GTI diagnostics kit.
Lupus
type inhibitor was not detected, and fibrinogen and all other measured
factor levels were normal. Other lab testing revealed: Hemoglobin 75
g/L, Platelets 110 x10e9, creatinine
139 umol/L, ANA-, RF-, Hep B and Hep C negative, cryoglobulin and
agglutinin testing negative. Based on these investigations, the patient
was diagnosed with Acquired von Willebrand syndrome (AVWS). No
coagulation factor replacement was required and desmopressin (DDAVP)
challenge test demonstrated a good increase in von Willebrand factor
and factor VIII levels, without rapid clearance (Factor levels were
measured at 30 mins, 1 and 4 hours) (Table 1).
The patient was treated with prednisone at 1 mg/Kg with slow dose
tapering. No further bleeding has been reported and normalization of
von Willebrand factor levels is maintained at 12 months. (Figure 1).
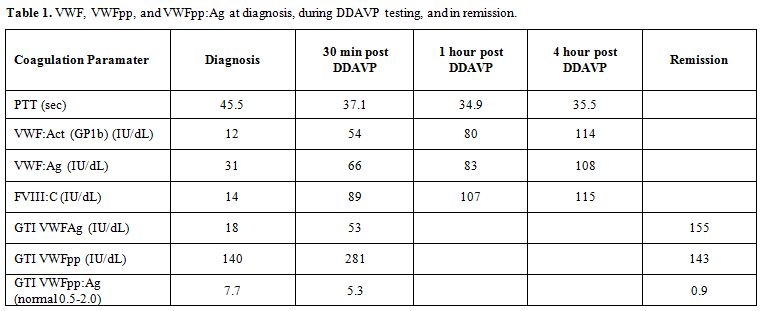 |
Table 1. VWF, VWFpp, and VWFpp:Ag at diagnosis, during DDAVP testing, and in remission. |
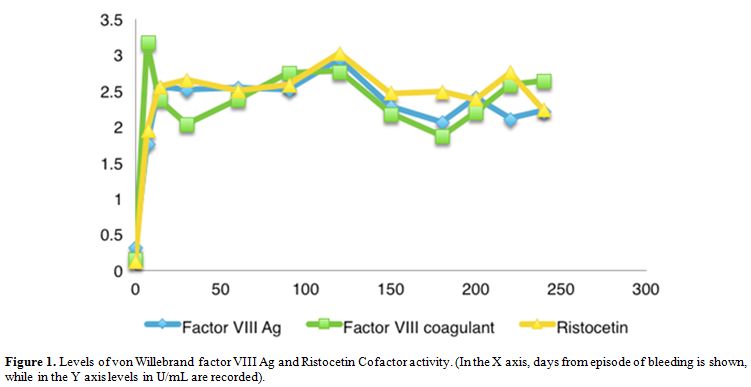 |
Figure 1. Levels of von Willebrand factor
VIII Ag and Ristocetin Cofactor activity. (In the X axis, days from
episode of bleeding is shown, while in the Y axis levels in U/mL are
recorded). |
Discussion
Acquired
von Willebrand syndrome (AVWS) is a rare hemorrhagic disorder that
usually occurs in patients with no previous personal or family history
of bleeding. [7] According to the ISTH registry, 48%
and 15% of 186 AVWS cases that qualified for the registry were
associated with lymphoproliferative and myeloproliferative disorders.[8]
Among lymphoproliferative disorders AVWS is most commonly associated
with Monoclonal Gammopathy of Undetermined Significance (MGUS) in up to
23% of cases.[9] In our patient, the pathogenic
aetiology of underlying AVWS seemed to be increased clearance of
FVIII-VWF complex due to non-neutralizing autoantibodies to VWF given
the lack of inhibitory effect on normal plasma on VWF:Act and FVIII:C
mixing studies (37 C x 2 hours of incubation). Our patient demonstrated
a decrease VWF Ag:Act ratio (0.45), similar to type 2 von Willebrand
Disease (VWD) and loss of high and intermediate molecular weight (HMW)
multimers, which has been described in aVWS.[8] (Figure 2)
VWFpp levels and VWFpp:Ag ratio were measured and demonstrated
increased ratio at diagnosis (7.7, normal range 0.5-2.0) and normal
ratio (0.9) in remission suggesting increase clearance of VWF had a
role in AWVS for our patient. Interestingly, DDAVP was able to
stimulate enough secretion of VWF from endothelial Wiebel-Palade bodies
to overcome the clearance of VWF by the autoantibody, resulting in
normalization of VWF:Ag and VWF:Act levels, and reappearance of HMW
multimers. The decrease (but still abnormal) VWFpp:Ag ratio (5.3) also
reflects the increased secretion stimulated by DDAVP relative to
clearance that remained constant. VWFpp is cleaved in the trans-Golgi
but remains stored together with mature VWF in platelet α-granules and
endothelial cell Weibel-Palade bodies in equimolar amounts. After
release, VWFpp dissociates from the mature VWF subunit, circulates with
a steady-state half-life, and serves as a marker of VWF secretion.[10] A high VWF:pp/Ag ratio has been proposed by Scott et al.,[11] as a simple method to distinguish AVWS due to decreased VWF synthesis from that due to increased clearance. Federici et al.[8]
characterized in their paper some differences in AVWS caused by
IgG-MGUS versus IgM MGUS. Although the paper reported on only 8
IgG-MGUS and 2 IgM MGUS, they reported a higher vWF:pp/Ag compared to
controls in IgG MGUS but not in IgM MGUS. In addition, a normal
multimeric pattern was seen in the IgG MGUS cases but selective loss of
large and intermediate multimers in the IgM-MGUS cases.[12]
Our case of IgM MGUS associated AWVS differs in that high VWFpp:Ag was
seen, although the loss of high and intermediate molecular weight
multimers is similar to that reported by Federici et al.[8]
 |
Figure 2. Multimer gel image demonstrated the presence of high molecular weight multimers. |
The
treatment goals in AVWS are to control acute bleeds, to prevent
bleeding in high-risk situations, and to obtain long-term
remission. In this case, DDAVP was able to stimulate enough
endogenous secretion of VWF to overcome the clearance caused by the
anti-VWF antibody, and maintain normal VWF levels at 1 and 4 hours
following DDAVP administration. In other cases of AVWS, DDAVP is often
ineffective for treatment as the anti-VWF antibody is either
neutralizing, or the clearance of VWF:Ag by the anti-VWF antibody is
much greater than the amount of endogenous VWF:Ag that can be secreted
by Weibel-Palade bodies. However, in our case, the anti-VWF antibody is
non-neutralizing and clearance by this antibody is not so rapid such
that the DDAVP-stimulated release of endogenous VWF can overcome the
anti-VWF antibody-mediated clearance. This is evidenced by the
increased VWF levels following DDAVP stimulation and the decreased
VWFpp:Ag ratio.
Bleeding and bruising are common in immunoglobulin
light chain (AL) amyloidosis and can occur through a number of
mechanisms, thus AVWS is not often considered when bleeding symptoms
occur. Kos et al,[13] recently reported a small
series of cases where the association of active AL amyloid and the
appearance of AWVS was noted. In our current report, in contrast, this
association was observed even in the setting of complete haematological
response. To our knowledge this is the first case report of
post-transplant IgM lambda secondary MGUS treated with high dose
prednisone. However, a previous report by Lazarchick et al.,[14]
described a 41 y/o male who after undergoing allogeneic bone
marrow transplantation for the treatment of acute myeloid leukemia was
admitted with worsening chronic GVHD and subsequently was found to have
markedly reduction of von Willebrand factor antigen consistent with a
diagnosis of AVWS. Our current report also illustrates the
emerging complications for patients with AL amyloidosis receiving
bortezomib-containing regimens prior to stem cell transplantation.
Further data in this regard is needed to better understand the
mechanisms by which these complications occur and how to minimize the
morbidity related to these events.
Acknowledgements
We
would like to acknowledge Paula James' laboratory in Kingston, ON for
performing VWF propeptide assays, and Gary Sinclair's Molecular
hematology laboratory for performing the multimer analysis.
References
- Rosenzweig M, Giralt S, Landau H. Light-chain
amyloidosis: SCT, novel agents and beyond. Bone Marrow Transplant.
2013;48(8):1022-1027. https://doi.org/10.1038/bmt.2012.199 PMid:23103675

- Jimenez-Zepeda
VH, Franke N, Reece DE, et al. Autologous stem cell transplant is an
effective therapy for carefully selected patients with AL amyloidosis:
experience of a single institution. Br J Haematol. 2014;164(5):722-728.
https://doi.org/10.1111/bjh.12673 PMid:24266428
- Roy V. Autologous stem cell transplant for Al amyloidosis. Bone Marrow Res. 2012;2012:238961. https://doi.org/10.1155/2012/238961 PMid:22675637 PMCid:PMC3361989
- Comenzo RL. Amyloidosis. Curr Treat Options Oncol. 2006;7(3):225-236. https://doi.org/10.1007/s11864-006-0015-8 PMid:16615878
- Dispenzieri
A, Buadi F, Kumar SK, et al. Treatment of Immunoglobulin Light Chain
Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy
(mSMART) Consensus Statement. Mayo Clin Proc. 2015;90(8):1054-1081. https://doi.org/10.1016/j.mayocp.2015.06.009 PMid:26250727
- Sanchorawala
V, Brauneis D, Shelton AC, et al. Induction Therapy with Bortezomib
Followed by Bortezomib-High Dose Melphalan and Stem Cell
Transplantation for Light Chain Amyloidosis: Results of a Prospective
Clinical Trial. Biol Blood Marrow Transplant. 2015;21(8):1445-1451. https://doi.org/10.1016/j.bbmt.2015.04.001 PMid:25858810
- Voisin
S, Hamidou M, Lefrancois A, Sigaud M, Mahe B, Trossaert M. Acquired von
Willebrand syndrome associated with monoclonal gammopathy: a
single-center study of 36 patients. Medicine (Baltimore).
2011;90(6):404-411. https://doi.org/10.1097/MD.0b013e3182397166
- Federici
AB RJ, Bucciarelli P, Budde U, van Genderen PJ, Mohri H, Meyer D,
Rodeghiero F, Sadler JE. Acquired von Willebrand syndrome: data from an
international registry. Thromb Haemost. 2000;84(2):345-349.
PMid:10959711
- Franchini M, Lippi G. Acquired von Willebrand syndrome: an update. Am J Hematol. 2007;82(5):368-375. https://doi.org/10.1002/ajh.20830 PMid:17133419
- Lee
A, Sinclair G, Valentine K, James P, Poon MC. Acquired von Willebrand
syndrome: von Willebrand factor propeptide to von Willebrand factor
antigen ratio predicts remission status. Blood. 2014;124(5):e1-3. https://doi.org/10.1182/blood-2014-02-557132 PMid:24951428 PMCid:PMC4729537
- Scott
JPV, E. A.; Schroeder, T.; Foster, P. A.; Gill, J. C.; Montgomery, R.
R. he Von Willebrand factor propolypeptide, Von Willebrand antigen II,
distinguishes acquired Von Willebrand syndrome due to decreased
synthesis of Von Willebrand factor from AvWS due to increased clearance
of vWF. Blood. 1995;86(10 (Suppl 1)).
- Federici
AB, Stabile F, Castaman G, Canciani MT, Mannucci PM. Treatment of
acquired von Willebrand syndrome in patients with monoclonal gammopathy
of uncertain significance: comparison of three different therapeutic
approaches. Blood. 1998;92(8):2707-2711. PMid:9763553
- Kos
CA, Ward JE, Malek K, et al. Association of acquired von Willebrand
syndrome with AL amyloidosis. Am J Hematol. 2007;82(5):363-367. https://doi.org/10.1002/ajh.20829 PMid:17205535
- Lazarchick
J, Green C. Acquired von Willebrand's disease following bone marrow
transplantation. Ann Clin Lab Sci. 1994;24(3):211-215.
PMid:8048792
[TOP]