Nikhil Rabade1#, Goutham Raval1#, Shruti Chaudhary1#, PG Subramanian1, Rohan Kodgule1, Swapnali Joshi1, Prashant Tembhare1, Syed K. Hasan1, Hasmukh Jain2, Manju Sengar2, Gaurav Narula2, Shripad Banavali2, Pratibha Amare Kadam3, Dhanalaxmi Shetty3, Sumeet Gujral1 and Nikhil Patkar1.
1 Hematopathology laboratory, Department of Pathology, Tata Memorial Centre, Mumbai.
2 Department of Medical Oncology, Tata Memorial Centre, Mumbai.
3 Department of Cancer Cytogenetics, Tata Memorial Centre, Mumbai
Corresponding
author: Dr.
Nikhil Patkar. Clinician Scientist and Assistant Professor.
Hematopathology lab, ground floor, CCE building, Tata Memorial center,
ACTREC, Kharghar, Navi Mumbai. E-mail:
nvpatkar@gmail.com
Published: January 1, 2018
Received: August 24, 2017
Accepted: November 6, 2017
Mediterr J Hematol Infect Dis 2018, 10(1): e2017027 DOI
10.4084/MJHID.2018.002
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Atypical
breakpoints and variant APL cases involving alternative chromosomal
aberrations are seen in a small subset of acute promyelocytic leukemia
(APL) patients. Over seven different partner genes for RARA have been
described. Although rare, these variants prove to be a diagnostic
challenge and require a combination of advanced cytogenetic and
molecular techniques for accurate characterization. Heterogeneity
occurs not only at the molecular level but also at clinico-pathological
level influencing treatment response and outcome. In this case series,
we describe the molecular heterogeneity of APL with a focus on seven
variant APL cases from a single tertiary cancer center in India over a
period of two and a half years.
We discuss five cases with ZBTB16-RARA fusion and two novel PML-RARA variants, including a Bcr3 variant involving fusion of PML exon4 and RARA exon3 with an additional 40 nucleotides originating from RARA intron2, another involving exon 6 of PML and exon 3 of RARA with addition of 126 nucleotides, which mapped to the central portion of RARA intron 2. To the best of our knowledge, this is the first case series of this kind from India.
|
Introduction
APL results from balanced reciprocal translocation, t(15;17)(q22;q12) which leads to fusion of promyelocytic leukemia (PML) and retinoic acid receptor alpha (RARA) genes.[1,2] The fusion is commonly caused by breakpoints in intron 2 of RARA and any one of the three breakpoint cluster regions in PML, intron 6 (Bcr1), exon 6 (Bcr2) or intron 3 (Bcr3) leading to long, variable and short isoforms respectively.[3] The PML-RARA
fusion protein not only leads to block in differentiation and leukemic
transformation in APL but also mediates the response to all-trans
retinoic acid (ATRA) therapy.[1,2]
Approximately 8% of APL cases lack the characteristic t(15;17)(q22;q12) translocation on cytogenetic testing.[3] These include cases with cryptic translocations or those with variant RARA translocations. Variant translocations involve fusion of RARA with one of eleven partner genes which include PLZF or ZBTB16 (chromosome 11q23), NPM (5q35), NuMa (11q13), STAT5b (17q21.1-21.2), PRKAR1A (17q24), FIP1L1 (4q12) and BCOR (X).[4] Recently OBFC2A, NABP1 and IRF2BP2 have also been reported to fuse with RARA. ZBTB16-RARA fusion is a rare entity seen in a minority of patients with APL. Unlike classical APL, variants involving ZBTB16 lack the differentiation response to ATRA therapy and carry an unfavorable prognosis.[5]
Identification
of the above mention fusion genes requires a combination of FISH,
conventional karyotyping and molecular techniques, which are not
readily available in most diagnostic laboratories in India. The
scarcity of diagnostic centers possessing these advanced techniques,
coupled with the rarity of this disease has resulted in lack of data
describing such cases from India. Thus, we illustrate the molecular
heterogeneity of APL from a tertiary cancer center in India over a
period of two and a half years. We describe the clinicopathological
features of variant APL cases along with two novel PML-RARA variants.
Materials and Methods
Cellular
morphology was assessed by Wright-stained peripheral blood/bone marrow
aspirate smears. A ten-color antibody panel was used for RQ-PCR flow
cytometric immunophenotyping (FCI). Bone marrow was processed using
bulk lyse-stain-wash method and analyzed on Beckman Coulter Navios flow
cytometer (Beckman Coulter, USA) using the Kaluza analysis software.
FISH was performed on interphase cells using a dual fusion probe for
detecting PML-RARA fusion (Abbott Molecular LSI), and a RARA break apart rearrangement probe (Abbott Molecular LSI) for RARA variant detection.
RNA extraction and cDNA synthesis.
RNA was extracted from bone marrow samples after red blood cell (RBC)
lysis using RNA blood mini kit (Qiagen, Germany). cDNA was obtained
from RNA using the ‘High capacity’ reverse transcriptase cDNA synthesis
kit (Applied Biosystems, CA, USA)
Fusion transcript detection and Real-time quantitative PCR (RT-qPCR). ZBTB16-RARA
fusion transcript was identified using a previously described real-time
quantitative PCR(RT-qPCR) assay on a Roche Light Cycler 96.[6] The delta-delta Ct method was applied to determine the post-induction ZBTB16-RARA fusion transcript copy number.[7] Each sample was run in triplicate along with ABL1 as the control gene.
The PML-RARA
fusion transcript was identified by using the reverse transcriptase PCR
(RT-PCR) BIOMED-1 protocol as described by van Dongen et al..[8] Plasmid standards for the novel PML-RARA variant, ranging from 106 to 101
were prepared in-house by cloning RT-PCR amplicons (see below). We
followed Europe against cancer (EAC) guidelines for primer, probe
design and RT-qPCR assay for quantifying PML-RARA fusion transcripts.[9]
Sequencing and cloning. The PML-RARA variant
RT-PCR products were purified with a SapExo solution (Life
Technologies, CA, USA) and incubated at 37 degrees C for 15 min
followed by 80C for 15 mins. Purified products were cloned into a pJET
1.2 vector by using the Clone JET PCR Cloning kit (Thermo Scientific,
USA) to give a final volume of 20 ul of cloned product. TOP HAT
DH5alpha competent cells (Invitrogen, CA, USA) were transformed with
10ul of cloned product and selected for recombinants by antibiotic
selection. DNA from the resultant clones was extracted by using a
QIAprep Spin Miniprep Kit (Qiagen, Germany) and quantified by NanoDrop
2000 spectrophotometer. The PCR product and the cloned product were
sequenced by Sanger sequencing. The following sequences from Ensemble
were used as reference: PML-001 (ENST00000268058.7) and RARA-007
(ENST00000425707.7).
Long-range genomic DNA PCR. Long-range PCR for the PML-RARA
variant was performed by using TaKaRa LA Taq kit according to the
manufacturer’s protocol. Briefly, 2 ul of genomic DNA (gDNA) was used
as a template and subjected to PCR at 94C for 1 min, 98C for 10 sec
& 68C for 15 mins (30 cycles) and 72C for 10 mins. One forward
primer spanning PML exon 3 and eight reverse primers for RARA intron 2
were used for PCR.[10] The PCR products were run on a
1% agarose gel and DNA from the required band was extracted by using
QIAquick Gel Extraction kit (Qiagen, Germany). The extracted DNA was
subjected to Sanger sequencing using the same primers. Results
A
total of 180 new cases of APL were diagnosed at our institute between
January 2015 and June 2017. These included 34 pediatric and 146 adult
cases with a median age of 10 years (range 2 – 16 years) and 34 years
(range 17 – 71 years) respectively. There were 20 males and 14 females
(male: female ratio – 1.4:1) in the pediatric age group, 78 males and
68 females (male: female ratio – 1.1:1) in the adult age group.
Distribution of cases as per the transcript type is discussed in Figure 1. The purview of this case series is in five cases with ZBTB16-RARA fusion and two PML-RARA variants along with their clinical and morphological details.
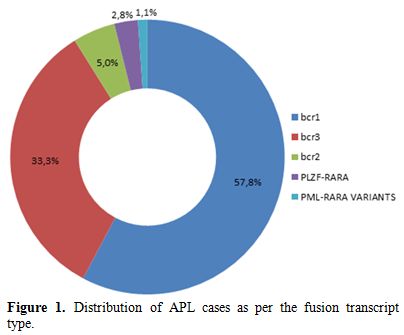 |
Figure 1. Distribution of APL cases as per the fusion transcript type. |
Case 1.
A 15-year-old boy complained of abdominal pain, weakness and
intermittent fever. Laboratory investigation values are depicted in Table 1.
Bone marrow examination revealed abnormal promyelocytes with regular,
round to oval nuclei, (lacking the classical bilobed appearance) dense
cytoplasmic granularity, the absence of Auer rods but strong
cytochemical myeloperoxidase (MPO) positivity (Figure 2, A & B).
Few Pelger-like neutrophils were also observed along with maturing
myeloid cells. The patient did not develop any bleeding manifestations.
FCI revealed cells having high side scatter and dim CD45 expression,
heterogeneous CD13, and homogeneous bright CD33, along with lack of
CD34, HLA-DR, and CD56 expression. FISH was negative for t(15;17). The
RARA break-apart probe detected a variant RARA translocation. PCR for PML-RARA fusion transcripts was negative as well. ZBTB16 as the partner gene for variant RARA
was confirmed with PCR. The patient received induction with Idarubicin
and arsenic trioxide (ATO). Forty-five days post induction remission
was not achieved.
FISH showed variant RARA positivity in 52% cells. RT-qPCR showed mean threshold cycle (Ct) value of 26.78 against a baseline Ct value of 21.42 for ZBTB16-RARA
and 21.03 for ABL1. The patient further received three cycles of high
dose cytarabine (HiDAC) following which complete remission was achieved
with no detectable ZBTB16-RARA fusion transcripts.
Case 2.
A 38-year-old man with complaints of easy fatigability, dyspnea, and
intermittent fever was referred with a suspicion of APL for molecular
testing. FISH detected a variant RARA translocation which was confirmed to be ZBTB16-RARA
by PCR. However, the morphological features differed from the previous
case. The abnormal promyelocytes with regular nuclei showed the
presence of Auer rods but had abundant cytoplasm with scanty
granularity along with the presence of Pelger-like neutrophils (Figure 2, C & D).
The
patient received ATO based induction therapy but responded poorly and
failed to achieve remission. The post induction Ct value (25.02) by
RT-qPCR was similar to the baseline one (23.65). However, the patient
died shortly after completing two months of therapy.
Case 3.
A 45-year-old man was referred for molecular testing to confirm the
morphological suspicion of APL. Fever and easy fatigability were the
only presenting symptoms. Morphological features were similar to case
1, as highlighted by the presence of abnormal promyelocytes containing
round nuclei with regular nuclear contours, abundant granulated
cytoplasm and a lack of Auer rods. In contrast to classical APL cases
maturing myeloid cells were also present along with a few hypolobated
or Pelger-like neutrophils. Dual fusion FISH for PML-RARA was negative, but the break-apart probe revealed RARA variant translocation. ZBTB16-RARA
fusion was confirmed by PCR. RT-qPCR showed a baseline mean Ct value of
21.64. Although the patient was started on induction with ATO, he was
lost to follow up subsequently.
Case 4.
The patient is a 36-year-old gentleman with relapsed APL referred to
our institute for further management. A detailed history revealed that
the patient was first presented with fever and rash in February 2016
and morphological features on peripheral smear raised the suspicion of
APL. However, PML-RARA was not detected by molecular testing (details of PML-RARA or variant RARA
testing not available). The patient was started on induction therapy
with daunorubicin and cytarabine followed by three cycles of HiDAC. The
patient achieved remission after the 1st
cycle of HiDAC. He also received two cycles of decitabine and arsenic
trioxide before being referred to us. The patient relapsed in January
2017 with confirmation of t(11;17) by FISH. On presentation, the
patient was asymptomatic and clinical examination did not show any
abnormality. CBC and FCI findings are detailed in Table 1.
Bone marrow smears showed 22% abnormal promyelocytes and 33%
differentiating myeloid cells. Morphological features of the abnormal
promyelocytes were similar to case 1. Immunophenotype was similar to
case 1 except the CD117 expression in the present case. PCR was used to
confirm the presence of ZBTB16-RARA fusion.
Baseline Ct value by RT-qPCR was 21.39. The patient was counseled for
bone marrow transplantation but was put on palliative care, given
non-affordability for transplantation.
Case 5.
The patient was a 22-year-old student with a history of fever and body
ache for the past 2-3 months. Pallor and tenderness over the rib cage
were the only positive findings on clinical examination. Laboratory
findings are detailed in Table 1.
Peripheral smear examination showed 90% abnormal promyelocytes with
strong cytochemical myeloperoxidase positivity. Promyelocyte morphology
resembled case 1. In addition, maturing myeloid cells, hypogranular and
hypolobated neutrophils were also present (Figure 2, E & F).
The patient was immediately started on ATO based on the morphological
suspicion of APL. The patient did not develop any bleeding
manifestations. The immunophenotype was similar to case 1 and 4,
however, unlike those cases, CD56 was dim (Table 1). FISH did not show t(15;17), but the break-apart probe revealed RARA variant translocation. After 11 days of ATO and following confirmation of ZBTB16-RARA fusion
by PCR, the patient was shifted to daunorubicin and cytarabine-based
induction therapy. Remission was not achieved post induction (Table 1), and the patient was started on consolidation therapy with HiDAC.
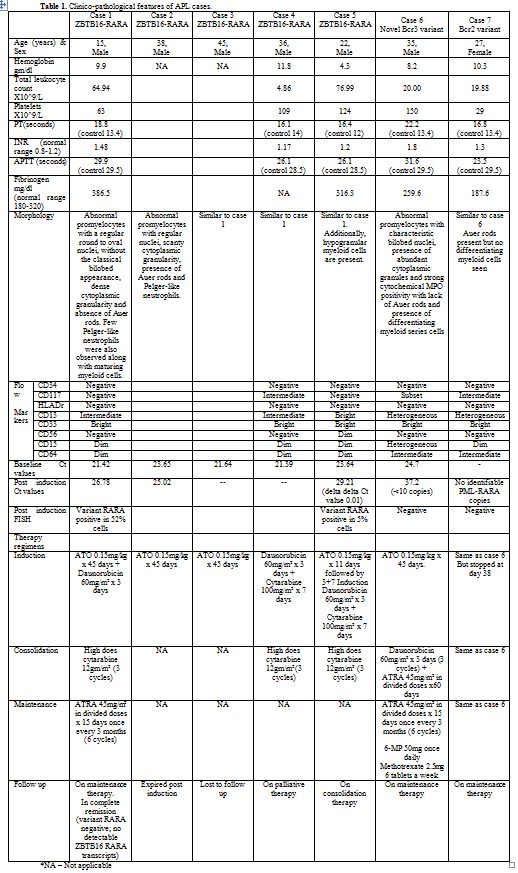 |
Table 1.
Clinico-pathological features of APL cases. |
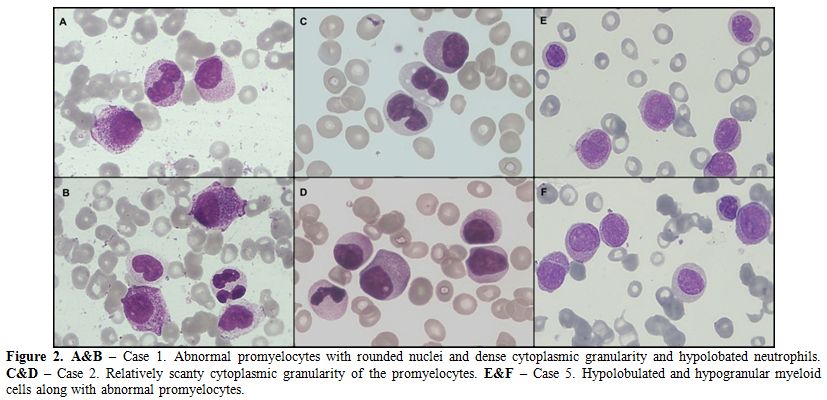 |
Figure 2. A&B – Case 1. Abnormal promyelocytes with rounded nuclei and dense cytoplasmic granularity and hypolobated neutrophils. C&D – Case 2. Relatively scanty cytoplasmic granularity of the promyelocytes. E&F – Case 5. Hypolobulated and hypogranular myeloid cells along with abnormal promyelocytes.
|
Case 6.
A 35-year-old male was referred to our Institute with complaints of
high grade, intermittent fever and back pain for one month. Laboratory
features are detailed in Table 1.
Peripheral smear revealed 29% abnormal promyelocytes with
characteristic morphology and presence of differentiating myeloid
series cells (Figure 3). The
patient did not develop any bleeding manifestations, in spite of
elevated PT and APTT values. Fluorescence in situ hybridization using a
dual fusion probe confirmed the presence of PML-RARA fusion in 93% of cells. The patient was classified as high risk as per the Sanz criteria[11]
and started on induction therapy with ATO. Post-induction bone marrow
examination for response assessment revealed that the patient had
achieved morphological and cytogenetic remission. Consolidation therapy
included three cycles of daunorubicin and ATRA. The patient is
currently doing well and is in maintenance therapy.
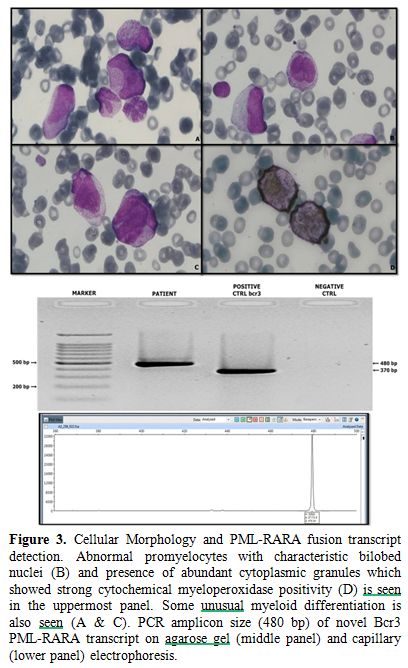 |
Figure 3. Cellular Morphology and PML-RARA
fusion transcript detection. Abnormal promyelocytes with characteristic
bilobed nuclei (B) and presence of abundant cytoplasmic granules which
showed strong cytochemical myeloperoxidase positivity (D) is seen in
the uppermost panel. Some unusual myeloid differentiation is also seen
(A & C). PCR amplicon size (480 bp) of novel Bcr3 PML-RARA
transcript on agarose gel (middle panel) and capillary (lower panel)
electrophoresis. |
Agarose gel electrophoresis for PML-RARA
transcript identification revealed a PCR amplicon size of approximately
480bp, which was larger than the expected Bcr3 fusion transcript size
(~370bp) (Figure 4). The PCR
amplicons were further subjected to Sanger sequencing and revealed an
atypical, novel fusion pattern between exon 4 of PML and exon 3 of RARA genes (Figure 4). Interestingly, an additional 40 nucleotides, mapped to intron 2 of RARA, were also coded as part of the mRNA fusion transcript (Figure 4).
The addition of exon 4 and 40 nucleotides from intron 2 explains the
increase in the size of the Bcr3 transcript type by 111 base pairs
(corresponding to the addition of 37 amino acids in the protein).
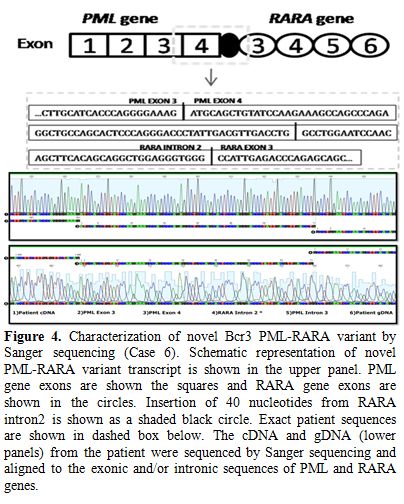 |
Figure 4. Characterization of novel Bcr 3
PML-RARA variant by Sanger sequencing (Case 6). Schematic
representation of novel PML-RARA variant transcript is shown in the
upper panel. PML gene exons are shown the squares and RARA gene exons
are shown in the circles. Insertion of 40 nucleotides from RARA intron
2 is shown as a shaded black circle. Exact patient sequences are shown
in dashed box below. The cDNA and gDNA (lower panels) from the patient
were sequenced by Sanger sequencing and aligned to the exonic and/or
intronic sequences of PML and RARA genes. |
Thus,
this novel Bcr3 variant transcript is in-frame and codes for a fusion
protein which is longer than expected PML-RARA fusion transcript
product. Cloning of the PCR product into a vector and Sanger sequencing
confirmed the above sequencing results (Figure 3). The same plasmid was serially diluted from 106 to 101
copies to be used as RT-qPCR standards. Baseline RT-qPCR revealed high
copies (13070) with amplification occurring at a Ct value of 24.7.
Post-induction bone marrow evaluation, after 48 days of ATO therapy,
revealed fewer than ten copies corresponding to a Ct value of 37.2.
Long-range genomic DNA PCR followed by Sanger sequencing identified a
breakpoint immediately downstream to exon 4 of PML gene (PML nucleotide
no: 74024927) and distal to the 11,789th base (RARA nucleotide no: 40343186) within intron 2 of RARA gene (Figure 4). The first 40 nucleotides of RARA intron 2 (starting from 11,790th base) alone were coded in the protein and were part of this novel fusion transcript.
Case 7.
A 26-year-old female was referred with a history of headache and right
iliac fossa pain for 2 months. Clinical examination revealed an
ill-defined, palpable mass in the right iliac fossa. There was no
history or evidence of any bleeding tendency on clinical examination.
Abdominal ultrasonography revealed the presence of a 3.3x2.2 cm
hemorrhagic cyst within the right ovary. Peripheral smear examination
showed 92% abnormal promyelocytes with classical morphology, strong MPO
positivity and the presence of Auer rods. The patient was classified as
high risk as per Sanz criteria[11] and started on ATO
based on the morphological suspicion of APL. FCI revealed typical
features of APL; with high side scatter, lack of CD34 and HLA-DR
expression, homogeneous bright CD33 and heterogeneous CD13 expression
(detailed in table 1). Dual fusion, dual color FISH showed evidence of PML-RARA
fusion. RT-PCR for transcript identification yielded an amplicon of ~
100 bases, higher than that of the expected Bcr1 transcript (390
bases). Sanger sequencing revealed breakpoints within exon 6 of PML and exon 3 of RARA (RARA nucleotide no: 40351910) with the addition of 126 nucleotides, which mapped to the central portion of RARA intron 2 (Figure 5). Therefore the patient was classified as a Bcr2 variant. PML exon 6 was truncated at nucleotide 248, 11 base pairs (bp) upstream of the normal exon 6/intron 6 boundary (PML
nucleotide no: 74033403). The insertion of 126 nucleotides from intron
2 can explain the increase in size of the transcript type on
electrophoresis. This novel Bcr2 variant transcript is in-frame and
codes for a longer PML-RARA fusion transcript product.
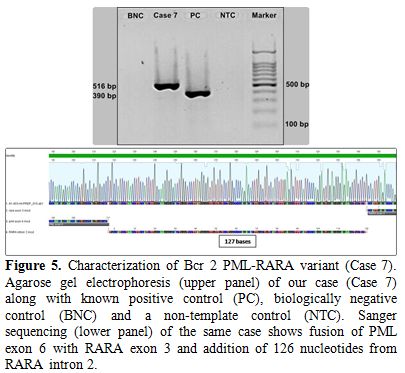 |
Figure 5. Characterization of Bcr 2
PML-RARA variant (Case 7). Agarose gel electrophoresis (upper panel) of
our case (Case 7) along with known positive control (PC), biologically
negative control (BNC) and a non-template control (NTC). Sanger
sequencing (lower panel) of the same case shows fusion of PML exon 6
with RARA exon 3 and addition of 126 nucleotides from RARA intron 2. |
Induction
therapy was withheld after 38 days due to suspicion of ATO induced
hepatotoxicity. However, investigations revealed acute viral hepatitis
due to hepatitis E virus (IgM HEV positive). Bone marrow
examination for response evaluation revealed complete morphological and
cytogenetic remission. RT-qPCR for PML-RARA
was also negative. The patient further received consolidation with
ATRA, daunorubicin and was started on maintenance therapy. RT-qPCR post
consolidation did not detect any PML-RARA copies.
Discussion
This
case series highlights the heterogeneity in molecular characteristics
of APL cases at a tertiary cancer center in India. APL’s variant
involving ZBTB16 is rare with a frequency of approximately 0.8% described in world literature.[4] However, the frequency of occurrence of ZBTB16-RARA
in our study was much higher than previously reported (2.8%). Distinct
morphological and immunophenotypic features have been described in
association with ZBTB16-RARA,[12]
which include round to oval nuclei with a regular nuclear membrane,
hypogranular cytoplasm, lack of Auer rods or Faggot cells and increased
number of Pelger-like neutrophils. Given these findings, Sainty et al.
have proposed a new morphological variant of APL, i.e. ‘M3r’ with a
threshold of more than 30% cells with above-described morphology as a
defining feature. Dense cytoplasmic granularity was seen in all but one
of our cases. Pelger-like neutrophils were consistent in three of the
five cases and the fifth case harbored hypogranular, almost dyspoietic
looking myeloid cells. Interestingly, these morphological features are
a combination of classical APL and ZBTB16-RARA
APL findings. Even within this rare subset of APL cases, we observed a
wide variation in the morphological features. In addition, lack of CD56
expression (except case 5) was also the only exception to the
immunophenotype associated with ZBTB16-RARA. Although Sainty et al. have reported strong CD56 expression in four and weak expression in one out of a total 6 cases with ZBTB16-RARA fusion, we found weak CD56 expression in just one of our cases.[12]
The morphology and immunophenotypic features of NPM1 mutated AML; such
as low or absent expression of immaturity associated antigens such as
CD34 and HLA-DR, can also be associated with APL, especially with ZBTB16-RARA fusion.[13,14]
None of our cases expressed CD34 or HLA-DR. Hence, relying on
morphology and immunophenotype alone, differentiation between the two
entities can be difficult and requires molecular diagnostic techniques
for confirmation. CD117 was not consistently expressed in our cases as
well, with only one of three ZBTB16-RARA cases being positive. The novel PML-RARA
Bcr3 variant described here showed classical APL morphology but with
the presence of differentiating myeloid cells, a feature which is
unusual in classic APL cases.
Bcr1 and Bcr3 breakpoints are seen
in 55% and 40% of APL cases respectively. Two Indian studies have
reported contrasting findings with bcr1 and bcr3 frequencies. Whereas
Chatterjee et al.[15] have reported lower bcr1(42.7%) but higher bcr2(14.8%) frequencies; Sazwal et al. (64%)[16] and Dutta et al. (72.7%)[17]
have reported over-representation of the bcr3 subtype in Indian
patients. The frequency of bcr1 (58%), bcr3 (33%) and bcr2 (5%)
subtypes detected at our center was similar to previously published
western literature.[3] Breakpoints in Bcr2 leading to
the variable (V) isoform are detected in up to 5% patients and have
been reported to be more common in the pediatric age group. The Bcr2
variant results from a breakpoint in exon 6 of PML (rarely exon 5) and may show insertion of genomic DNA from RARA intron 2. The size of the inserted RARA intron 2 segments may vary between 3 to 127 nucleotides.[18,19] The loss of distal part of PML exon 6 has been reported to negatively influence the response to treatment.[18,19] We report an adult, Bcr2 variant case showing the addition of 126 nucleotides between PML exon 6 and RARA exon 3 (Figure 5).
Our patient responded well to ATO based induction and in spite of
complications showed a good response and remains in complete remission.
We also report a unique Bcr3 variant with breakpoint at the junction of exon 4/intron 4 and intron 2 of RARA along with the addition of 40 nucleotides originating from a central portion of RARA intron 2 (Figure 4).
To the best of our knowledge, such a case has not been reported
previously. Jeziskova et al. also report a unique case with similar
breakpoint but with a smaller intronic insertion (9 nucleotides).[20]
Our patient showed a slightly delayed response to ATO based therapy,
achieving complete molecular remission post consolidation as compared
to the patient described by Jeziskova et al., who was in complete
molecular remission post-ATRA based induction therapy.
Classical and cytogenetic variant APL’s have distinct natural histories. APL with PML-RARA fusion
has a favorable outcome when treated with ATRA or arsenic trioxide.
Pharmacological doses of retinoic acid (in the form of ATRA) unbind the
N-Cor corepressor from RAR-RXR (RARA-retinoic
X receptor) complex and allow transcription of target genes, ultimately
leading to myeloid differentiation. The inability of ATRA to dissociate
ZBTB16 from the corepressor complex and epigenetic factors such as Polycomb group complexes contribute to the resistance of ZBTB16-RARA APL to conventional therapy.[21] The reduction in ZBTB16-RARA copy
number in our cases was determined using relative quantification based
RT-qPCR or the delta-delta Ct method. There was no significant change
in our treated patients between the baseline and post-induction
Ct-values, signifying resistance to ATRA/ATO based therapy. The
efficacy of combination therapy with anthracyclines and cytarabine
along with ATRA in inducing remission has been previously reported.[22]
Rohr et al. describe two cases in which the patients did not achieve
remission following induction with ATRA and anthracycline combination
but achieved partial remission after reinduction in one of the cases.[23] Even though our case (case 1) was not in remission post induction, no ZBTB16-RARA transcripts were detected by RT-qPCR post consolidation with high dose cytarabine.
Although
the clinical features and risk of developing life-threatening
disseminated intravascular coagulation is similar to classical APL, ZBTB16-RARA, however, shows distinct morphological features and a lack of response to ATRA/ATO.[5,12] ZBTB16-RARA
diagnosis offers a challenge that can be met with a combination of
morphological and molecular testing. We report a series of five cases
of ZBTB16-RARA associated APL from India with unusual morphological and immunophenotypic features. Cases with atypical breakpoints in PML-RARA represent sporadic events. We also report a unique Bcr3 PML-RARA variant hitherto unreported in the literature.
Acknowledgements
Dr.
Nikhil Patkar is supported by an intermediate fellowship of the
Wellcome Trust/Department of Science & Biotechnology, Govt of India
(India Alliance Grant: IA/CPHI/14/1/501485)
References
- Grimwade D. The pathogenesis of acute promyelocytic
leukemia: evaluation of the role of molecular diagnosis and monitoring
in the management of the disease. Br J Haematol. 1999;106(3):591-613. https://doi.org/10.1046/j.1365-2141.1999.01501.x PMid:10468848

- Grimwade
D, Lo Coco F. Acute promyelocytic leukemia: a model for the role of
molecular diagnosis and residual disease monitoring in directing
treatment approach in acute myeloid leukemia. Leukemia.
2002;16(10):1959-73. https://doi.org/10.1038/sj.leu.2402721 PMid:12357347
- Grimwade
D, Biondi A, Mozziconacci MJ, Hagemeijer A, Berger R, Neat M, et al.
Characterization of acute promyelocytic leukemia cases lacking the
classic t(15;17): results of the European Working Party. Groupe
Francais de Cytogenetique Hematologique, Groupe de Francais
d'Hematologie Cellulaire, UK Cancer Cytogenetics Group and BIOMED 1
European Community-Concerted Action "Molecular Cytogenetic Diagnosis in
Haematological Malignancies". Blood. 2000;96(4):1297-308.
PMid:10942371
- Adams
J, Nassiri M. Acute Promyelocytic Leukemia: A Review and Discussion of
Variant Translocations. Arch Pathol Lab Med. 2015;139(10):1308-13. https://doi.org/10.5858/arpa.2013-0345-RS PMid:26414475
- Grimwade
D, Mistry AR, Solomon E, Guidez F. Acute promyelocytic leukemia: a
paradigm for differentiation therapy. Cancer Treat Res.
2010;145:219-35. https://doi.org/10.1007/978-0-387-69259-3_13 PMid:20306254
- Jovanovic
JV, Rennie K, Culligan D, Peniket A, Lennard A, Harrison J, et al.
Development of real-time quantitative polymerase chain reaction assays
to track treatment response in retinoid resistant acute promyelocytic
leukemia. Front Oncol. 2011;1:35. https://doi.org/10.3389/fonc.2011.00035 PMid:22655241 PMCid:PMC3356041
- Livak
KJ, Schmittgen TD. Analysis of relative gene expression data using
real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.
Methods. 2001;25(4):402-8. https://doi.org/10.1006/meth.2001.1262 PMid:11846609
- van
Dongen JJ, Macintyre EA, Gabert JA, Delabesse E, Rossi V, Saglio G, et
al. Standardized RT-PCR analysis of fusion gene transcripts from
chromosome aberrations in acute leukemia for detection of minimal
residual disease. Report of the BIOMED-1 Concerted Action:
investigation of minimal residual disease in acute leukemia. Leukemia.
1999;13(12):1901-28. https://doi.org/10.1038/sj.leu.2401592 PMid:10602411
- Gabert
J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, et
al. Standardization and quality control studies of 'real-time'
quantitative reverse transcriptase polymerase chain reaction of fusion
gene transcripts for residual disease detection in leukemia - a Europe
Against Cancer program. Leukemia. 2003;17(12):2318-57. https://doi.org/10.1038/sj.leu.2403135 PMid:14562125
- Hasan
SK, Mays AN, Ottone T, Ledda A, La Nasa G, Cattaneo C, et al. Molecular
analysis of t(15;17) genomic breakpoints in secondary acute
promyelocytic leukemia arising after treatment of multiple sclerosis.
Blood. 2008;112(8):3383-90. https://doi.org/10.1182/blood-2007-10-115600 PMid:18650449 PMCid:PMC2954750
- Sanz
MA, Lo Coco F, Martin G, Avvisati G, Rayon C, Barbui T, et al.
Definition of relapse risk and role of nonanthracycline drugs for
consolidation in patients with acute promyelocytic leukemia: a joint
study of the PETHEMA and GIMEMA cooperative groups. Blood.
2000;96(4):1247-53. PMid:10942364
- Sainty
D, Liso V, Cantu-Rajnoldi A, Head D, Mozziconacci MJ, Arnoulet C, et
al. A new morphologic classification system for acute promyelocytic
leukemia distinguishes cases with underlying PLZF/RARA gene
rearrangements. Blood. 2000;96(4):1287-96. PMid:10942370
- Ossenkoppele
GJ, van de Loosdrecht AA, Schuurhuis GJ. Review of the relevance of
aberrant antigen expression by flow cytometry in myeloid neoplasms. Br
J Haematol. 2011;153(4):421-36. https://doi.org/10.1111/j.1365-2141.2011.08595.x PMid:21385170
- Falini
B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al.
Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal
karyotype. N Engl J Med. 2005;352(3):254-66. https://doi.org/10.1056/NEJMoa041974 PMid:15659725
- Chatterjee
T, Gupta S, Sharma S, Ganguli P. Distribution of Different PML/RARalpha
bcr Isoforms in Indian Acute Promyelocytic Leukemia (APL) Patients and
Clinicohematological Correlation. Mediterr J Hematol Infect Dis.
2014;6(1):e2014004. https://doi.org/10.4084/mjhid.2014.004 PMid:24455113 PMCid:PMC3894845
- Sazawal
S, Hasan SK, Dutta P, Kumar B, Kumar R, Kumar L, et al.
Over-representation of bcr3 subtype of PML/RARalpha fusion gene in APL
in Indian patients. Ann Hematol. 2005;84(12):781-4. https://doi.org/10.1007/s00277-005-1095-4 PMid:16132910
- Dutta
P, Sazawal S, Kumar R, Saxena R. Does acute promyelocytic leukemia in
Indian patients have biology different from the West? Indian J Pathol
Microbiol. 2008;51(3):437-9. https://doi.org/10.4103/0377-4929.42555 PMid:18723985
- Slack
JL, Willman CL, Andersen JW, Li YP, Viswanatha DS, Bloomfield CD, et
al. Molecular analysis and clinical outcome of adult APL patients with
the type V PML-RARalpha isoform: results from intergroup protocol 0129.
Blood. 2000;95(2):398-403. PMid:10627441
- Reiter
A, Saussele S, Grimwade D, Wiemels JL, Segal MR, Lafage-Pochitaloff M,
et al. Genomic anatomy of the specific reciprocal translocation
t(15;17) in acute promyelocytic leukemia. Genes Chromosomes Cancer.
2003;36(2):175-88. https://doi.org/10.1002/gcc.10154 PMid:12508246
- Jeziskova
I, Razga F, Gazdova J, Doubek M, Jurcek T, Koristek Z, et al. A case of
a novel PML/RARA short fusion transcript with truncated transcription
variant 2 of the RARA gene. Mol Diagn Ther. 2010;14(2):113-7. https://doi.org/10.1007/BF03256361 PMid:20359255
- Spicuglia
S, Vincent-Fabert C, Benoukraf T, Tiberi G, Saurin AJ, Zacarias-Cabeza
J, et al. Characterisation of genome-wide PLZF/RARA target genes. PLoS
One. 2011;6(9):e24176. https://doi.org/10.1371/journal.pone.0024176 PMid:21949697 PMCid:PMC3176768
- George
B, Poonkuzhali B, Srivastava VM, Chandy M, Srivastava A. Hematological
and molecular remission with combination chemotherapy in a patient with
PLZF-RARalpha acute promyelocytic leukemia (APML). Ann Hematol.
2005;84(6):406-8. https://doi.org/10.1007/s00277-004-0979-z PMid:15592671
- Rohr
SS, Pelloso LA, Borgo A, De Nadai LC, Yamamoto M, Rego EM, et al. Acute
promyelocytic leukemia associated with the PLZF-RARA fusion gene: two
additional cases with clinical and laboratorial peculiar presentations.
Med Oncol. 2012;29(4):2345-7. https://doi.org/10.1007/s12032-011-0147-y PMid:22205181
[TOP]