Ashley Rose1, Leidy Isenalumhe2, Magali Van den Bergh2 and Lubomir Sokol2.
1 Department of Internal Medicine, College of Medicine, University of South Florida, Tampa, Florida, USA.
2 Department of Malignant Hematology, Moffitt Cancer Center, Tampa, Florida, USA.
Corresponding
author: Lubomir Sokol, M.D., Ph.D. Department of Malignant Hematology. 12902 Magnolia Drive, Tampa, FL 33612, USA. E-mail:
lubomir.sokol@moffitt.org
Published: June 21, 2018
Received: December 30, 2017
Accepted: May 9, 2018
Mediterr J Hematol Infect Dis 2018, 10(1): e2018036 DOI
10.4084/MJHID.2018.036
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
We
report five patients with human immunodeficiency virus-1/acquired
immunodeficiency syndrome (HIV-1/AIDS) who developed T-cell large
granular lymphocytic proliferation (T-LGLP) or leukemia (T-LGLL). None
of the patients fulfilled criteria for diagnosis of diffuse
infiltrative lymphocyte syndrome (DILS) or HIV-associated CD8+
lymphocytosis syndrome at the time of diagnosis of LGL. The
immunophenotype of malignant T-cells was identical in three patients
with co-expression of CD3, CD8, CD57, and T-cell receptor (TCR)
alpha/beta. Three out of five patients were also diagnosed with clonal
disorders of B-cell origin including diffuse large B-cell lymphoma,
Burkitt’s lymphoma, and monoclonal gammopathy of undetermined
significance (MGUS). Two patients developed cytopenias due to
T-LGLL prompting initiation of therapy. Our study suggests that chronic
viral infection with HIV can contribute to the evolution of T-LGLP.
Clinical and laboratory characteristics of T-LGLP associated with
HIV-1/AIDS resemble those of immunocompetent patients.
|
Introduction
T-cell
large granular lymphocytic leukemia (T-LGLL) is a rare, clonal
lymphoproliferative disorder of mature T-cells manifesting with
peripheral blood cytopenias, splenomegaly, and increased incidence of
autoimmune disorders.[1,2] The diagnosis is based on
clinicopathological characteristics. Early reports suggested a
mandatory clonal LGL count of > 2x109/L in peripheral blood and duration of lymphocytosis >6 months.[3]
However, subsequent work postulated that diagnosis is possible in
patients with lower LGL count manifesting with characteristic clinical
features.[4,5] The spectrum of disease ranges from
asymptomatic large granular lymphocytic proliferation to symptomatic
leukemia which requires treatment. Long-term follow-up is recommended
to distinguish between the two.
The association of T-LGLL with
retroviral infections has been described previously. Two reports of
patients with HIV-1 associated with T-cell or natural killer (NK) LGL
have been previously documented.[6,7]
We
report a cohort of patients with HIV-1/AIDS who subsequently or
concurrently developed T-LGLP or T-LGLL. Clinical and laboratory
characteristics are analyzed and discussed.
Case 1
A
43-year-old Caucasian male presented with fatigue, sweats, weight loss,
musculoskeletal pain, and recurrent pneumonia in November 2006. His
complete blood count (CBC) revealed a white blood cell count (WBC) 19.6
x109/L, hemoglobin (Hb) 8.38 g/dL, platelet count (plt) 242 x109/L, absolute neutrophil count (ANC) 0.760 x109/L, and absolute lymphocyte count (ALC) 17.1 x109/L.
Bone marrow biopsy (BMB) showed a hypercellular marrow (70%) with
trilineage hematopoiesis and diffuse lymphocytic infiltration
comprising 40% of the total cellularity. Lymphocytes were
predominantly small to medium size with irregular nuclei and abundant
granular cytoplasm. Flow cytometry of bone marrow aspirate
revealed an atypical T-cell population co-expressing CD3, CD5, CD8,
CD16, CD57 and TCR alpha/beta, consistent with T-LGL. Cytogenetics were
normal. Serum protein electrophoresis showed elevated gamma globulin
level of 57 g/L and M spike of 31 g/L. Immunofixation confirmed IgG
kappa monoclonal gammopathy of undetermined significance
(MGUS). CT scan, PET/CT scan, and bone survey were unremarkable.
HIV-1 antibody screen and western blot were positive. CD4 count was 0.30 x109/L
and HIV-1 RNA viral load was 43,064 copies/mL. The patient was started
on highly active antiretroviral therapy (HAART), and after five months
of therapy, his viral load became undetectable. Repeat BMB and flow
cytometry confirmed persistent infiltration of marrow with T-LGL. Due
to persistent moderate neutropenia and three episodes of pneumonia,
treatment with cyclosporine A (CSA) was initiated for symptomatic
T-LGLL. The patient’s most recent laboratory studies and response to
therapy are not available.
Case 2
A
39-year-old Caucasian male who was diagnosed with HIV-1 in 1994
developed oral mucositis in January 2007. WBC revealed leukopenia with
an ANC close to 0 x109/L,
and normal Hb and plt. He was non-compliant with HAART until
February 2007 when he resumed emtricitabine, tenofovir, atazanavir, and
ritonavir, along with G-CSF 480 mcg SC biweekly due to severe
neutropenia.
In April 2007, his CBC revealed a WBC 2.96 x109/L, Hb 8.81 g/dL, plt 264 x109/L, ANC 0.26 x109/L, and ALC 1.35 x109/L.
Peripheral flow cytometry revealed an atypical T-cell population with
abundant granulated cytoplasm co-expressing CD3, CD5 weakly, CD7, CD8,
CD57 and TCR alpha/beta. Molecular studies revealed clonally rearranged
TCR beta gene, consistent with T-LGL. BMB and cytogenetics were normal.
Repeat BMB in August 2007 demonstrated a hypercellular marrow
(70%) with low level infiltration of T-LGL. Flow cytometry of bone
marrow aspirate was positive for a clonal T-LGL population with the
identical immunophenotype as seen in peripheral blood.
HIV-1 RNA viral load was undetectable, and the absolute CD4 count was 0.045 x109/L. CT scan showed no adenopathy or hepatosplenomegaly. ANC stabilized above 1.0 x 109/L with G-CSF treatment, then remained normal for more than two years after discontinuation of G-CSF. Case 3
A 47-year-old
Caucasian male was diagnosed with HIV in 2000. He was treated with
emtricitabine, tenofovir, lopinavir, and ritonavir, but became
non-compliant with therapy from 2006 until 2008.
In August 2008, his CD4 count was 0.021x109/L,
and viral load was 53,000 copies/mL. He was restarted on his HAART
regimen with improvement in CD4 counts, ranging from 0.090x109/L to 0.100 x109/L. He
was diagnosed with stage 3AE AIDS-associated diffuse large B-cell
lymphoma (DLBCL) of the oral cavity in May 2008. He underwent therapy
with five cycles of rituximab, cyclophosphamide, doxorubicin,
vincristine, and prednisone (R-CHOP) with intrathecal prophylactic
methotrexate. He achieved complete remission.
In September 2009, he developed lymphocytosis of 7.19 x109/L. Peripheral
flow cytometry revealed an abnormal T-cell population co-expressing
CD3, CD8, CD57, TCR alpha/beta, CD5 weakly, +CD7 weakly, and
HLA-DR. TCR gamma gene was clonally rearranged. His CBC showed
otherwise normal blood counts. He was clinically asymptomatic and has
been managed with observation.
Case 4
A 51-year-old
Caucasian man presented with unintentional weight loss, diarrhea, and
acute renal failure in January 2009. His CBC revealed absolute
lymphocytosis >10 x109/L. Flow
cytometry on peripheral blood was consistent with a CD8+
lymphoproliferative disorder. Esophagogastroduodenoscopy and
colonoscopy with biopsies revealed non-specific inflammatory changes
from the distal esophagus through the colon. Immunohistochemistry
showed a colonic infiltration with an atypical lymphoid population
co-expressing CD2, CD3, CD5, CD8 and TCR alpha/beta.
His CBC in February 2009 showed WBC 19 x109/L, Hb 9.8 g/dL, plt 156 x109/L, ANC 5.6 x109/L, and ALC 12.3 x109/L.
BMB was normocellular with focal interstitial infiltration of CD8+
cytotoxic T-lymphocytes without aberrant antigen expression.
Cytogenetics were normal. TCR beta gene was clonally rearranged. HIV-1
ELISA and confirmatory western blot were positive. HIV-1 viral load was
251,189 copies/mL and CD4 count was 0.53x109/L
CMV PCR revealed 400 copies/mL suggesting CMV reactivation. PET/CT
revealed diffuse hypermetabolic lymphadenopathy and splenomegaly.
Cervical lymph node excisional biopsy was consistent with follicular
hyperplasia. Flow cytometry revealed no evidence of an atypical clonal
T- or B-cell population. TCR beta gene rearrangement studies on
lymphonodal tissue were positive. Repeat flow cytometry on
peripheral blood showed the presence of CD8+ lymphocytosis without
aberrant immunophenotype, despite rearrangement of both TCR beta and
gamma genes.
The patient started efavirenz, emtricitabine,
and tenofovir in April 2009. His CBC normalized and GI symptoms
subsided. Repeat CT scans in February 2010 showed resolution of
lymphadenopathy. However, repeat peripheral flow cytometry in
February 2011 revealed a new clonal atypical T-cell population
co-expressing CD3, CD8, CD57, TCR alpha/beta, and weakly CD5. TCR
beta gene was clonally rearranged. Absolute LGL count was 0.773 x 109/L. The patient has been clinically asymptomatic and has been managed with observation. Case 5
A 58-year-old male
presented with unintentional weight loss, night sweats, and fatigue in
2009. He was found to have extensive abdominal and retroperitoneal
lymphadenopathy and was diagnosed with stage 3B Burkitt’s lymphoma and
HIV. At the time of diagnosis, his CD4 count was 0.050x109/L,
and viral load was 69,000 copies/mL. He was started on
cyclophosphamide, vincristine, doxorubicin, dexamethasone, cytarabine,
and methotrexate (hyperCVAD) as well as emtricitabine, tenofovir,
lopinavir, and ritonavir. He achieved complete remission, and his viral
load became undetectable. Lopinavir was switched to efavirenz due to
side effects. His viral load remained undetectable with CD4 counts >
1.0x109/L.
He was noted to have mild lymphocytosis on routine labs in August 2016 with CBC showing WBC 11 x 109/L, Hb 9.31 g/dL, plt 222 x109/L, ALC 4.09 x 109/L, and ANC 5.86 x 109/L.
Peripheral flow cytometry showed increased clonal CD8+/CD57+ large
granular lymphocytic T-cells with an absolute LGL count of 0.92 x 109/L.
TCR beta and gamma genes were clonally rearranged. The patient has been
clinically asymptomatic without cytopenias and has not required
treatment.Discussion
A hallmark of HIV
infection is a depletion of infected helper CD4+ cells resulting in an
increased incidence of opportunistic infections and AIDS-defining
malignancies.[8] The introduction of HAART
therapy results in a significantly decreased incidence of such
malignancies, as well as an improved patient outcome.[9,10]
HIV-associated mature T-cell malignancies comprised only 3% of all
AIDS-related lymphomas in a single institutional study in the US.[11]
However, significantly higher frequency (27%) was observed in a study
from South America, suggesting geographical and ethnic differences.[12]
In
contrast to the decrease in absolute CD4+ lymphocytes, transient
expansion of CD8+ cells has been detected early in the course of HIV
infection due to host immune response.[13]
Sustained expansions of the CD8+ T-cell population have been reported
in two conditions associated with HIV: diffuse infiltrative
lymphocytosis syndrome (DILS) and HIV-associated CD8+ lymphocytosis
syndrome.[14]
DILS was initially described in
1989 by Itescu et al. as a sicca syndrome in HIV infection. TCR gene
rearrangement was detected in a significant proportion of patients with
DILS, suggesting a clonal origin in these cells.[15]
The immunophenotype of these virally expanded CD8+ T-cells is similar
to memory and effector T-cells with co-expression of CD8, CD11a, CD11c,
and CD57.[15] A characteristic feature of DILS is a
CD8+ lymphocytic infiltration of salivary glands, and less frequently,
other visceral organs.[15] None of our patients
presented with salivary gland infiltration. A recent report suggested
that the incidence of DILS has decreased over time due to the
introduction of HAART.[16] Four out of five patients
in our series did not fulfill criteria for DILS at any time of our
observation; they developed the expansion of clonal LGLs 2 to 13 years
after diagnosis of HIV/AIDS. Case 4 manifested initially with
lymphocytosis, infiltration of colon and bone marrow with clonal CD8+
T-cells, suggesting DILS before the diagnosis of HIV. The CD8+ clone
associated with DILS disappeared from his circulation almost two years
before a new immunophenotypically distinct clonal CD8+ population
occurred in peripheral blood. His HIV infection was well controlled
with HAART at the time he developed a new clonal T cell LGL
population.
There are few documented reports of
patients with HIV and T-cell or NK-cell LGLP. Smith et al. reported 18
patients with HIV-1 who had persistent expansions of T-cell LGLs for 6
to 30 months. However, only five patients revealed clonal TCR gene
rearrangement, and no cytopenias or LGL infiltration of bone marrow
were reported.[14] Ghrenassia et al. reported 14
patients, three of which had HIV/AIDS, with CD8+ T-cell expansion. Six
patients had non-clonal, symptomatic organ infiltration and nine
patients had at least one cytopenia. Some patients with cytopenias were
found to have rearrangements consistent with T-LGL, while others did
not.[17]
A large retrospective study of
patients with LGLL found that 45.6% never required therapy. Of those,
peripheral blood LGL population percentage ranged from <0.5 to
>2.0 x 109/L. At the time of diagnosis, 24.6% of patients with LGLL had levels <0.5 x 109/L. Patients with intermediate LGL counts (0.5-2.0 x 109/L) required the highest mean number of therapies compared to those with high (>2.0 x 109/L) or low (<0.5x 109/L) LGL counts.[2]
Increased
incidence of B-cell dysregulation resulting in the development of
autoimmune disorders and B-cell malignancies were reported in patients
with LGL leukemia as well as patients with DILS, which could suggest a
causative role of chronic viral antigenic stimulation or the presence
of a putative autoantigen. Two separate studies identified a high
frequency of B-cell dyscrasias in patients with T-LGLL; MGUS, chronic
lymphocytic leukemia, Hodgkin and non-Hodgkin lymphoma were reported in
20% to 43% of T-LGLP patients.[18,19] Another study
described 20 patients with a dual diagnosis of T-LGLP and either a
B-cell or plasma cell lymphoproliferative disorder.[20]
Interestingly, three of five patients in our series developed B-cell
malignancies. Since HIV-positive patients also have an increased
incidence of B-cell lymphoproliferative disorders, both HIV/AIDS and
T-LGLL could be implicated in the development of B-cell malignancies.
In three patients, the diagnosis of HIV-1 infection was made prior to
the development of T-LGLP. The median time from diagnosis of HIV/AIDS
to the diagnosis of T-LGLP was two years (range 0-13 years). Conclusions
We describe five
unique patients with HIV/AIDS who developed persistent expansions of
clonal T-cell LGLs while their HIV infection was controlled with HAART (Table 1). All patients fulfilled earlier or more recent criteria for the diagnosis of LGLL (Table 2).
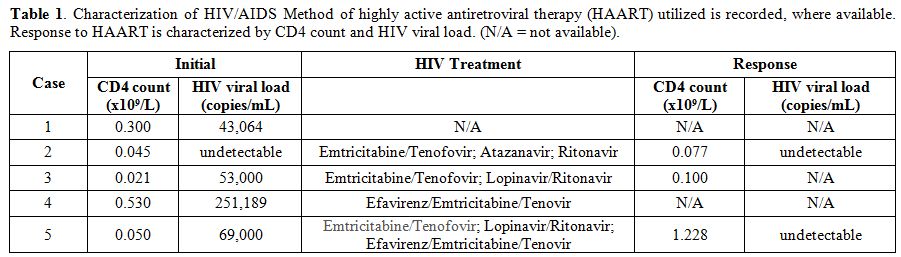 |
Table 1.
Characterization of HIV/AIDS Method of highly active
antiretroviral therapy (HAART) utilized is recorded, where available.
Response to HAART is characterized by CD4 count and HIV viral load.
(N/A = not available). |
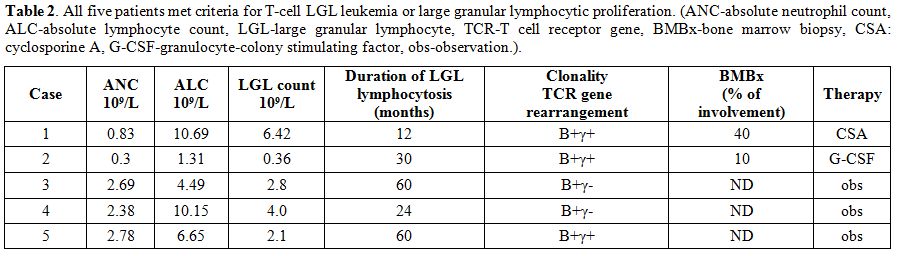 |
Table 2. All five patients met criteria
for T-cell LGL leukemia or large granular lymphocytic proliferation.
(ANC-absolute neutrophil count, ALC-absolute lymphocyte count,
LGL-large granular lymphocyte, TCR-T cell receptor gene, BMBx-bone
marrow biopsy, CSA: cyclosporine A, G-CSF-granulocyte-colony
stimulating factor, obs-observation.). |
All
patients demonstrated expansion of an immunophenotypically abnormal
clonal T-cell large granular lymphocytic population which persisted for
more than six months. Furthermore, two out of five patients developed
sustained neutropenia requiring therapy, which is the most common
cytopenia diagnosed in patients with LGLL. The rest of patients
demonstrated an indolent course of disease which was previously
reported in 30-50% of immunocompetent patients with LGLL. Our
observation expands a spectrum of T-cell large granular lymphocyte
disorders associated with HIV/AIDS, and supports the hypothesis that a
chronic antigenic stimulation with viral antigens could be implicated
in the etiopathogenesis of T-LGLP and T-LGLL.
References
- Sokol, L. and T.P. Loughran, Jr., Large granular lymphocyte leukemia. Oncologist, 2006. 11(3): p. 263-73. https://doi.org/10.1634/theoncologist.11-3-263 PMid:16549811

- Van
den Bergh, M., et al., A Single Institutional Experience of 261
Patients with Large Granular Lymphocytic Leukemia. Clinical Lymphoma,
Myeloma and Leukemia, 2017. 17: p. S379. https://doi.org/10.1016/j.clml.2017.07.225
- Loughran, T.J., Clonal diseases of large granular lymphocytes [see comments]. Blood, 1993. 82(1): p. 1-14. PMid:8324214
- Semenzato,
G., et al., The lymphoproliferative disease of granular lymphocytes:
updated criteria for diagnosis. Blood, 1997. 89(1): p. 256-260.
PMid:8978299
- Swerdlow,
S.H., et al., WHO Classification of Tumours of Haematopoietic and
Lymphoid Tissues. 2017: International Agency for Research on Cancer.
- Pulik,
M., et al., CD3+ CD8+ CD56− clonal large granular lymphocyte leukaemia
and HIV infection. British journal of haematology, 1997. 98(2): p.
444-445. https://doi.org/10.1046/j.1365-2141.1997.1913009.x PMid:9266947
- Ghali,
V., et al., Expansion of large granular lymphocytes (natural killer
cells) with limited antigen expression (CD2+, CD3−, CD4−, CD8−, CD16+,
NKH− 1‐) in a human immunodeficiency virus‐positive homosexual man.
Cancer, 1990. 65(10): p. 2243-2247. https://doi.org/10.1002/1097-0142(19900515)65:10<2243::AID-CNCR2820651014>3.0.CO;2-D
- Moir,
S., T.-W. Chun, and A.S. Fauci, Pathogenic mechanisms of HIV disease.
Annual Review of Pathology: Mechanisms of Disease, 2011. 6: p. 223-248.
https://doi.org/10.1146/annurev-pathol-011110-130254 PMid:21034222
- Diamond,
C., et al., Changes in acquired immunodeficiency syndrome‐related
non‐Hodgkin lymphoma in the era of highly active antiretroviral
therapy. Cancer, 2006. 106(1): p. 128-135. https://doi.org/10.1002/cncr.21562 PMid:16329140
- Lim,
S.-T., et al., Prognostic factors in HIV-related diffuse large-cell
lymphoma: before versus after highly active antiretroviral therapy.
Journal of Clinical Oncology, 2005. 23(33): p. 8477-8482. https://doi.org/10.1200/JCO.2005.02.9355 PMid:16230675
- Arzoo,
K.K., et al., T-cell lymphoma in HIV-infected patients. JAIDS Journal
of Acquired Immune Deficiency Syndromes, 2004. 36(5): p. 1020-1027. https://doi.org/10.1097/00126334-200408150-00004 PMid:15247554
- Collins,
J.A., et al., High proportion of T-cell systemic non-Hodgkin lymphoma
in HIV-infected patients in Lima, Peru. JAIDS Journal of Acquired
Immune Deficiency Syndromes, 2005. 40(5): p. 558-564. https://doi.org/10.1097/01.qai.0000185135.54920.79 PMid:16284532
- Brugnoni,
D., et al., The primary response to HIV infection is characterized by
an expansion of activated CD8+ CD28− cells. Aids, 1996. 10(1): p.
104-105. https://doi.org/10.1097/00002030-199601000-00017 PMid:8924239
- Smith,
P.R., et al., Benign monoclonal expansion of CD8+ lymphocytes in HIV
infection. Journal of clinical pathology, 2000. 53(3): p. 177-181. https://doi.org/10.1136/jcp.53.3.177 PMid:10823134 PMCid:PMC1731162
- Itescu,
S., et al., Certain HLA-DR5 and-DR6 major histocompatibility complex
class II alleles are associated with a CD8 lymphocytic host response to
human immunodeficiency virus type 1 characterized by low lymphocyte
viral strain heterogeneity and slow disease progression. Proceedings of
the National Academy of Sciences, 1994. 91(24): p. 11472-11476. https://doi.org/10.1073/pnas.91.24.11472
- Basu,
D., et al., Changing spectrum of the diffuse infiltrative lymphocytosis
syndrome. Arthritis Care & Research, 2006. 55(3): p. 466-472. https://doi.org/10.1002/art.21980
- Ghrenassia,
E., VRoulin L, Aline-Fardin A, Marzac C, Féger F, Gay J, PacanowskiJ,
Hertig A, Coppo P. The Spectrum of Chronic CD8+ T-Cell Expansions:
Clinical Features in 14 Patients. PloS one, 2014. 9(3): p. e91505. https://doi:10.1371/journal.pone.0091505
- Viny,
A.D., et al., Chronic B-cell dyscrasias are an important clinical
feature of T-LGL leukemia. Leukemia & lymphoma, 2008. 49(5): p.
932-938. https://doi.org/10.1080/10428190801932635
- Isenalumhe,
L., Frequency of Additional Malignancies in Patients with Large
Granular Lymphocytic Leukemia (LGLL): A Single Institutional
Experience. Clinical Lymphoma, Myeloma and Leukemia, 2017. 17: p. S380.
- Van
den Bergh, M., et al., A Single Institution Analysis of Patients with
Dual Diagnosis of T-Cell Large Granular Lymphocytic Leukemia and either
a Plasma Cell and/or a B-Cell Lymphoproliferative Disorder. Clinical
Lymphoma, Myeloma and Leukemia, 2016. 16: p. S117. https://doi.org/10.1016/j.clml.2016.07.169
[TOP]