Holly Lee1, Peter Duggan2, Paola Neri2, Jason Tay2, and Victor H Jimenez-Zepeda2.
1 Department of Internal Medicine, Cumming School of Medicine, University of Calgary, Calgary, Canada.
2 Tom Baker Cancer Center, Department of Medical Oncology and Hematology, Calgary, AB, Canada.
Correspondence to: Holly Lee, Department of Internal Medicine, Cumming
School of Medicine, University of Calgary, Calgary, Canada. E-mail:
holly.lee@ahs.ca
Published: January 1, 2019
Received: September 13, 2018
Accepted: November 17, 2018
Mediterr J Hematol Infect Dis 2019, 11(1): e2019007 DOI
10.4084/MJHID.2019.007
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Monoclonal
gammopathy of renal significance (MGRS) defines renal diseases
resulting from the nephrotoxic effects of monoclonal proteins secreted
from non-malignant clonal B cells or plasma cells, that do not meet
criteria for multiple myeloma, Waldenstrom's macroglobulinemia, chronic
lymphocytic leukemia, or lymphomas. Renal disease in MGRS can result
from monoclonal immunoglobulin deposition to different parts of the
kidney and includes a wide spectrum of glomerular, tubulointerstitial
and vascular renal diseases. Recognizing MGRS is important because
renal outcomes are poor and treatments targeting the underlying clonal
disease have been associated with improved renal survival. In this case
report, we present a case of a patient with proliferative
glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID)
subtype of MGRS who underwent a phased clone directed treatment of
induction and extended maintenance therapy to achieve renal response.
|
Introduction
Monoclonal
gammopathy of renal significance (MGRS) is an entity that was defined
in 2012 to describe a spectrum of renal diseases resulting from the
nephrotoxic effects of paraproteinemia from non-malignant clonal B
cells or plasma cells, and by definition does not meet criteria for
multiple myeloma, chronic lymphocytic leukemia, or malignant lymphoma.[1]
While renal disease such as myeloma light chain cast nephropathy or
renal damage from hyperviscosity syndrome in Waldenstrom’s
macroglobulinemia reflect the nephrotoxicity of the monoclonal proteins
produced by the underlying malignant tumor burden, renal conditions of
MGRS occur independent of the size or progression of the underlying
clonal disease.[2] The clonal size of MGRS is small
and most often resembles the state of monoclonal gammopathy of
undetermined significance (MGUS).[3]
MGRS can result from deposition of monoclonal proteins, dysregulation of complements, and activation of humoral factors.[4]
Monoclonal proteins can affect any parts of the kidney including
glomerular, tubulointerstitial and vascular compartments. Glomerular
lesions from monoclonal protein deposition is classified into 1)
organized deposition patterns of AL amyloidosis, cyroglobulinemic
glomerulonephritis (GN), and immunotactoid GN, and 2) unorganized
deposition including monoclonal immunoglobulin deposition disease,
proliferative glomerulonephritis with monoclonal immunoglobulin
deposits (PGNMID), and C3 GN with monoclonal gammopathy.
Tubulointerstitial diseases such as light chain proximal tubulopathy
and renal vascular diseases from deposition of amyloid fibrils and
crystalglobulinemia/ cryocrystalglobulinemia also constitute MGRS.[2,4]
Proliferative
glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) has
renal biopsy features of glomerular monoclonal immunoglobulin
deposition, and of the different immunoglobulin subtypes, IgG is most
commonly involved. Biopsy often reveals a proliferative or
membranoproliferative pattern, and some cases have reported mesangial
proliferation as well.[5-7] The distinction between
MGUS and MGRS is important given that the effect of monoclonal proteins
in MGRS is far from undetermined or benign.[1] Case
studies have shown that PGNMID renal injury has high rates of
progression, with up to 22% of patients progressing to end stage renal
disease.[8]
The management of MGRS highlights the
importance of timely diagnosis and initiation of therapy targeting the
underlying clonal disorder to improve renal outcomes.[3]
Here we report a case of a patient with PGNMID subtype of MGRS who
underwent clone directed treatment in a phased approach. After initial
induction therapy, the patient had complete renal response (defined as
proteinuria 0.5 g/day or less, albuminemia level > 30 g/L, and no
more than 10% decrease in eGFR from baseline[9]) which
lasted 6 months before she had recurrent proteinuria. She required re-
induction followed by ongoing maintenance treatment with bortezomib.
This case highlights the importance of long term follow up and a role
for maintenance therapy in MGRS management. Case Presentation
A
61-year-old female presented for assessment of anemia and microscopic
hematuria. She had no other significant medical comorbidities. At
presentation, her hemoglobin was 90 g/L and her creatinine was 79
umol/L (eGFR 69.6 mL/min/1.73m2,
creatinine clearance 62 mL/min). She described a history of fatigue and
mild pedal edema, but denied other constitutional symptoms. Review of
systems on history was otherwise unremarkable.
Investigations
revealed serum free light chains of kappa 51.6 mg/L, lambda 29.4 mg/L,
and elevated kappa to lambda ratio of 1.76 (normal range 0.26-1.65).
Serum protein electrophoresis and immunofixation did not reveal
evidence of monoclonal peak. Immunoglobulin levels were IgA 2.68, IgG
8.20, IgM 1.17 g/L. Serum albumin was 32 g/L and calcium level was 2.2
mmol/L. Initial urine studies showed proteinuria of 2.01 g/day. Urine
protein electrophoresis and immunofixation did not reveal evidence of
monoclonal peak. On blood work, her hepatitis screen was negative and
she had negative cryoglobulins, anti-GBM, ANCA, ANA, anti-dsDNA, and
rheumatoid factor levels. Her C3 and C4 were normal. HIV testing was
not done at the time of diagnosis.
For work up of her significant
proteinuria, she underwent an ultrasound guided renal biopsy with a
total of four passes with an 18-gauge biopsy needle to the lower pole
of the left kidney. Under the dissecting microscope, samples were taken
for plastics, electron microscopy, and immunofluorescence. The biopsy
revealed membranoproliferative glomerulonephritis (MPGN) with IgG kappa
deposition in granular and non-linear pattern, non-Randall type. On
microscopic analysis, most of the glomeruli showed marked cellular
proliferation with a lobular pattern and diffuse mesangial and
endocapillary proliferation with basement membrane duplication. There
was mild patchy interstitial fibrosis and tubular atrophy of 15% of the
cortex, with no significant interstitial inflammation.
Immuofluorescence microscopy revealed IgG 4+ finely granular and short
pseudo-linear stain along the basement membrane with lobular
accentuation. There was 2+ kappa stain. Stains for IgA, lambda and
fibrinogen were negative. There were trace IgM, 3+ C3c and 3+ C1q
stains. On electron microscopy, there were innumerable small
subendothelial and rare subepithelial deposits, as well as extensive,
but not total foot process effacement.
Bone marrow biopsy showed
3% plasma cells by immunohistochemistry, and flow cytometry showed a
slight bimodal population with lambda light chain excess. Congo red
staining on the marrow sample was negative. Cardiac MRI, echocardiogram
and skeletal survey did not demonstrate evidence of multiple myeloma or
AL amyloidosis.
Overall, the patient was diagnosed with PGNMID
subtype of MGRS. During initial clinical monitoring, she developed
worsening proteinuria of up to 3.54 g/day (Figure 1). Treatment was started with six cycles of cyclophosphamide po 300 mg/m2, bortezomib sc 1.3 mg/m2,
and dexamethasone po 40 mg once weekly (CyBorD). At the end of
induction, her proteinuria decreased to 0.17 g/day. At this time, the
treatment was stopped and she was followed clinically. Six months after
the last dose of bortezomib, she had recurrent proteinuria that peaked
at 2.8 g/day. She went on to receive three more cycles of CyBorD
followed by bortezomib sc 1.3 mg/m2
maintenance (without dexamethasone) every two weeks for the first three
months, then every three weeks, and then monthly afterwards.
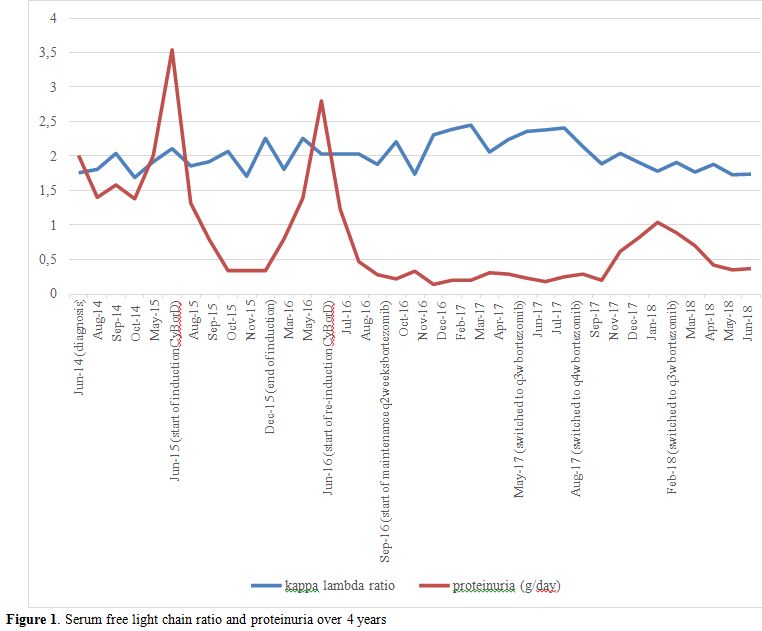 |
Figure 1. Serum free light chain ratio and proteinuria over 4 years |
Hematological
response was monitored by following light chain measurements per AL
amyloidosis response criteria. CR was defined as negative serum and
urine immunofixation and normal FLC ratio, VGPR defined as difference
in free light chains (dFLC) <40 mg/L, PR defined as dFLC decrease
>50%, and no response.[3,10] Renal response was measured using the KDIGO practice guideline on glomerulonephritis.[9,11]
Complete renal response was defined as proteinuria 0.5 g/day or less,
albuminemia level > 30 g/L, and no more than 10% decrease in eGFR
from baseline value.[9]
With re-induction and
maintenance treatment, she maintained hematological VGPR and met one of
the criteria for complete renal response with near resolution of
proteinuria (0.18 – 0.29 g/day). During recent follow up on monthly
bortezomib treatment, her proteinuria showed an increasing trend up to
1 g/day, and the bortezomib maintenance therapy frequency was switched
back to once every three weeks. Her creatinine remained in the 60-80
umol/L throughout the course of treatment. She does not have peripheral
neuropathies or gastrointestinal side effects and is tolerating ongoing
bortezomib therapy.
Discussion
Proliferative
glomerulonephritis with monoclonal immunoglobulin deposits (PGNMID) was
first described by Nasr et al. in 2004 when 10 patients were identified
who had renal biopsy findings that showed unclassifiable proliferative
glomerulonephritis with monoclonal IgG deposition.[12]
Biopsies most commonly showed diffuse proliferative or
membranoproliferative glomerulonephritis pattern on light microscopy.
Immunofluorescence studies revealed immune deposits staining for a
single IgG subclass and a single light chain isotype. On electron
microscopy, mesangial, subendothelial, and/ or subepithelial granular
electron-dense deposits were found.[12]
In
addition to a renal biopsy consistent with MPGN with IgG kappa
restricted deposits, our patient had the classical clinical presenting
features of PGNMID which included microhematuria and proteinuria.
Patients can also present with nephrotic syndrome or end stage renal
disease.[12] The clinical course of PGNMID was
reported by Nasr et al., who retrospectively assessed 37 PGNMID
patients with mean follow up time of 30 months, and found that 38% had
complete or partial recovery, 37% had persistent renal dysfunction, and
22% progressed to ESRD.[8] There is a risk of recurrence of disease in renal allograft in PGNMID patients who undergo renal transplantation.[6,13]
Furthermore, Steiner et al. reported that in their retrospective
observation study comparing 2891 MGUS patients versus biopsy proven 44
MGRS patients, there was a significantly higher rate of progression to
multiple myeloma in MGRS patients than in MGUS patients (18% vs 3%).[14]
As randomized controlled studies or prospective studies are not yet
available in the field of MGRS, treatment is mainly based on expert
consensus opinion and clinical experiences.[3]
Targeting the underlying clone is the central aspect to therapy, in
addition to managing the consequences of chronic kidney disease and end
stage renal disease. In the case of PGNMID, recommendations on
initiation of therapy are based on the stage of chronic kidney disease
and the degree of proteinuria.[3] At diagnosis, our patient had stage 2 CKD with 2.01 g of proteinuria per day, meeting indications for starting treatment.
The
decision to initiate treatment in MGRS can be challenging, particularly
when there is undetectable corresponding dysproteinemia or clear
evidence of clonal disease in the bone marrow. Among the different
types of renal diseases with monoclonal IgG deposition, PGNMID has one
of the lowest rates of detection of corresponding dysproteinemia. In
the 10 patients with PGNMID reported by Nasr et al., 5 had detectable
serum monoclonal protein.[12] In another cohort study
of 37 patients with PGNMID, only 10 patients had dysproteinemia at
diagnosis and 1 patient developed a serum monoclonal peak 3 years after
initial presentation.[8] Bhutani et al. showed that in
addition to the low detection rates in the serum, evidence of clonal
disease in the bone marrow biopsies were found in only 25% of the
patients with monoclonal immunoglobulin proliferative
glomerulonephritis, and concluded that current clonal disease detection
techniques may be inadequate to capture the low clonal tumor size in
MGRS.[15] Without detectable hematological
involvement, objective assessment of disease response to therapy
largely relies on monitoring renal markers.
Our patient had
evidence of dysproteinemia with excess kappa chain on serum free light
chain ratio, kappa restriction on renal biopsy, and lambda chain excess
on bone marrow flow cytometry. Given her diagnosis of MGRS with
progressive proteinuria of up to 3.54 g/day, she was started on
treatment with CyBorD. Her disease response was assessed monthly by
assessing free light chain measurements per AL amyloidosis response
criteria,[3,10] and the renal response was measured using the KDIGO practice guideline.[9,11] Studies have shown that hematological response corresponds with renal response.[3]
Chauvet et al. showed that the depth of hematological response is
associated with renal survival in MGRS. In their retrospective report
on 50 patients with monoclonal gammopathy-associated C3 glomerulopathy
(C3G), patients who had complete hematological response or very good
partial response had higher rates of renal survival compared to those
with no hematological response or partial response.[11]
Notably
in our patient, the renal response changed while the hematological
response remained constant. With treatment, she remained in VGPR
hematological response with no further improvement to complete
hematological response, while her proteinuria decreased from 3.54 g/day
to 0.17 g/day, meeting one of the criteria for complete renal response.
Serum protein electrophoresis and immunofixation did not reveal
evidence of monoclonal protein during follow up, and there was
persistent abnormal serum free light chain ratio with dFLC <40 mg/L.
When she had a recurrence of proteinuria 6 months after induction
treatment, this was again not reflected in her hematological markers as
she continued to remain in hematologic VGPR. In this case, the
hematological response did not appear to capture disease recovery or
relapse. As discussed in previous reports, it is possible that the
sensitivity of the current monoclonal protein detection assays
including serum free light chains or immunofixation may not be
sensitive enough to capture the low levels of dysproteinemia or the
small changes in the serum protein quantity.[15]
At
the time of renal relapse, the decision on re- induction and
maintenance treatment for this patient was made after seeking expert
clinical opinion. This is a case of MGRS treatment using induction and
prolonged maintenance bortezomib therapy. Through a single case report,
it is certainly not possible to establish a role for extended therapy.
Long term follow up with larger study population is required to
validate the extended treatment approach. It would be interesting to
assess how the time to relapse post-clone directed therapy impacts
renal outcomes. We note that our patient had renal relapse within 6
months after completion of therapy. In multiple myeloma, early relapse
of disease within a year of treatment portends poorer prognosis.[16]
Not only the depth but also the sustainability of disease response and
its impact on renal survival need to be studied. Furthermore, it would
be important to standardize the definition of relapse as well as
indications and therapy options for relapsed MGRS.
Acknowledgements
All authors contributed to the collection, analysis, interpretation of data, and the writing of the manuscript.
References
- Leung, N., F. Bridoux, C.A. Hutchison, S.H. Nasr,
P. Cockwell, J.P. Fermand, A. Dispenzieri, K.W. Song, and R.A. Kyle.
Monoclonal gammopathy of renal significance: when MGUS is no longer
undetermined or insignificant. Blood, 2012; 120(22): p. 4292-5. https://doi.org/10.1182/blood-2012-07-445304 PMid:23047823
- Rosner,
M.H., A. Edeani, M. Yanagita, I.G. Glezerman, and N. Leung.
Paraprotein-Related Kidney Disease: Diagnosing and Treating Monoclonal
Gammopathy of Renal Significance. Clin J Am Soc Nephrol, 2016; 11(12):
p. 2280-2287. https://doi.org/10.2215/CJN.02920316 PMid:27526705 PMCid:PMC5142062
- Fermand,
J.P., F. Bridoux, R.A. Kyle, E. Kastritis, B.M. Weiss, M.A. Cook, M.T.
Drayson, A. Dispenzieri, and N. Leung. How I treat monoclonal
gammopathy of renal significance (MGRS). Blood, 2013; 122(22): p.
3583-90. https://doi.org/10.1182/blood-2013-05-495929 PMid:24108460
- Leung, N., M.E. Drosou, and S.H. Nasr. Dysproteinemias and Glomerular Disease. Clin J Am Soc Nephrol, 2018; 13(1): p. 128-139. https://doi.org/10.2215/CJN.00560117 PMid:29114004
- Al-Hussain,
T., M.H. Hussein, H. Al Mana, and M. Akhtar. Renal involvement in
monoclonal gammopathy. Adv Anat Pathol, 2015; 22(2): p. 121-34. https://doi.org/10.1097/PAP.0000000000000056 PMid:25664947
- Nasr,
S.H., S. Sethi, L.D. Cornell, M.E. Fidler, M. Boelkins, F.C. Fervenza,
F.G. Cosio, and V.D. D'Agati. Proliferative glomerulonephritis with
monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nephrol,
2011; 6(1): p. 122-32. https://doi.org/10.2215/CJN.05750710 PMid:20876681 PMCid:PMC3022233
- Lusco,
M.A., A.B. Fogo, B. Najafian, and C.E. Alpers. AJKD Atlas of Renal
Pathology: Proliferative Glomerulonephritis With Monoclonal
Immunoglobulin Deposits. Am J Kidney Dis, 2016; 67(3): p. e13-5. https://doi.org/10.1053/j.ajkd.2016.01.003 PMid:26916379
- Nasr,
S.H., A. Satoskar, G.S. Markowitz, A.M. Valeri, G.B. Appel, M.B.
Stokes, T. Nadasdy, and V.D. D'Agati. Proliferative glomerulonephritis
with monoclonal IgG deposits. J Am Soc Nephrol, 2009; 20(9): p.
2055-64. https://doi.org/10.1681/ASN.2009010110 PMid:19470674 PMCid:PMC2736767
- Radhakrishnan,
J. and D.C. Cattran. The KDIGO practice guideline on
glomerulonephritis: reading between the (guide)lines--application to
the individual patient. Kidney Int, 2012; 82(8): p. 840-56. https://doi.org/10.1038/ki.2012.280 PMid:22895519
- Palladini,
G., A. Dispenzieri, M.A. Gertz, S. Kumar, A. Wechalekar, P.N. Hawkins,
S. Schonland, U. Hegenbart, R. Comenzo, E. Kastritis, M.A. Dimopoulos,
A. Jaccard, C. Klersy, and G. Merlini. New criteria for response to
treatment in immunoglobulin light chain amyloidosis based on free light
chain measurement and cardiac biomarkers: impact on survival outcomes.
J Clin Oncol, 2012; 30(36): p. 4541-9. https://doi.org/10.1200/JCO.2011.37.7614 PMid:23091105
- Chauvet,
S., V. Fremeaux-Bacchi, F. Petitprez, A. Karras, L. Daniel, S. Burtey,
G. Choukroun, Y. Delmas, D. Guerrot, A. Francois, M. Le Quintrec, V.
Javaugue, D. Ribes, L. Vrigneaud, B. Arnulf, J.M. Goujon, P. Ronco, G.
Touchard, and F. Bridoux. Treatment of B-cell disorder improves renal
outcome of patients with monoclonal gammopathy-associated C3
glomerulopathy. Blood, 2017; 129(11): p. 1437-1447. https://doi.org/10.1182/blood-2016-08-737163 PMid:28069603
- Nasr,
S.H., G.S. Markowitz, M.B. Stokes, S.V. Seshan, E. Valderrama, G.B.
Appel, P. Aucouturier, and V.D. D'Agati. Proliferative
glomerulonephritis with monoclonal IgG deposits: a distinct entity
mimicking immune-complex glomerulonephritis. Kidney Int, 2004; 65(1):
p. 85-96. https://doi.org/10.1111/j.1523-1755.2004.00365.x PMid:14675039
- Lorenz,
E.C., S. Sethi, N. Leung, A. Dispenzieri, F.C. Fervenza, and F.G.
Cosio. Recurrent membranoproliferative glomerulonephritis after kidney
transplantation. Kidney Int, 2010; 77(8): p. 721-8. https://doi.org/10.1038/ki.2010.1 PMid:20130531
- Steiner,
N., G. Göbel, P. Suchecki, W. Prokop, H. Neuwirt, and E. Gunsilius.
Monoclonal gammopathy of renal significance (MGRS) increases the risk
for progression to multiple myeloma: an observational study of 2935
MGUS patients. Oncotarget, 2018; 9(2): p. 2344-56. https://doi.org/10.18632/oncotarget.23412 PMid:29416776 PMCid:PMC5788644
- Bhutani,
G., S.H. Nasr, S.M. Said, S. Sethi, F.C. Fervenza, W.G. Morice, P.J.
Kurtin, F.K. Buadi, D. Dingli, A. Dispenzieri, M.A. Gertz, M.Q. Lacy,
P. Kapoor, S. Kumar, R.A. Kyle, S.V. Rajkumar, and N. Leung.
Hematologic characteristics of proliferative glomerulonephritides with
nonorganized monoclonal immunoglobulin deposits. Mayo Clin Proc, 2015;
90(5): p. 587-96. https://doi.org/10.1016/j.mayocp.2015.01.024 PMid:25939936
- Kumar,
S., S.T. Mahmood, M.Q. Lacy, A. Dispenzieri, S.R. Hayman, F.K. Buadi,
D. Dingli, S.V. Rajkumar, M.R. Litzow, and M.A. Gertz. Impact of early
relapse after auto-SCT for multiple myeloma. Bone Marrow Transplant,
2008; 42(6): p. 413-20. https://doi.org/10.1038/bmt.2008.180 PMid:18587435
[TOP]