Zhuo Gao1, Yini Wang1, Jingshi Wang1, Jia Zhang1 and Zhao Wang1.
1
Department of Hematology, Beijing Friendship Hospital, Capital Medical University, Beijing, China.
Correspondence to: Zhao Wang. Department of Hematology,
Beijing Friendship Hospital, Capital Medical University, 95 Yong An
Road, Xicheng District, Beijing 100050, China. Tel/Fax:
+86-10-63138303. E-mail:
wangzhao@ccmu.edu.cn
Published: January 1, 2019
Received: August 8, 2018
Accepted: November 26, 2018
Mediterr J Hematol Infect Dis 2019, 11(1): e2019008 DOI
10.4084/MJHID.2019.008
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
differentiation of primary hemophagocytic lymphohistiocytosis (pHLH)
and macrophage activation syndrome (MAS) poses a challenge to
hematologists. The aim of this study was (1) to compare the levels of
soluble ST2 (sST2), sCD163, IFN-γ, IL-10, IL-18, TNF-α and Serum
soluble interleukin-2 receptor (sCD25)in patients with pHLH and MAS and
(2) to investigate whether they can help differentiate the two
diseases. A total of 52 participants were recruited in this study,
including 12 pHLH patients, 20 MAS patients, and 20 healthy subjects.
We measured the levels of sST2, sCD163 and sCD25 in serum by ELISA. The
serum levels of IFN-γ, IL-10, IL-18, and TNF-α were detected using a
Luminex 200 instrument. The serum levels of sST2 and sCD163 in MAS
patients were markedly higher than that in pHLH patients (363.13 ±
307.24 ng/ml vs 80.75 ± 87.04 ng/ml, P = 0.004; 3532.72 ± 2479.68 ng/ml
vs 1731.96 ± 1262.07 ng/ml, P = 0.046). There was no significant
difference in the expression of IFN-γ (306.89 ± 281.60 pg/ml vs 562.43
± 399.86 pg/ml), IL-10 (20.40 ± 30.49 pg/ml vs 8.3 ± 13.14 pg/ml),
IL-18 (463.33 ± 597.04
pg/ml vs 1247.82 ± 1318.58 pg/ml), TNF-α
(61.48 ± 84.69 pg/ml vs 106.10 ± 77.21 pg/ml), and sCD25 (21062.1 ±
18515.26 pg/ml vs 11074.78 ± 11149.96 pg/ml) between pHLH and MAS.
Patients with pHLH and MAS show some differences in cytokine profiles.
The elevated levels of IFN-γ, IL- 10, TNF-α, IL-18, and sCD25 can
contribute to the diagnosis of HLH, but may not discriminate pHLH from
MAS. Levels of sST2 and sCD163 may serve as markers to distinguish pHLH
from MAS.
|
Introduction
Hemophagocytic
lymphohistiocytosis (HLH) is a clinical syndrome manifested by fever,
hepatosplenomegaly, cytopenia, and hemophagocytosis in bone marrow,
liver, spleen or lymph nodes. HLH is classified into primary HLH (pHLH)
and secondary HLH (sHLH). The pHLH is triggered by genetic mutations
whereas sHLH is mainly associated with infection, cancer or autoimmune
diseases.[1] According to the diagnostic guidelines HLH-2004,[2] the diagnosis for pHLH requires definite evidence of a genetic defect.
Nevertheless,
genetic sequencing is time-consuming and may unnecessarily delay the
administration of specific therapy. Several indicators such as natural
killer (NK) cell function, perforin (PRF1), granzyme, SLAM- Associated
protein (SAP), an X-linked inhibitor of apoptosis protein (XIAP),
MUNC13-4, Syntaxin11, LYST, and ITK protein levels may provide a quick
prediction of pHLH. However, the tests for those indicators are
difficult to popularize in a short time. Thus the identification of
pHLH is quite difficult, if not impossible, to achieve. MAS is a
subtype of sHLH with an approximate mortality rate of 8-22%.[3-5]
It is a severe complication of rheumatic diseases caused by excessive
activation and expansion of T lymphocytes and macrophages that exhibit
hemophagocytic activity.[6] MAS is a serious,
potentially fatal state associated with many rheumatologic diseases
including systemic juvenile idiopathic arthritis (SJIA), adult-onset
still disease (AOSD), rheumatoid arthritis, systemic lupus
erythematosus (SLE), Kawasaki disease (KD), and dermatomyositis.[7-12]
It is clinically characterized by fever, hepatosplenomegaly,
lymphadenectasis, severe anemia, liver dysfunction, disseminated
intravascular coagulation (DIC) and central nervous system involvement.[13]
HLH,
regardless of its subtypes, has an aggressive clinical course with a
high mortality rate. In spite of recent advances in the treatment of
HLH, therapeutic regimens are often empirically based, and patients’
responses to therapies differ sharply. Better prognosis relies heavily
on early diagnosis, timely and comprehensive treatments. Therapy for
HLH should center on the suppression of the hyperinflammatory state and
treatment of any existing HLH triggers.[14,15] In
pHLH, hematopoietic stem cell transplantation (HSCT) will be eventually
needed for a full recovery, whereas a pulse of high-dose
corticosteroids with or without cyclosporine A (CsA) will work in most
MAS patients.[15] Thus, the differentiation of the
two subtypes is essential not just for the assessment of a patient’s
condition but also for the subsequent selection of appropriate
treatment. Unfortunately, apart from the similar clinical
manifestations with pHLH patients, patients with MAS may also present
decreased NK cell activity, lower perforin expression, and
single-nucleotide polymorphisms of UNC13D and PRF1 genes.[16] Therefore, it poses a great challenge for us to make a distinction between them with the absence of genetic testing.
In
recent years, the role of cytokines in HLH has gained extensive
attention and studies on cytokines may open up new avenues in the
diagnosis and differential diagnosis of HLH. Increasing evidence
suggests that IL- 10, IFN-γ, IL-18, TNF-α, and sCD25 play an important
role in the pathogenesis of HLH.[17-21] Besides, Rood[22]
indicates that disruption of ST2 signaling in the murine model of
Family HLH (FHL) can influence the immune regulation and the blockade
of ST2 may be a novel therapeutic strategy for FHL. In addition, CD163
has been shown to serve as a potential biomarker for HLH and relevant
diseases.[23] Unfortunately, few studies have been focused on the expression of these cytokines in the subtypes of HLH.
In
this study, we measured the expression levels of sST2, sCD163, IL-10,
IFN-γ, IL-18, TNF-α, and sCD25 and analyzed their role as possible
markers to distinguish pHLH from MAS.
Patients and Methods
Patients. Diagnosis of HLH was based on the criteria set in the HLH-2004.[2] MAS patients were identified according to the international consensus published in 2011,[24]
and all the MAS patients meet the HLH-2004 criterion. Patients were
confirmed as pHLH based on the evidence of a documented molecular
mutation. The evaluation of treatment efficacy was described in our
previous study.[25] A total of 32 patients was
recruited in this study, including 12 pHLH patients, 20 AOSD associated
MAS patients presented at the Department of Hematology at Beijing
Friendship Hospital from January 2015 to March 2018. Additionally, 20
healthy subjects were invited to participate as controls. This study
was approved by the ethics committee of Beijing Friendship Hospital,
and informed consent forms were signed by all of the subjects prior to
participation in this study. All experiments were performed in
accordance with the approved guidelines and regulations.
Sample collection.
Peripheral venous blood was collected in the serum-separating tube from
patients with pHLH, MAS and healthy controls. The blood was centrifuged
at 3000 rpm for 10 min, and the serum was collected and stored at -80°
until undergoing analysis.
Detection of cytokines.
The Luminex 200 instrument was applied to detect the expression of
IFN-γ, IL-10, IL- 18 and TNF-α cytokines (eBioscience, EPX340-12167-
901). The expression of Human serum sST2 (R&D, CAT# DST200), sCD163
(R&D, CAT# DC163) and Human sCD25/IL-2R (Diaclone, CAT#850.500.096)
were measured using ELISA.
Gene sequencing.
The exon and related cleavage products of HLH-related genes were
obtained by using specific-primer design and PCR on DNA extracted from
mononuclear cells. This was followed by bi- directional Sanger
sequencing.
Statistical analysis.
Statistical analysis was performed using the SPSS19.0 software. All
normally distributed data were represented by means ± standard
deviations, and comparison of multiple samples between groups was
performed by one-way analysis of variance (ANOVA). All data that were
not distributed normally were represented by median and range, and
comparison of multiple samples between groups was performed by Wilcoxon
rank sum test. P values< 0.05 were considered indicative of statistically differences.
Results
Characteristics of the enrolled participants.
Twelve patients diagnosed as pHLH were recruited with an average age of
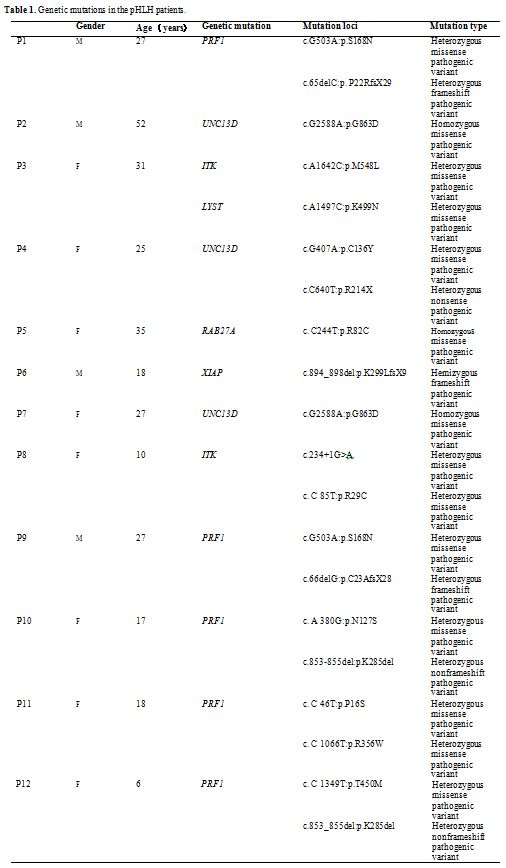
24 years. The details of the genetic mutations were summarized in Table1.
 |
Table
1. Genetic mutations in the pHLH patients. |
Seven
cases (58.33%) of pHLH have compound heterozygous pathogenic variants,
including 5 cases involved PRF1, one case involved UNC13D, and one case
involved ITK. Other mutated genes include homozygous pathogenic
variants of UNC13D in 2 patients (16.67%), homozygous pathogenic
variants of RAB27A in 1 patient (8.33%), hemizygous pathogenic variants
in 1 patient (8.33%), and pathogenic variants in both ITK and LYST in 1
patient (8.33%).
The clinical symptoms of pHLH patients were
characterized by fever, hepatosplenomegaly, hemophagocytosis in bone
marrow, and lymphadenectasis. Fever was observed in all 12 pHLH
patients (100%); Splenomegaly and hemophagocytosis in bone marrow were
found in both 11 patients (91.67%); 9 patients (75%) had
lymphadenectasis; Other clinical features included rash (25%),
pharyngalgia (8.33%) and arthralgia (16.67%). Besides, five patients
(41.67%) had central nervous system involvement. The clinical
characteristics of all participants are shown in Table 2.
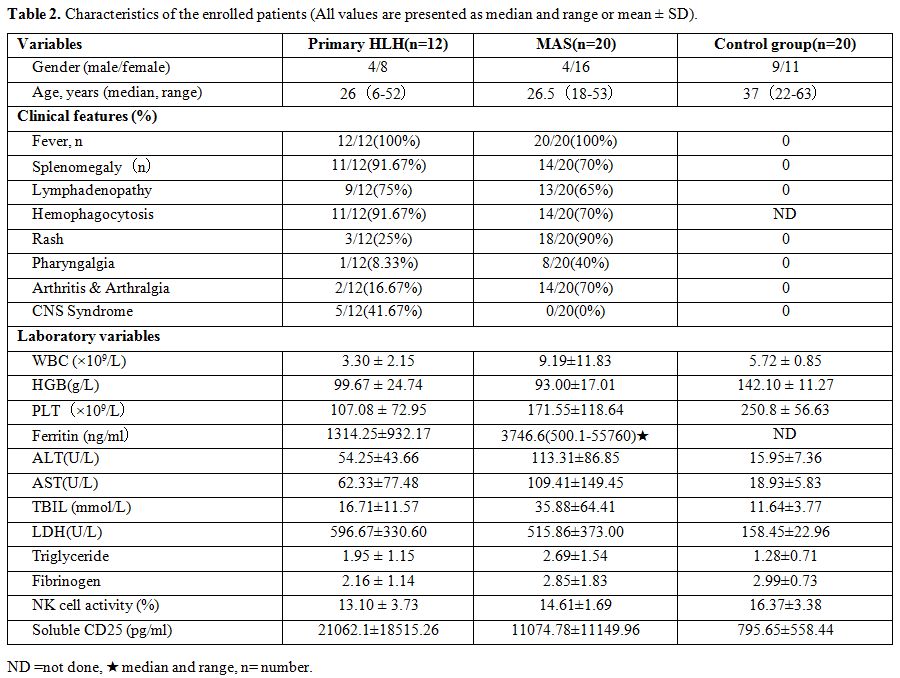 |
Table 2. Characteristics of the enrolled patients (All values are presented as median and range or mean ± SD). |
Twenty
AOSD associated MAS patients in the active stage were enrolled,
including four males and 16 females. The median age of those patients
was 26 years. In MAS patients, fever was observed in all 20 patients
(100%); splenomegaly and hemophagocytosis in bone marrow were found in
both 14 patients (70%), 13 patients (65%) had lymphadenectasis; up to
18 (90%) patients had rash, and the incidence of pharyngalgia and
arthralgia was 40% and 70%. No patients suffered central nervous system
involvement.
Treatment and outcomes.
8 cases (66.67%) of pHLH received HLH-94/2004 regimen as initial
treatment; 4 cases (33.33%) of pHLH received dexamethasone as initial
treatment. Three cases (25%) have no response to the initial treatment
after two weeks. The DEP regimen[25] was used as
salvage therapy for those three patients, and all of them (25%)
achieved partial response (PR) or complete response (CR). Allogeneic
hematopoietic stem cell transplantation (Allo-HSCT) was performed in
six patients (50%) within the remission stage. Among the six patients
who received Allo-HSCT, two died, and the remaining 4 HLH patients
survived. The death of the two patients was attributed to
graft-versus-host disease (GVHD) and severe pneumonia, respectively.
All of the six patients who did not receive Allo-HSCT relapsed, and
four patients died (Table 3).
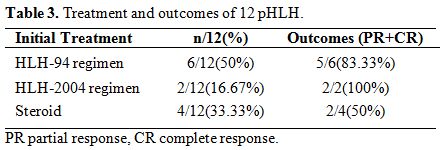 |
Table 3. Treatment and outcomes of 12 pHLH. |
14
cases (70%)of MAS received HLH-94/2004 regimen as initial treatment; 2
cases have no response to this treatment, and one patient died. Three
patients (15%) underwent the regime of steroid combined CsA. DEP
regimen was carried out in 2 patients (10%). All the five patients
achieved PR after two weeks. One patient (5%) died before the initial
treatment was performed (Table 4).
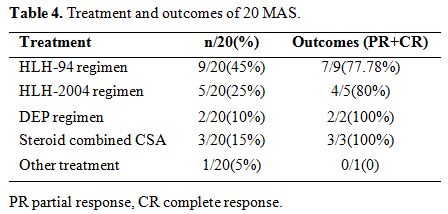 |
Table 4. Treatment and outcomes of 20 MAS. |
Different levels of sST2 and sCD163 in pHLH and MAS.
In order to identify the potential markers of cytokines in HLH, we
analyzed the levels of sST2 in the pHLH, MAS and control
groups (80.75 ± 87.04 ng/ml, 363.13
± 307.24 ng/ml and 19.05 ± 8.31 ng/ml, respectively), as shown in Figure 1A,
it appears that levels of sST2 were significantly increased in MAS
patients in comparison with that in pHLH (P = 0.004) and healthy
controls (P = 0.001). Next, sCD163 levels in pHLH, MAS and healthy
controls were detected and analyzed. Median sCD163 levels in patients
with MAS were higher compared with patients with pHLH and were elevated
compared with healthy controls (3532.72 ± 2479.68 ng/ml, 1731.96 ±
1262.07 ng/ml and 393.94 ± 148.72 ng/ml, respectively).
Strikingly, statistically significant differences of sCD163 levels were
also observed between the pHLH and MAS groups (P = 0.046) (Figure 1B).
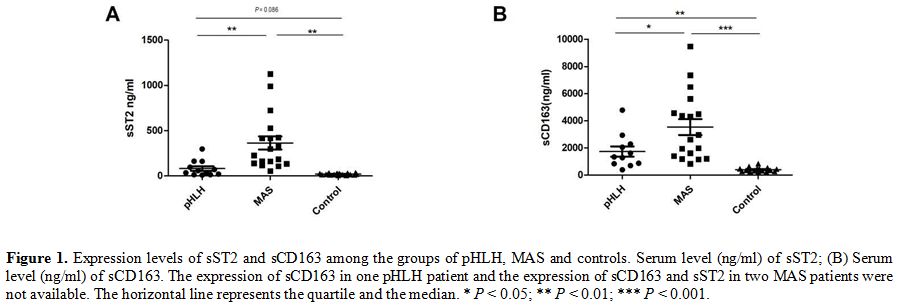 |
Figure 1. Expression
levels of sST2 and sCD163 among the groups of pHLH, MAS and controls.
Serum level (ng/ml) of sST2; (B) Serum level (ng/ml) of sCD163. The
expression of sCD163 in one pHLH patient and the expression of sCD163
and sST2 in two MAS patients were not available. The horizontal line
represents the quartile and the median. * P < 0.05; ** P < 0.01; *** P < 0.001. |
Comparison of serum levels of IFN-γ, IL-10, IL-18 and TNF-α among groups of patients pHLH, MAS and healthy control.
To explore the role of IFN-γ, IL-10, IL- 18, and TNF-α in the
development of HLH, we measured and compared the expressions of those
cytokines in the groups. In patients with pHLH and MAS, there was a
trend toward higher levels of IFN-γ, IL-10, IL-18 and TNF-α than that
in healthy controls (306.89 ± 281.60 pg/ml, 20.40 ± 30.49 pg/ml, 463.33
± 597.04 pg/ml, 61.48 ± 84.69 pg/ml in pHLH; 562.43 ± 399.86 pg/ml, 8.3 ± 13.14 pg/ml, 1247.82 ± 1318.58 pg/ml, 106.10 ± 77.21 pg/ml in MAS, respectively). IL-10
could not be detected in the majority of healthy subjects, and a barely
detectable concentration of IL-10 was observed in
only three healthy participants (0.11 pg/ml, 0.11 pg/ml and 6.19
pg/ml). TNF-α could be detected in 4 healthy persons (22.94 pg/ml,
40.37 pg/ml, 9.06
pg/ml and 5.22 pg/ml) and IFN-γ could be detected in only one healthy
serum sample (2.14 pg/ml). Similarly, IL-18 levels are also difficult
to detect in the serum of healthy group, and we only detected the
expression of IL-18 in 7 persons (16.79 pg/ml,70.71 pg/ml,69.26
pg/ml,43.38 pg/ml,2.71 pg/ml,4.82 pg/ml and 6.72 pg/ml, respectively).
These data clearly show a cytokine storm in pHLH, MAS with raised
levels of IFN-γ, IL-10, IL-18, and TNF-α but no significant differences
were found between pHLH and MAS (Figure 2).
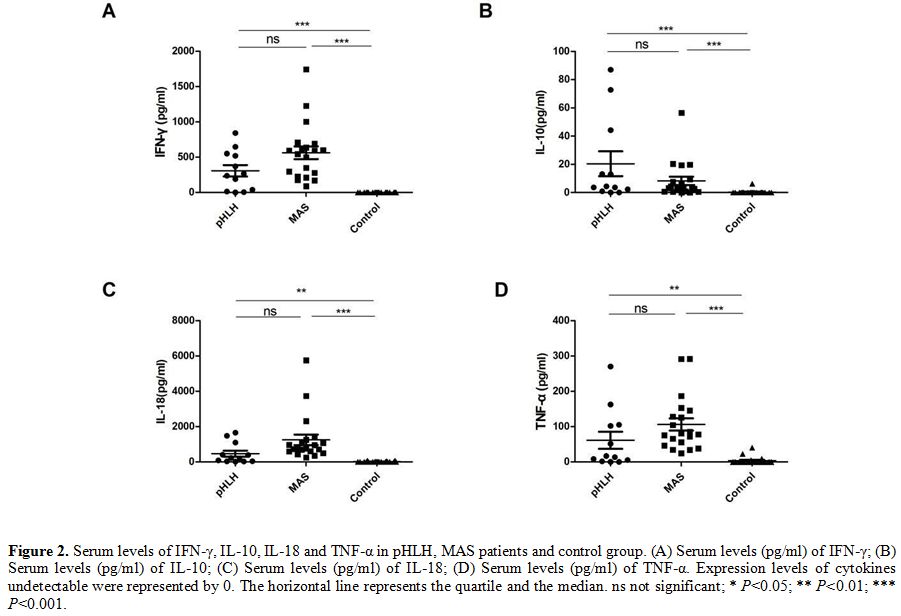 |
Figure 2. Serum levels of
IFN-γ, IL-10, IL-18 and TNF-α in pHLH, MAS patients and control group.
(A) Serum levels (pg/ml) of IFN-γ; (B) Serum levels (pg/ml) of IL-10;
(C) Serum levels (pg/ml) of IL-18; (D) Serum levels (pg/ml) of TNF-α.
Expression levels of cytokines undetectable were represented by 0. The
horizontal line represents the quartile and the median. ns not
significant; * P<0.05; ** P<0.01; *** P<0.001. |
sCD25.
In patients with pHLH and MAS, the level of sCD25 before initial
treatment was significantly increased compared to healthy controls
(21062.1 ± 18515.26 pg/ml in pHLH, 1074.78 ± 11149.96 pg/ml in MAS, and 795.65 ± 558.44 pg/ml in control), but there was no a significant difference between pHLH and MAS groups(P
= 0.07). Two weeks after treatment, we found an obvious decline in the
expression of sCD25 in both pHLH and MAS groups (21062.1 ± 18515.26
pg/ml vs 9835.98 ± 4015.52 pg/ml, 11074.78 ± 11149.96 pg/ml vs 1766.88
± 1358.93 pg/ml, respectively).
This result may indicate that sCD25 is closely
associated with the activity of the disease (Figure 3).
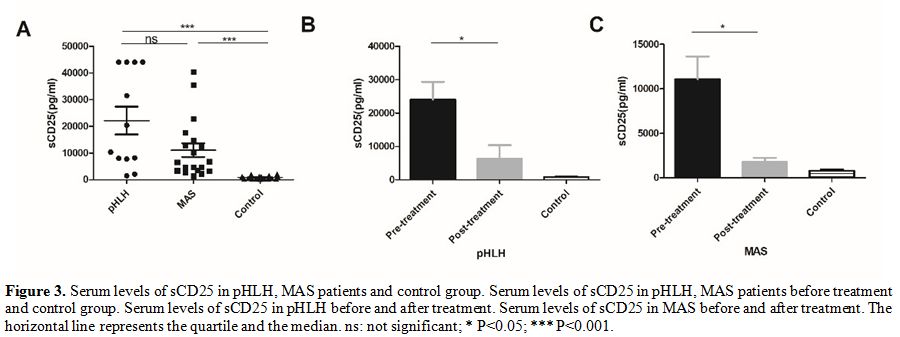 |
Figure 3. Serum levels of
sCD25 in pHLH, MAS patients and control group. Serum levels of sCD25 in
pHLH, MAS patients before treatment and control group. Serum levels of
sCD25 in pHLH before and after treatment. Serum levels of sCD25 in MAS
before and after treatment. The horizontal line represents the quartile
and the median. ns: not significant; * P<0.05; *** P<0.001. |
Discussion
pHLH,
a severe subtype of HLH, is triggered by genetic mutations that induce
dysfunctions of NK and cytotoxic T lymphocytes (CTLs). The remaining NK
cells and CTLs can’t eradicate the antigens effectively. Thus
persistent antigen presentation leads to the over-activation of CTLs.
The excessive cellular activation and expansion induce macrophages to
releases large amounts of inflammatory factors, causing a cytokine
storm.[26] Hitherto, hematopoietic stem cell transplantation (HSCT) is the optimal treatment for pHLH.[15,27-29] MAS, as previously discussed, is categorized as a form of secondary HLH.[5]
The development of MAS is also featured by a cytokine storm, with the
presentation of numerous proinflammatory cytokines. Hence, pHLH and MAS
bears great similarity in the cytokine profiles.
Accumulated
evidence suggests that many cytokines play a pivotal role in the
pathogenesis of HLH. In our study,we initially investigated the
expression of sST2, sCD163, sCD25, MIP-1α, SDF-1α, IL-1α, IL-1 β, IL-2,
IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-12p70, IL-13, IL-15,
IL-17A, IL-18, IL-21, IL-22, IL-23, IL-27, IL-31, IL-1 RA, RANTES,
IFN-γ, GM-CSF, TNF-α, MIP-1β, IFN-α, MCP-1, TNF-β, GRO-α and Eotaxin in
pHLH, MAS and healthy group. The majority of those cytokines, however,
did not affect the diagnosis and differential diagnosis of HLH.
IFN-γ
is considered to be uniquely essential for the development of HLH and
evidence suggests that neutralization of IFN-γ has a dramatic effect on
the survival of mice with HLH.[17] IL-10 is generally
thought to be an immunosuppressive molecule and has a protective effect
on inflammatory responses. However, excessive IL-10 may accelerate the
progression of HLH via inhibiting activations of lymphocytes which are
crucial for immune homeostasis.[18] In line with this, the study of Xiao-Jun[21]
implies that the elevated levels of IFN-γ and IL-10 with only modestly
elevated IL-6 has high diagnostic accuracy for HLH. IL-18 is also
involved in the pathophysiology of HLH and Takada H[19]
reveals that IL-18 levels significantly correlate with the activity of
HLH. TNF-α has long been illustrated to be associated with the
occurrence and development of many diseases. Henter[20]
demonstrates that TNF levels are augmented in active FHL and may
contribute to the pathogenesis of the disease. So, accumulated evidence
indicates that those cytokines are of paramount importance in HLH. In
our study, the expressions of all those cytokines increase in both pHLH
and MAS groups compared to the healthy group, which is consistent with
the above-published studies. Interestingly, there is no significant
difference in the expressions of those cytokines between pHLH and MAS,
indicating that the levels of IFN-γ, IL-10, IL-18 and TNF-α cannot
contribute to the differentiation of pHLH and MAS.
We further analyzed the cytokines with different
expression levels in pHLH and MAS. sCD163 is the soluble form of CD163,
which acts as the hemoglobin- haptoglobin scavenger receptor. CD163
expresses mainly on the membranes of activated monocytes/macrophages
and is regarded as a macrophage-specific marker for inappropriate
macrophage activation in inflammatory diseases.[23,30]
Upregulation of
CD163 on the surface of activated monocytes or
macrophages can facilitate the process of phagocytosis; therefore some
scholars consider it as a potential biomarker for HLH and relevant
diseases.[23] ST2, expressed in Th2 cells, belongs to
the IL-1 receptor family and is a receptor of IL-33. ST2 can express as
a membrane-bound form (ST2L) or a secreted form (sST2) and has been
clearly implicated as a regulator of both the development and effect
phases of Th2-type responses.[31] Th2 cells mainly
secrete IL-4, IL-5 and IL- 13, and those cytokines regulate immune
response by activating B cells to produce antibodies or by deactivating
and reprogramming macrophages.[31-33] sST2 can mediate this response through downregulating the pro-inflammatory effect of macrophages.[31,34] Previous studies have shown that the expression of sST2 elevated in systemic juvenile idiopathic arthritis and MAS.[35] And the blockage of the sST2 pathway can facilitate the treatment of HLH in Perforin deficient mice.
Serum
soluble interleukin-2 receptor(sCD25) is the most studied
cytokine/cytokine receptor to date in HLH and is one of the diagnostic
criteria set in the HLH-2004 criterion. In our study, we found that the
level of sCD25 is higher in both pHLH and MAS group compared to the
control group, but no significant differences were found between pHLH
and MAS groups. Moreover, the level of sCD25 decreased when the
patients received treatment. This data indicates the importance of
sCD25 as a diagnostic and disease marker in HLH (including MAS).[36,37]
Conclusions
In
our study, sCD163 and sST2 levels in pHLH were significantly lower than
that in MAS. We suggest that sST2 and sCD163 may serve as markers to
distinguish pHLH from MAS. The elevated sCD163 levels in MAS patient
may indicate that the macrophage activation in MAS is higher than that
in pHLH. Furthermore, we speculate that sST2 may act as a
counterbalance to the over-activated macrophages and, consequently, the
over-activation of macrophage in MAS results in the higher levels of
sST2. Nevertheless, the specific molecular mechanism for the
elevated sST2 levels in MAS needs to be further investigated.
The
elevated levels of IFN-γ, IL-10, IL-18, TNF-α, and sCD25 can contribute
to the diagnosis of HLH, but may not discriminate pHLH from MAS. Levels
of sST2 and sCD163 in MAS were significantly higher than that in pHLH,
sST2 and sCD163 may serve as markers to distinguish pHLH from MAS.
Acknowledgments
This
work was supported by Beijing Municipal Administration of Hospitals
Clinical Medicine Development of Special Funding (ZYLX201702), Beijing
Municipal Administration of Hospitals’ Ascent Plan (DFL20180101),
Beijing Natural Science Foundation (7181003).
References
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166(2): 95-109. https://doi.org/10.1007/s00431- 006-0258-1 PMid:17151879
- Henter
JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S,
McClain K, Webb D, Winiarski J, Janka G. HLH-2004: Diagnostic and
therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr
Blood Cancer. 2007; 48(2): 124-31. https://doi.org/10.1002/pbc.21039 PMid:16937360
- Stephan
JL, Kone-Paut I, Galambrun C, Mouy R, Bader-Meunier B, Prieur AM.
Reactive haemophagocytic syndrome in children with inflammatory
disorders. A retrospective study of 24 patients. Rheumatology (Oxford).
2001; 40(11): 1285-92. https://doi.org/10.1093/rheumatology/40.11.1285
- Sawhney
S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially
fatal complication of rheumatic disorders. Arch Dis Child. 2001; 85(5):
421-6. https://doi.org/10.1136/adc.85.5.421 PMid:11668110 PMCid:PMC1718981
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012; 63: 233-46. https://doi.org/10.1146/annurev- med 041610-134208 PMid:22248322
- Schulert
GS, Grom AA. Macrophage activation syndrome and cytokine-directed
therapies. Best Pract Res Clin Rheumatol. 2014; 28(2): 277-92. https://doi.org/10.1016/j.berh.2014.03.002 PMid:24974063 PMCid:PMC4074772
- Avcin
T, Tse SM, Schneider R, Ngan B, Silverman ED. Macrophage activation
syndrome as the presenting manifestation of rheumatic diseases in
childhood. J Pediatr. 2006; 148(5): 683-6. https://doi.org/10.1016/j.jpeds.2005 12.070 PMid:16737887
- Kim
JM, Kwok SK, Ju JH, Kim HY, Park SH. Reactive hemophagocytic syndrome
in adult Korean patients with systemic lupus erythematosus: a
case-control study and literature review. J Rheumatol. 2012; 39(1):
86-93. https://doi.org/10.3899/jrheum.110639 PMid:22174206
- Parodi
A, Davi S, Pringe AB, Pistorio A, Ruperto N, Magni-Manzoni S, Miettunen
P, Bader-Meunier B, Espada G, Sterba G, Ozen S, Wright D, Magalhães CS,
Khubchandani R, Michels H, Woo P, Iglesias A, Guseinova D, Bracaglia C,
Hayward K, Wouters C, Grom A, Vivarelli M, Fischer A, Breda L, Martini
A, Ravelli A; Lupus Working Group of the Paediatric Rheumatology
European Society. Macrophage activation syndrome in juvenile systemic
lupus erythematosus: a multinational multicenter study of thirty-eight
patients. Arthritis Rheum. 2009; 60(11): 3388-99. https://doi.org/10.1002/art.24883 PMid:19877067
- Muise
A, Tallett SE, Silverman ED. Are children with Kawasaki disease and
prolonged fever at risk for macrophage activation syndrome? Pediatrics.
2003; 112(6 Pt 1): e495. https://doi.org/10.1542/peds.112 6.e495 PMid:14654653
- Al-Eid
W, Al-Jefri A, Bahabri S, Al-Mayouf S. Hemophagocytosis complicating
Kawasaki disease. Pediatr Hematol Oncol. 2000; 17(4): 323-9. https://doi.org/10.1080/088800100276316 PMid:10845231
- Atteritano
M, David A, Bagnato G, Beninati C, Frisina A, Iaria C, Bagnato G,
Cascio A. Haemophagocytic syndrome in rheumatic patients. A systematic
review. Eur Rev Med Pharmacol Sci. 2012; 16(10): 1414-24. PMid:23104659
- Ramanan AV, Schneider R. Macrophage activation syndrome--what's in a name!. J Rheumatol. 2003; 30(12): 2513-6. PMid:14719185
- Campo M, Berliner N. Hemophagocytic Lymphohistiocytosis in Adults. Hematol Oncol Clin North Am. 2015; 29(5): 915-25. https://doi.org/10.1016/j.hoc.2015 06.009 PMid:26461151
- Janka
GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and
treatment. Hematology Am Soc Hematol Educ Program. 2013; 2013: 605-11. https://doi.org/10.1182/asheducation- 2013 1.605 PMid:24319239
- Ravelli
A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part
of systemic juvenile idiopathic arthritis: diagnosis, genetics,
pathophysiology and treatment. Genes Immun. 2012; 13(4): 289-98. https://doi.org/10.1038/gene.2012.3 PMid:22418018
- Jordan
MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic
lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are
essential for the disorder. Blood. 2004; 104(3): 735-43. https://doi.org/10.1182/blood-2003-10-3413 PMid:15069016
- Tang
Y, Xu X, Song H, Yang S, Shi S, Wei J, Pan B, Zhao F, Liao C, Luo C.
Early diagnostic and prognostic significance of a specific Th1/Th2
cytokine pattern in children with haemophagocytic syndrome. Br J
Haematol. 2008; 143(1): 84-91. https://doi.org/10.1111/j.1365- 2141.2008.07298.x PMid:18673367
- Takada H, Nomura A, Ohga S, Hara T. Interleukin-18 in hemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2001; 42(1- 2): 21-8. https://doi.org/10.3109/10428190109097673 PMid:11699209
- Henter
JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U.
Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood.
1991; 78(11): 2918-22. PMid:1954380
- Xu
XJ, Tang YM, Song H, Yang SL, Xu WQ, Zhao N, Shi SW, Shen HP, Mao JQ,
Zhang LY, Pan BH. Diagnostic accuracy of a specific cytokine pattern in
hemophagocytic lymphohistiocytosis in children. J Pediatr 2012;
160(6):984-90. https://doi.org/10.1016/j.jpeds.2011 11.046 PMid:22226576
- Rood
JE, Rao S, Paessler M, Kreiger PA, Chu N, Stelekati E, Wherry EJ,
Behrens EM. ST2 contributes to T-cell hyperactivation and fatal
hemophagocytic lymphohistiocytosis in mice. Blood. 2016; 127(4): 426-35. https://doi.org/10.1182/blood-2015-07-659813 PMid:26518437 PMCid:PMC4731846
- Schaer
DJ, Schleiffenbaum B, Kurrer M, Imhof A, Bächli E, Fehr J, Moller HJ,
Moestrup SK, Schaffner A. Soluble hemoglobin- haptoglobin scavenger
receptor CD163 as a lineage-specific marker in the reactive
hemophagocytic syndrome. Eur J Haematol. 2005; 74(1): 6-10. https://doi.org/10.1111/j.1600-0609.2004.00318.x PMid:15613100
- Davi
S, Consolaro A, Guseinova D, Pistorio A, Ruperto N, Martini A, Cron RQ,
Ravelli A; MAS Study Group. An international consensus survey of
diagnostic criteria for macrophage activation syndrome in systemic
juvenile idiopathic arthritis. J Rheumatol. 2011; 38(4): 764-8. https://doi.org/10.3899/jrheum.100996 PMid:21285158
- Wang
Y, Huang W, Hu L, Cen X, Li L, Wang J, Shen J, Wei N, Wang Z.
Multicenter study of combination DEP regimen as a salvage therapy for
adult refractory hemophagocytic lymphohistiocytosis. Blood. 2015;126
(19):2186-92. https://doi.org/10.1182/blood-2015-05-644914 PMid:26289641 PMCid:PMC4635114
- Brisse
E, Wouters CH, Matthys P. Advances in the pathogenesis of primary and
secondary haemophagocytic lymphohistiocytosis: differences and
similarities. Br J Haematol. 2016; 174(2): 203-17. https://doi.org/10.1111/bjh.14147 PMid:27264204
- Fu
L, Wang J, Wei N, Wu L, Wang Y, Huang W, Zhang J, Liu J, Wang Z.
Allogeneic hematopoietic stem-cell transplantation for adult and
adolescent hemophagocytic lymphohistiocytosis: a single center
analysis. Int J Hematol. 2016;104(5):628-35. https://doi.org/10.1007/s12185-016 2062-7 PMid:27431489
- Jin
Z, Wang Y, Wang J, Zhang J, Wu L, Gao Z, Lai W, Wang Z. Primary
hemophagocytic lymphohistiocytosis in adults: the utility of family
surveys in a single-center study from China. Orphanet J Rare Dis.
2018;13(1):17. https://doi.org/10.1186/s13023-017-0753-7 PMid:29357941 PMCid:PMC5778699
- Li
Z, Wang Y, Wang J, Zhang J, Wang Z. Successful haploidentical stem cell
transplantation for three adults with primary hemophagocytic
lymphohistiocytosis. Bone Marrow Transplantation 2017;52(2):330-3. https://doi.org/10.1038/bmt 2016.284 PMid:27775696
- Greisen
SR, Moller HJ, Stengaard-Pedersen K, Hetland ML, Hørslev-Petersen K,
Junker P, Østergaard M, Hvid M, Deleuran B. Macrophage activity
assessed by soluble CD163 in early rheumatoid arthritis: association
with disease activity but different response patterns to synthetic and
biologic DMARDs. Clin Exp Rheumatol. 2015; 33(4): 498-502. PMid:25962601
- Trajkovic
V, Sweet MJ, Xu D. T1/ST2--an IL-1 receptor-like modulator of immune
responses. Cytokine Growth Factor Rev. 2004; 15(2-3): 87-95. https://doi.org/10.1016/j.cytogfr.2004.02.004 PMid:15110792
- Mosmann TR, Sad S. The expanding universe of T-cell subsets: Th1, Th2 and more. Immunol Today. 1996; 17(3): 138-46 https://doi.org/10.1016/0167-5699(96)80606-2
- Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996; 383(6603): 787-93. https://doi.org/10.1038/383787a0 PMid:8893001
- Sweet
MJ, Leung BP, Kang D, Sogaard M, Schulz K, Trajkovic V, Campbell CC, Xu
D, Liew FY. A novel pathway regulating lipopolysaccharide-induced shock
by ST2/T1 via inhibition of Toll-like receptor 4 expression. J Immunol.
2001; 166(11): 6633-9. https://doi.org/10.4049/jimmunol.166.11.6633 PMid:11359817
- Ishikawa
S, Shimizu M, Ueno K, Sugimoto N, Yachie A. Soluble ST2 as a marker of
disease activity in systemic juvenile idiopathic arthritis. Cytokine.
2013;62(2):272-7. https://doi.org/10.1016/j.cyto.2013.03.007 PMid:23561929
- Lin
M, Park S, Hayden A, Giustini D, Trinkaus M, Pudek M, et al. Clinical
utility of soluble interleukin-2 receptor in hemophagocytic syndromes:
a systematic scoping review. Annals of Hematology 2017;96:1241-51. https://doi.org/10.1007/s00277-017-2993-y PMid:28497365
- Hayden
A, Lin M, Park S, Pudek M, Schneider M, Jordan MB, et al. Soluble
interleukin-2 receptor is a sensitive diagnostic test in adult HLH.
2017;1:2529-34.
[TOP]