Paramee Phanrahan1,2, Supawadee Yamsri2, Nattiya Teawtrakul3, Goonnapa Fucharoen2, Kanokwan Sanchaisuriya2 and Supan Fucharoen2..
1 Medical Science Program, Graduate School, Khon Kaen University.
2
Centre for Research and Development of Medical Diagnostic Laboratories,
Faculty of Associated Medical Sciences, Khon Kaen University.
3 Department of Internal Medicine, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand.
Correspondence to: Dr. Supan Fucharoen. Centre for Research and
Development of Medical Diagnostic Laboratories, Faculty of Associated
Medical Sciences, Khon Kaen University, Khon Kaen, Thailand 40002.
Tel/Fax +66-43-202083. E-mail:
supan@kku.ac.th
Published: July 01, 2019
Received: February 12, 2019
Accepted: May 17, 2019
Mediterr J Hematol Infect Dis 2019, 11(1): e2019038 DOI
10.4084/MJHID.2019.038
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background: The finding of many Thai Hb E-β0-thalassemia
patients with non-transfusion dependent thalassemia (NTDT) phenotype
without co-inheritance of α-thalassemia has prompted us to investigate
the existence of other genetic modifying factors.
Methods:
Study was done on 122 adult Thai patients with NTDT Hb E-β-thalassemia
patients without co-inheritance of α-thalassemia. Multiple
single-nucleotide polymorphisms (SNPs) associated with γ-globin gene
expression including the Gγ-XmnI
of HBG2 gene, rs2297339, rs4895441, and rs9399137 of the HBS1L-MYB
gene, rs4671393 in the BCL11A gene, and G176AfsX179, T334R, R238H and
-154 (C-T) in the KLF1 gene were investigated using PCR and related
techniques.
Results: Heterozygous and homozygous for Gγ-XmnI
of HBG2 gene were detected at 70.5% and 7.4%, respectively. Further DNA
analysis identified the rs2297339 (C-T), rs4895441 (A-G), and rs9399137
(T-C) of HBS1L-MYB gene in 86.9%, 25.4%, and 23.0%, respectively. The
rs4671393 (G-A) of the BCL11A gene was found at 31.2%. For the KLF1
gene, only T334R was detected at 9.0%.
Conclusions:
It was found that these SNPs, when analyzed in combination, could
explain the mild phenotypic expression of all cases. These results
underline the importance of these informative SNPs on phenotypic
expression of Hb E-β-thalassemia patients.
|
Introduction
Thalassemia
is one of the most common genetic disorders worldwide, especially in
Southeast Asia. Thalassemia results from reduction or absence of globin
chain synthesis. Two main types divided by defected globin chains are
α-thalassemia and β-thalassemia. On the other hand, it can be divided
based on blood transfusion requirement into transfusion-dependent
thalassemia (TDT) and non-transfusion-dependent thalassemia (NTDT).[1] The most common thalassemia disease found in northeast Thailand is hemoglobin E-β-thalassemia (Hb E-β-thal).[2]
It has been shown that clinical severity of this disease is variable,
ranging from mild to severe transfusion-dependent thalassemia.[3-6] Patients with transfusion-dependent Hb E-β-thal
disease require lifelong regular blood transfusion for survival, while
NTDT patients generally have mild anemia and do not require regular
blood transfusion for survival. However, several severe complications
in NTDT have been noted including chronic hypoxia, pulmonary
hypertension, and thromboembolic events.[7]
Understanding of molecular features and accurate prediction of NTDT are
therefore essential to reduce the morbidity of the patients. Studies
have shown that type of β-thalassemia mutation alone is not enough to predict the clinical phenotype of the patients, and many patients with Hb E-β0-thalassemia are associated with NTDT phenotype.[8,9]
This indicates that other genetic factors might be involved in the
clinical expression of the patients. These include a coinheritance
of β-thalassemia
or the presence of genetic factors associated with increased production
of γ-globin chains for Hb F. It has been shown that at least three
major loci regulate this level of Hb F: HBG2 gene (Gγ-XmnI
polymorphism), HBS1L-MYB intergenic region and BCL11A gene.
Polymorphisms on these three loci were found to be responsible for Hb F
variation in patients with homozygous Hb E, β-thalassemia or sickle cell disease and in healthy Europeans.[10-14]
Preliminary
study on subjects with a mild form of thalassemia encountered among
couple at risk of having fetuses with thalassemia diseases in northeast
Thailand has been carried out. The result indicated that four
informative SNPs, including rs7482144 in HBG2 gene and rs2297339,
rs4895441 & rs9399137 of HBS1L-MYB gene were associated with high
Hb F levels in the patients.[9] Further studies on
homozygous Hb E identified the rs11886868 additionally in the BCL11A
gene and 4 SNPs in the Krüppel-like factor 1 (KLF1) gene (G176AfsX179, T334R, -154 (C-T) and R328H) to be associated with high Hb F level in homozygous Hb E.[15-17] It is likely therefore that these informative SNPs might be important genetic modifying factors among NTDT- Hb E-β0-thal patients. However, data on these SNPs among clinically well-defined cases of NTDT with Hb E-β-thal
patients in northeast Thailand is relatively limited. It has been known
that co-inheritance of α-thalassemia is associated with a mild
phenotype of the Hb E-β-thal disease. However, we have demonstrated previously that among Hb E-β0-thal patients associated with NTDT phenotypes, co-inheritance of β-thalassemia could explain the phenotypic expression only in a few cases.[18]
We report in this study, the existence of several genetic modifying
SNPs in the HBS1L-MYB, BCL11A, and KLF1 genes among 122 clinically
well-defined NTDT Hb E-β-thal patients in northeast Thailand.
Materials and Methods
Specimens.
Ethical approval of the study protocol was obtained from the
Institutional Review Board of the Khon Kaen University, Khon Kaen,
Thailand (HE561018). Archival DNA specimens were obtained from NTDT Hb
E-β-thal patients of our previous study.[18]
Altogether, specimens of 122 patients with complete hematological data
were obtained. All of them enrolled in the project “Epidemiologic study
of major complications in adolescence and adult patients with
thalassemia in northeast Thailand: the E-SAAN study” conducted at
Srinagarind Hospital, Khon Kaen University, Khon Kaen Hospital,
Mahasarakham hospital, and Udonthani hospital, all located in northeast
Thailand, from October 2012 to June 2014. Inclusion criteria were an
age of > 10 years and a diagnosis of thalassemia based on clinical
symptoms, e.g., anemia, pallor, hepatosplenomegaly, jaundice, skeleton
changes, growth and development deficiency, and a Hb levels of 6.0-10.0
g/dl, Hb and DNA analysis. Cases with abnormal Hb, iron deficiency
anemia, and other causes of anemia were excluded.[19]
Hematological and DNA analyses.
Hematological parameters were recorded at steady state (no blood
transfusion and no fever) using automated blood cell counter (Beckman
Coulter Co., Fullerton, California, USA). Hb analysis was done using
capillary electrophoresis (Capillarys 2; Sebia, Lisses, France) or high
-performance liquid chromatography (Variant II, Bio-Rad Laboratories,
Hercules, California, USA). Identification of β-thalassemia and the Hb
E mutations found in Thailand was performed in our laboratory using
allele-specific PCR assays and DNA sequencing. Identification of α0-thalassemia (SEA and THAI deletions), α+-thalassemia
(3.7 and 4.2 kb deletions), Hb Constant Spring and Hb Paksé genes are
routinely performed in our laboratory using multiplex gap PCR and
allele-specific PCR.[2]
SNP Genotyping.
Four KLF1 SNPs including G176AfsX179, -154 (C-T), T334R and R328H were
determined using allele-specific PCR assays and DNA sequencing as
described.[16,17] Representative gel electrophoresis of these SNPs genotyping was shown in Figure 1.
The rs4895441 (G-A) and rs9399137 (T-C) of HBS1L-MYB gene and rs4671393
(A-G) of BCL11A gene were determined using high resolution melting
(HRM) analysis on an Illumina Eco Real-Time PCR System (Illumina, CA,
USA). Primers G166 (5’ CACAACACTCCAGGGAGGCAG 3’) and G167 (5’
GGAGGCAGGGGGAATCTTAAT 3’) were used to produce an 84 bp fragment for
detection of rs4671393 (A-G) of BCL11A gene. The rs4895441 (G-A) of
HBS1L-MYB intergenic region was determined on a 157 bp fragment
generated using primers G156 (5’ GGGGGTAAGAAGGAAACCAG 3’) and G157 (5’
TCTGAGGGCCTTCGAACTTA 3’). The rs9399137 (T-C) of HBS1L-MYB intergenic
region was detected on a 136 bp fragment produced by primers G158 (5’
TCACCTTAAAAGGCGGTATTG 3’) and G159 (5’ TCAGAACTTATCCCAAGATTTTAAC 3’).
Representative temperature shifted curves, and corresponding difference
curves of these HRM assays were demonstrated in Figure 2. Identification of the Gγ-XmnI
of HBG2 gene and rs2297339 (C-T) of the HBS1L-MYB gene was done using
PCR-restriction fragment length polymorphism (PCR-RFLP) assay as
described.[8,9]
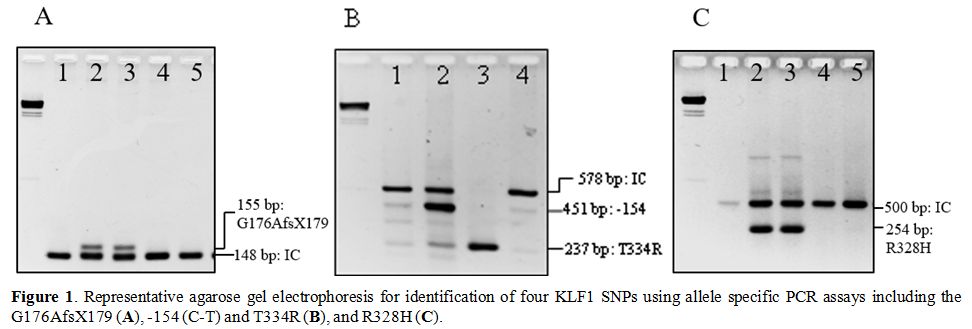 |
Figure1. Representative agarose gel
electrophoresis for identification of four KLF1 SNPs using allele
specific PCR assays including the G176AfsX179 (A), -154 (C-T) and T334R (B), and R328H (C). |
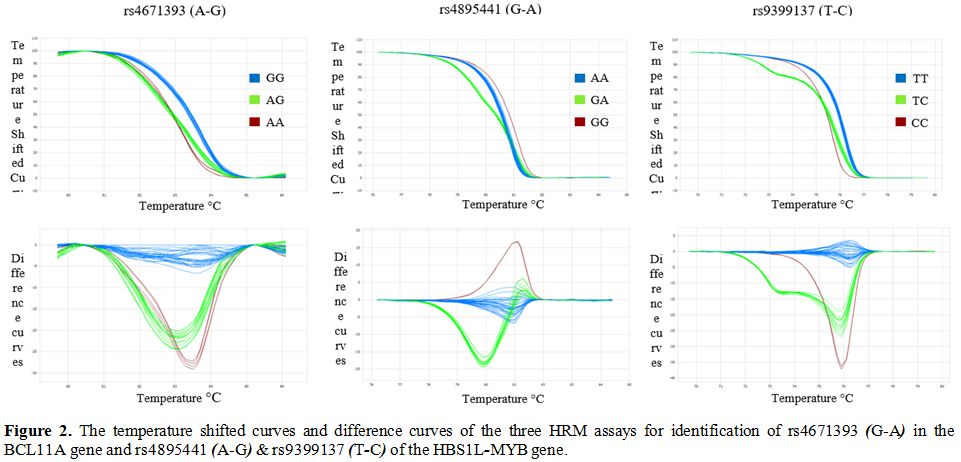 |
Figure
2. The temperature shifted curves and difference curves of the three HRM assays for identification of rs4671393 (G-A) in the BCL11A gene and rs4895441 (A-G) & rs9399137 (T-C) of the HBS1L-MYB gene. |
Statistical analysis.
The STATA statistical software version 10.0 (StataCorp, Tx, USA.) was
used for data analyses. Descriptive statistics, mean and standard
deviation, were used to describe all continuous variables, including
red blood cell indices and Hb F levels. Multiple regression analysis
was applied to demonstrate the effect of various SNPs on Hb F levels.
Statistical significance was set at P < 0.05.
Results
Table 1
listed the globin genotypes and associated hematological data of 122
patients studied. Most of them carried β0-thalassemia in trans to the
βE globin gene (n = 119). The remaining 3 of them carried the β+-thalassemia
mutation with the β-28 mutation. Similar hematological findings between
groups with different mutations were observed, but variability in Hb F
was noted. Table 2 summarized
the frequencies of 9 SNPs of the 4 genes observed among 122 NTDT
patients with Hb E-β-thalassemia. These included Gγ-XmnI
of the HBG2, G176AfsX179, T334R, -154 (C-T) and R328H of KLF1 gene,
rs11886868 of BCL11A gene and rs4895441, rs9399137 and rs2297339 of the
HBS1L-MYB. As shown in the table, heterozygosity (+/-) and homozygosity
(+/+) for Gγ-XmnI polymorphism of the HBG2 were detected in 86 (70.5%) and 9 (7.4%) cases, respectively.
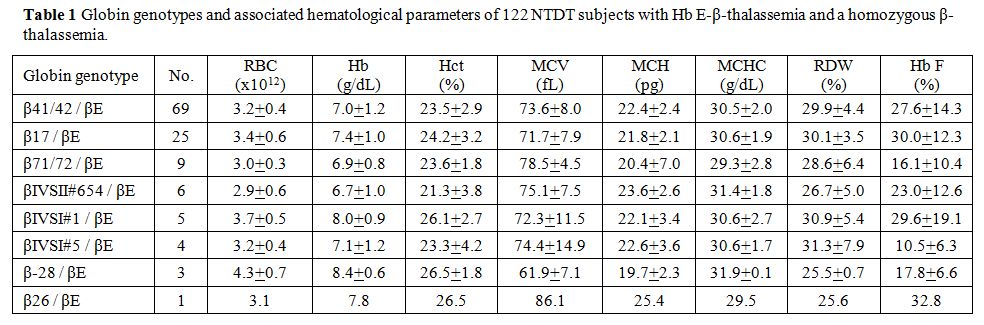 |
Table 1. Globin genotypes and associated hematological parameters of 122 NTDT subjects with Hb E-β-thalassemia. |
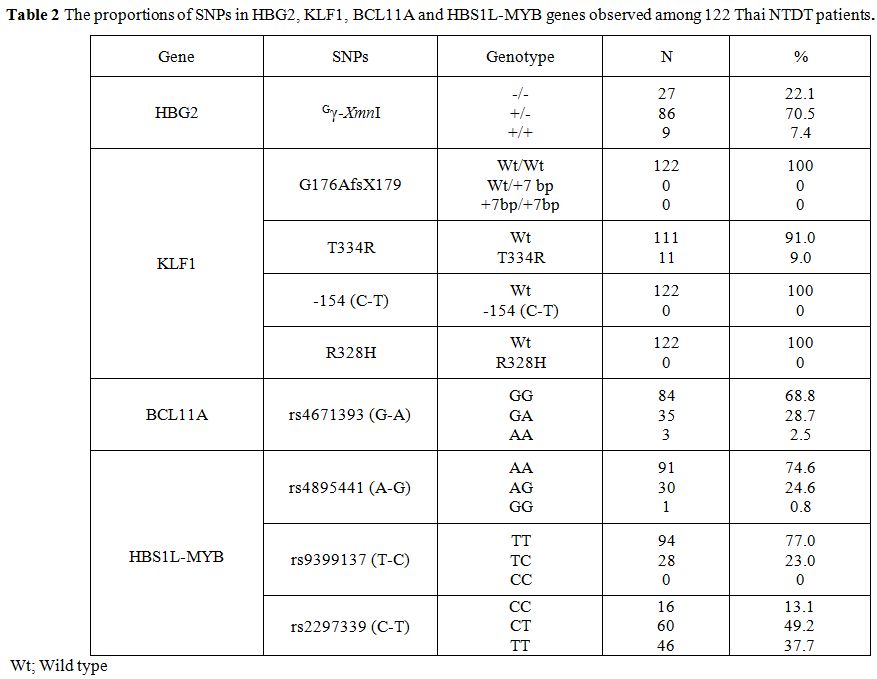 |
Table
2. The proportions of SNPs in HBG2, KLF1, BCL11A and HBS1L-MYB genes observed among 122 Thai NTDT patients. |
Among 4 SNPs of the
KLF1 gene examined, including the G176AfsX179, T334R, -154 (C-T) and
R238H, only T334R was detected. While no R328H, -154 (C-T) and
G176AfsX179 was observed, heterozygosity for the T334R was identified
in 11 (9.0%) of 122 cases. In contrast, a relatively higher proportion
of the rs4671393 (G-A) of the BCL11A, i.e., GG, GA, and AA varieties
were detected in 84 (68.8%), 35 (28.7%) and 3 (2.5%) cases,
respectively.
For the HBS1L-MYB gene, the proportions of AA, AG
and GG of the rs4895441 (A-G) were identified in 91 (74.6%), 30 (24.6%)
and 1 (0.8%) cases, respectively. Heterozygosity for the rs9399137
(T-C) was found in 28 (23.0%) cases. The most common SNP in this
HBS1L-MYB gene was found to be the rs2297339 (C-T) including CT and TT
which were identified in 60 (49.2%) and 46 (37.7%) cases, respectively.
Multiple
regression analysis was applied to demonstrate the effect of these SNPs
detected on Hb F levels of 122 subjects with Hb E-β-thal (Table 3). As shown in the table, statistical significance (P < 0.001) was observed only on the homozygosity (+/+) of the Gγ-XmnI polymorphism. However, a low proportion of this Gγ-XmnI
(+/+) in this group of Thai patients (9 of 122) makes it unlikely to be
the sole factor on phenotypic expression of these cases. In fact, we
observed that each patient carried at least one of these SNPs. Table 4 listed number of patients carrying 1-5 SNPs observed, and Figure 3
plots the proportions of subjects in correspondence with the number of
conferring SNPs in this study. As shown in the figure, while only 12 of
122 cases carried single SNP, the remaining subjects had 2-5 SNPs at
different genes, possibly indicating of interaction between these SNPs
in the phenotypic modification of the cases.
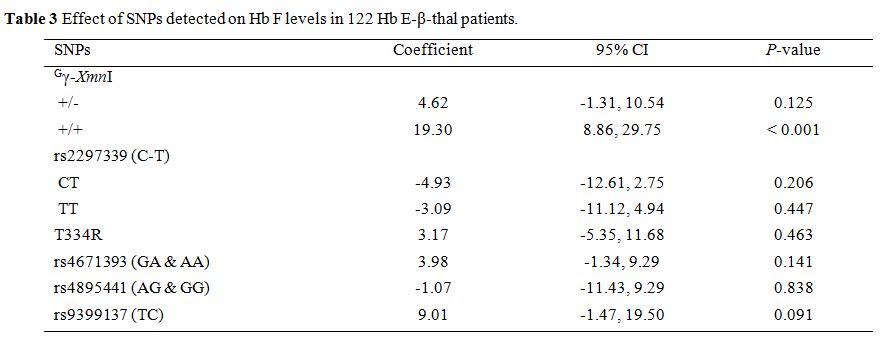 |
Table 3. Effect of SNPs detected on Hb F levels in 122 Hb E-β-thal patients. |
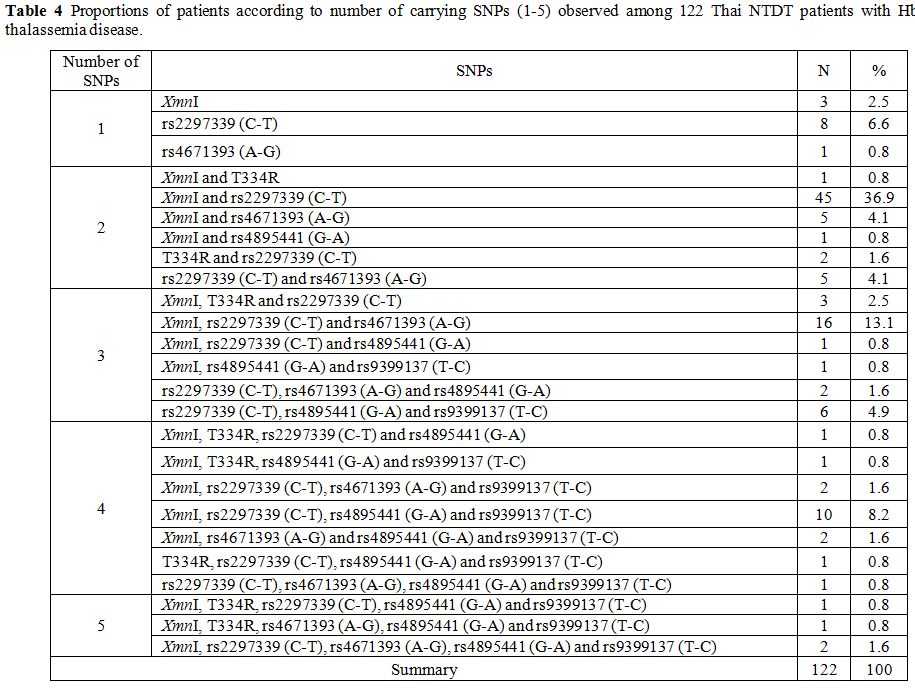 |
Table 4. Proportions of
patients according to number of carrying SNPs (1-5) observed among 122
Thai NTDT patients with Hb E-β-thalassemia disease. |
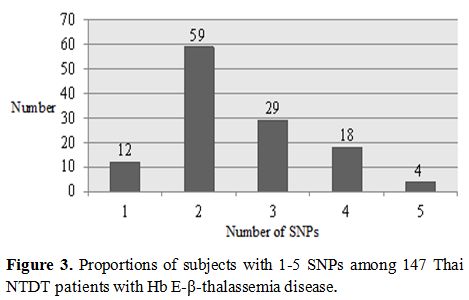 |
Figure 3. Proportions of subjects with 1-5 SNPs among 122 Thai NTDT patients with Hb E-β-thalassemia disease. |
Discussion
NTDT
refers to as thalassemia phenotype that does not require blood
transfusions for survival. Most of the patients have mild anemia, with
baseline Hb levels ranging from 7.0-9.0 g/dl and have a higher life
expectancy. However, they may still suffer from many complications if
not properly managed, including pulmonary hypertension and subsequent
thrombotic events. Diagnosis and understanding of the basis for NTDT
are therefore important.[7,20,21]
It has been known that major genetic modifying factor in β-thalassemia disease is a coinheritance of β-thalassemia
as this leads to a more balanced in α- and non-α- globin chains ratio.
However, this could not explain the phenotypic expression of all cases.
Multiple single nucleotide polymorphisms (SNPs) associated with high Hb
F expression have been identified in many populations on genes such as
the HBG2, BCL11A, HBS1L-MYB, and KLF1 genes.[22-25] The results from our study of 122 Thai NTDT Hb E-β-thalassemia patients without β-thalassemia revealed that all of them carried at least one SNPs in these modifying genes (Table 4).
While the majority of them (59 of 122) had two SNPs, the remaining
carried one (12 of 122), three (29 of 122), four (18 of 122) or five (4
of 122) SNPs as shown in Figure 3. These 9 genetic modifying SNPs on the Gγ-globin, HBS1L-MYB, BCL11A, and KLF1 genes are known to play important roles in modifying disease severity. Among them, the Gγ-XmnI
polymorphism was the most common SNP observed in our patients, i.e.,
70.5% in heterozygous and 7.4% in homozygous states. Study in Thai
homozygous Hb E has indicated a strong association between this
polymorphism and increased Hb F level. We also observed that the Gγ-XmnI (+/+) has a significant effect on the Hb F in Thai NTDT Hb E-β-thalassemia patients, as shown in Table 3. However, the finding of only 9 of 122 cases with homozygotic form (+/+) of this polymorphism (Table 2)
might underscore the importance of this SNP in Thai population and
point possibly to interaction with other genetic modifiers.
We
have previously documented in Thai subjects with homozygous Hb E that
four KLF1 SNPs including G176AfsX179, T334R, -154 (C-T) and R328H are
associated with increased Hb F expression.[16,17] In this study on 122 Thai NTDT Hb E-β-thalassemia patients, only one of them; the T334R was identified in heterozygote, the frequency of which was 9.0 % (Table 1).
Although KLF1 gene has been thought to play an essential role in the
clinical modification of the disease severity and homozygous for KLF1
mutation may be associated with mild thalassemia intermedia phenotype,[26] our result on Thai NTDT patients indicates that KLF1 gene alone may play a minimal role in Thai population.
In
contrast, a higher proportion of an A allele of the rs4671393 (G-A)
polymorphism of the BCL11A gene was detected among 122 Thai NTDT
patients i.e., 28.7% in heterozygote form and 2.5% in the homozygote.
This rs4671393 (G-A) polymorphism is associated with Hb F variation and
clinical events in sickle anemia.[27] As compared to
other genes, more prevalence of the G allele of rs4895441 (A-G), the C
allele of rs9399137 (T-C) and T allele of rs2297339 (C-T) of the
HBS1L-MYB intergenic region were observed among our Thai NTDT patients.
This data is consistent with a previous finding for Thai homozygous Hb
E.[15] Study on the Mediterranean β-thalassemia
intermedia patients has indicated a minor effect of the rs4671393 (G-A)
of the BCL11A and the rs4895441 (A-G) & rs9399137 (T-C) of
HBS1L-MYB intergenic region on phenotypic expression of the patients.[28]
Conclusions
Considering all the results, we found that among 122 Thai NTDT patients investigated, a total of 6 SNPs including Gγ-XmnI
of HBG2 gene, T334R of KLF1 gene, A allele of rs4671393 in BCL11A gene
and T allele of rs2297339, G allele of rs4895441 and C allele of
rs9399137 in HBS1L-MYB intergenic region, alone or in combination with
others could be used to explain the mild phenotypic expression of all
cases. Further study on NTDT subjects of other populations would be
required to prove that screening of these informative SNPs in the NTDT
patients is useful for clinical prediction and improving genetic
counseling of the patients.
Acknowledgment
This
work was supported by Centre for Research and Development of Medical
Diagnostic Laboratories (CMDL), Faculty of Associated Medical Sciences,
Khon Kaen University, Thailand.
References
- Weatherall DJ, Clegg JB. The thalassemia syndromes. 4th ed.; Oxford: Blackwell Science; 2001. https://doi.org/10.1002/9780470696705
- Yamsri
S, Sanchaisuriya K, Fucharoen G, Sae-Ung N, Ratanasiri T, Fucharoen S.
Prevention of severe thalassemia in northeast Thailand: 16 years of
experience at a single university center. Prenat Diagn 2010;30:540-546.
https://doi.org/10.1002/pd.2514 PMid:20509153
- Italia
K, Dabke P, Sawant P, Nadkarni A, Ghosh K, Colah RB. Hb E-β-thalassemia
in five Indian states. Hemoglobin 2016;40:310-315. https://doi.org/10.1080/03630269.2016.1201487 PMid:27623935
- George
E, Wong HB. Hb E beta +-thalassaemia in West Malaysia: clinical
features in the most common beta-thalassaemia mutation of the Malays
[IVS 1-5 (G-->C)], Singapore Med J 1993;34:500-503.
- Winichagoon
P, Fucharoen S, Chen P, Wasi P. Genetic factors affecting clinical
severity in beta-thalassemia syndromes. J Pediatr Hematol Oncol
2000;22:573-580. https://doi.org/10.1097/00043426-200011000-00026 PMid:11132233
- Fucharoen
S, Ketvichit P, Pootrakul P, Siritanaratkul N, Piankijagum A, Wasi P.
Clinical manifestation of beta-thalassemia/hemoglobin E disease. J
Pediatr Hematol Oncol 2000;22:552-557. https://doi.org/10.1097/00043426-200011000-00022 PMid:11132229
- Sleiman
J, Tarhini A, Bou-Fakhredin R, Saliba AN, Cappellini MD, Taher AT.
Non-transfusion dependent thalassemia: an update on complications and
management. Int J Mol Sci 2018;19:182. https://doi.org/10.3390/ijms19010182 PMid:29316681 PMCid:PMC5796131
- Nuntakarn
L, Fucharoen S, Fucharoen G, Sanchaisuriya K, Jetsrisuparb A, Wiangnon
S. Molecular, hematological and clinical aspects of thalassemia major
and thalassemia intermedia associated with Hb E-beta-thalassemia in
northeast Thailand. Blood Cells Mol Dis 2009;42:32-35. https://doi.org/10.1016/j.bcmd.2008.09.002 PMid:18951049
- Yamsri
S, Pakdee N, Fucharoen G, Sanchaisuriya K, Fucharoen S. Molecular
understanding of non-transfusion-dependent thalassemia associated with
hemoglobin E-beta-thalassemia in northeast Thailand. Acta Haematol
2016;136:233-239. https://doi.org/10.1159/000449120 PMid:27710960
- Nuinoon
M, Makarasara W, Mushiroda T, et al. A genome-wide association
identified the common genetic variants influence disease severity in
β0-thalassemia/hemoglobin E. Hum Genet 2010;127:303-314. https://doi.org/10.1007/s00439-009-0770-2 PMid:20183929
- Lettre
G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A,
HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels
and pain crises in sickle cell disease. Proc Natl Acad Sci USA
2008;105:11869-11874. https://doi.org/10.1073/pnas.0804799105 PMid:18667698 PMCid:PMC2491485
- Sedgewick
AE, Timofeev N, Sebastiani P, et al. BCL11A is a major Hb F
quantitative trait locus in three different populations with
beta-hemoglobinopathies. Blood Cells Mol Dis 2008;41:255-258. https://doi.org/10.1016/j.bcmd.2008.06.007 PMid:18691915 PMCid:PMC4100606
- Creary
LE, Ulug P, Menzel S, et al. Genetic variation on chromosome 6
influences F cell levels in healthy individuals of African descent and
Hb F levels in sickle cell patients. PLoS One 2009;4:e4218. https://doi.org/10.1371/journal.pone.0004218 PMid:19148297 PMCid:PMC2621086
- So
CC, Song YQ, Tsang ST, et al. The HBS1L-MYB intergenic region on
chromosome 6q23 is a quantitative trait locus controlling fetal
haemoglobin level in carriers of beta-thalassaemia. J Med Genet
2008;45:745-751. https://doi.org/10.1136/jmg.2008.060335 PMid:18697826
- Pakdee
N, Yamsri S, Fucharoen G, Sanchaisuriya K, Pissard S, Fucharoen S.
Variability of hemoglobin F expression in hemoglobin EE disease:
hematological and molecular analysis. Blood Cells Mol Dis
2014;53:11-15. https://doi.org/10.1016/j.bcmd.2014.02.005 PMid:24581976
- Tepakhan
W, Yamsri S, Fucharoen G, Sanchaisuriya K, S. Fucharoen S. Krüppel-like
factor 1 mutations and expression of hemoglobins F and A2 in homozygous
hemoglobin E syndrome. Ann Hematol 2015;94:1093-1098. https://doi.org/10.1007/s00277-015-2335-x PMid:25694242
- Tepakhan
W, Yamsri S, Sanchaisuriya K, Fucharoen G, Xu X, Fucharoen S. Nine
known and five novel mutations in the erythroid transcription factor
KLF1 gene and phenotypic expression of fetal hemoglobin in hemoglobin E
disorder. Blood Cells Mol Dis 2016;59:85-91. https://doi.org/10.1016/j.bcmd.2016.04.010 PMid:27282573
- Prayalaw
P, Teawtrakul N, Jetsrisuparb A, Pongudom S, G. Fucharoen S. Non
transfusion-dependent thalassemia in northeast Thailand. Acta Haematol
2016;135:15-20. https://doi.org/10.1159/000435802 PMid:26303193
- Teawtrakul
N, Pussadhamma B, Ungprasert P, et al. A risk score for predicting
pulmonary hypertension in patients with non-transfusion dependent
thalassemia in northeast Thailand: The E-SAAN score. Hematology
2015;20:416-420. https://doi.org/10.1179/1607845414Y.0000000211 PMid:25386747
- Teawtrakul
N, Jetsrisuparb A, Pongudom S, et al. Epidemiologic study of major
complications in adolescent and adult patients with thalassemia in
northeast Thailand: The E-SAAN study phase I. Hematology 2018;23:55-60.
https://doi.org/10.1080/10245332.2017.1358845 PMid:28759343
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica 2013;98:833-844. https://doi.org/10.3324/haematol.2012.066845 PMid:23729725 PMCid:PMC3669437
- Uda
M, Galanello R, Sanna S, et al. Genome-wide association study shows
BCL11A associated with persistent fetal hemoglobin and amelioration of
the phenotype of beta-thalassemia. Proc Natl Acad Sci USA
2008;105:1620-1625. https://doi.org/10.1073/pnas.0711566105 PMid:18245381 PMCid:PMC2234194
- Wahlberg
K, Jiang J, Rooks H, et al. The HBS1L-MYB intergenic interval
associated with elevated Hb F levels shows characteristics of a distal
regulatory region in erythroid cells. Blood 2009;114:1254-1262. https://doi.org/10.1182/blood-2009-03-210146 PMid:19528534
- Gallienne
AE, Dreau HM, Schuh A, Old JM, Henderson S. Ten novel mutations in the
erythroid transcription factor KLF1 gene associated with increased
fetal hemoglobin levels in adults. Haematologica 2012;97:340-343. https://doi.org/10.3324/haematol.2011.055442 PMid:22102705 PMCid:PMC3291586
- Thein SL. Molecular basis of β-thalassemia and potential therapeutic targets. Blood Cells Mol Dis 2018;70:54-65. https://doi.org/10.1016/j.bcmd.2017.06.001 PMid:28651846 PMCid:PMC5738298
- Rani
N, Jamwal M, Kaur J, et al. Homozygous KLF1 mutation c.901C>T
(p.Arg301Cys) resulting in mild thalassemia intermedia in an Indian: A
next-generation sequencing diagnosis. Blood Cells Mol Dis
2018;72:19-21. https://doi.org/10.1016/j.bcmd.2018.06.003 PMid:29980343
- Chaouch
L, Moumni I, Ouragini H, et al. rs11886868 and rs4671393 of BCL11A
associated with Hb F level variation and modulate clinical events among
sickle cell anemia patients. Hematology 2016;27:425-429. https://doi.org/10.1080/10245332.2015.1107275 PMid:27077760
- Nguyen
TK, Joly P, Bardel C, Moulsma M, Bonello-Palot N, Francina A. The XmnI
(G)gamma polymorphism influences hemoglobin F synthesis contrary to
BCL11A and HBS1L-MYB SNPs in a cohort of 57 beta-thalassemia intermedia
patient. Blood Cells Mol Dis 2010;45:124-127. https://doi.org/10.1016/j.bcmd.2010.04.002 PMid:20472475
[TOP]