Alessandro Buonomo1, Eleonora Nucera1 and Marianna Criscuolo2.
1 Allergy Unit, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy.
2
Department of Radiological and Hematological Sciences Fondazione,
Policlinico Universitario A. Gemelli, IRCCS Università Cattolica del
Sacro Cuore, Largo A. Gemelli, 100168, Rome, Italy.
Published: May 1, 2022
Received: March 2, 2022
Accepted: April 14, 2022
Mediterr J Hematol Infect Dis 2022, 14(1): e2022040 DOI
10.4084/MJHID.2022.040
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Management of Indolent and Smoldering
SM is focused on preventing anaphylactic reactions and identifying and
avoiding symptom triggers. Skin and gastrointestinal symptoms are
managed with H1- and H2-antihistamines. When skin symptoms are not
adequately controlled, leukotriene antagonists and oral psoralen
combined with ultraviolet therapy may be added. Proton pump inhibitors,
sodium cromolyn, and oral corticosteroids may be added for
gastrointestinal symptoms. Patients should be prescribed
self-injectable epinephrine and trained to treat recurrent
cardiovascular symptoms or anaphylaxis. Depression and cognitive
impairment require a psychiatric evaluation for tailored treatment.
Bone involvement is managed with bisphosphonates and eventually
interferon. Omalizumab is effective on all vasomotor symptoms,
including anaphylaxis, but not on respiratory, musculoskeletal, and
neuropsychiatric symptoms. A cytoreductive treatment is not recommended
unless anti-mediator therapy has failed. Venom immunotherapy is
mandatory for patients with Hymenoptera venom allergy.
There is no
curative option for patients with advanced SM. The available
therapeutic options include tyrosine-kinase inhibitors and cladribine,
with variable duration and extent of response. Imatinib mesylate was
the first drug approved for SM lacking the cKIT D816V mutation;
dasatinib and nilotinib are ineffective. Midostaurin is active on both
wild-type and mutant cKIT D816V, while Avapritinib is a selective cKIT
D816V inhibitor: they are approved for the treatment of advanced SM.
Cladribine is a purine analog with significant activity against
monocytes that were thought to have a common progenitor with mast
cells. Allogeneic stem cell transplantation is usually performed in
younger selected patients.
|
Introduction
Systemic
mastocytosis (SM) is a myeloproliferative neoplasm resulting from a
clonal expansion of morphologically and immunophenotypically abnormal
mast cells (MCs). A gain-of-function somatic mutation in the KIT gene, which codifies for a tyrosine kinase, is responsible for uncontrolled MC proliferation and survival.[1]
MCs are effector cells of an immune response, especially involved in
parasites infections control and allergic and anaphylactic reactions.
Mastocytosis
is characterized by protean clinical manifestations: the release of
several mediators and cytokines is responsible for symptoms that
involve skin, gastrointestinal tract, cardiovascular system, bone, and
neurological and psychological status.
Treatment of Indolent SM and Smoldering SM
Management
of Indolent SM (ISM) and Smoldering SM (SSM) is focused on the
prevention and treatment of anaphylactic reactions and symptom control (Table 1).
In case of severe symptoms refractory to anti-mediator therapy or bone
disease unresponsive to bisphosphonates, disease-modifying treatments
with cytoreductive agents may be attempted.
The first approach is
to identify symptom triggers and suggest avoidance strategies of
triggers, such as physical stimuli (heat, change of temperature,
pressure, cold, rubbing), exercise, sleep deprivation, emotions, drugs
(opiates, contrast media, succinylcholine, nonsteroidal
anti-inflammatory drugs, agents with tetrahydroisoquinoline such as
quinolones, atracurium, and rocuronium), alcohol, food, and Hymenoptera
stings.[2,3]
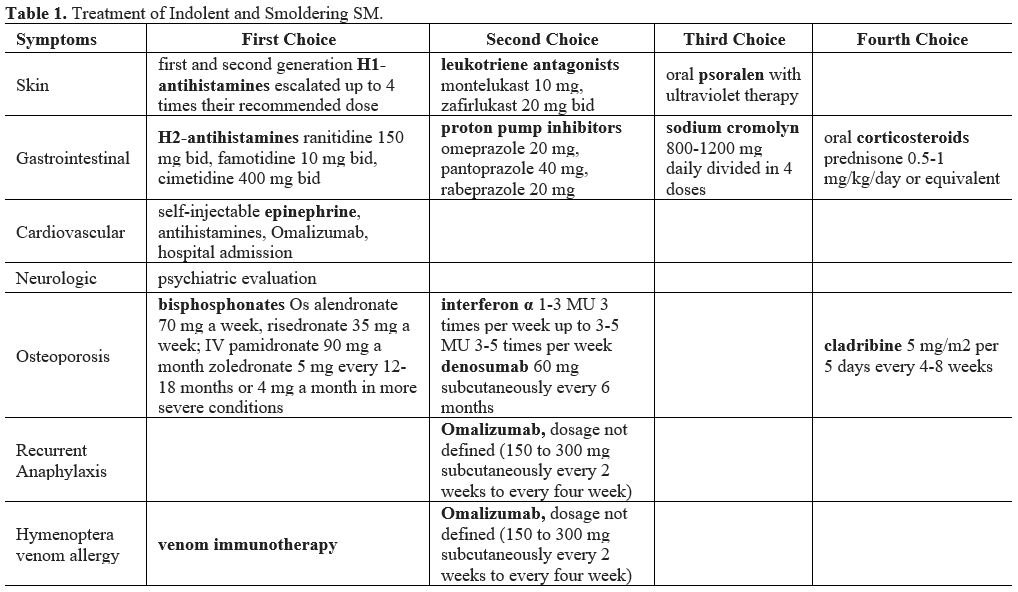 |
Table
1. Treatment of Indolent and Smoldering SM. |
Skin.
Flushing, urticaria (wheals), itching, angioedema, and dermatographism
are the most frequently reported skin symptoms in ISM and SSM.
H1-antihistamines are the first-line drugs in this setting:
first-generation sedating molecules (diphenhydramine, hydroxyzine,
ketotifen, doxepin, cyproheptadine) and second-generation non-sedating
molecules (cetirizine, levocetirizine, loratadine, desloratadine,
ebastine, rupatadine, bilastine, mozolastine) are available.
First-generation antihistamines should be used at bedtime, especially
when cutaneous symptoms impair sleeping quality. Dosage of
antihistamines (especially second-generation molecules) may be
escalated up to 4 times their recommended dose for chronic spontaneous
urticaria, with no relevant side effects. A recent review shows that
antihistamines can improve patients’ quality of life, reduce wheals and
itching on standardized provocation testing, and improve symptoms like
itching, flushing, tachycardia, and headache.[4]
When
H1-antihistamines do not adequately control skin symptoms, leukotriene
antagonists (montelukast 10 mg, zafirlukast 20 mg bid) may be added.
Unfortunately, Zileuton, a 5-lipoxygenase inhibitor, is not available
in Europe.
Aspirin can be used as a third-line treatment for skin
symptoms, starting from 81 mg bid to 500 mg bid if nonsteroidal
anti-inflammatory drugs are tolerated. Its effect should be monitored
by measuring prostaglandine2 metabolite excretion. On the other hand,
aspirin use may increase cysteinyl leukotrienes, causing/worsening
vasoconstriction and bronchospasm.[5]
Finally,
oral psoralen combined with ultraviolet therapy is reserved for
patients with cutaneous mastocytosis (urticarial pigmentosa) in case of
severe/resistant skin symptoms.[6,7]
Gastrointestinal tract.
Abdominal pain, cramping, nausea, vomiting, heartburn, gastroesophageal
reflux, and diarrhea may be present in both ISM and SSM. H2-antagonists
(ranitidine 150 mg bid, famotidine 10 mg bid, cimetidine 400 mg bid)
are the drug of choice in case of gastrointestinal symptoms.[1,3]
They inhibit gastric acid secretion via the H2-receptors and,
associated with H1-antihistamines, reduce the release of mediators from
MCs,[8] but they are less effective in controlling diarrhea.[9]
Proton
pump inhibitors (omeprazole 20 mg, pantoprazole 40 mg, rabeprazole 20
mg) are second line drugs in case H2-antihistamines are not effective.[1,3]
Sodium
cromolyn at a dose of 800-1200 mg daily divided into four doses is a
third-line treatment. It acts as a stabilizing agent on MC membranes
and reduces MC degranulation by interfering with Ca++
influx. In addition, sodium cromolyn has shown to be effective in the
management of gastrointestinal symptoms when compared to placebo.[10]
Oral
corticosteroids (prednisone 0.5-1 mg/kg/day or equivalent) are the
fourth-line drug for gastrointestinal symptoms. However, because of
their side effects, corticosteroids should be tapered as soon as
possible based on the patient response.[1,6]
Cardiovascular symptoms.
SM patients may have recurrent presyncope or syncope, hypotension,
tachycardia, or anaphylaxis. The prevalence of anaphylaxis in adults
with SM ranges from 22 to 49%, about 100 times higher than the general
population. Anaphylaxis may be provoked by a concomitant IgE-mediated
allergy (especially to Hymenoptera venoms) or may also be spontaneous.
Thus, all patients should be prescribed self-injectable epinephrine and
should be trained to treat attacks.[2,11]
Acute
episodes of anaphylaxis should be treated according to current
guidelines. The drug of choice is intramuscular epinephrine at a dosage
of 0.3-0.5 mg, followed by fluid replacement (saline or Ringer
lactate), 500-1000 mg of intravenous hydrocortisone and intravenous H1
H2-antihistamines.[12] Prevention of new episodes is
based on chronic treatment with H1- and H2-antihistamines and
leukotriene antagonists, while oral corticosteroids (prednisone 0.5-1
mg/kg/day) should be proposed only in very resistant patients.[1,3] Recently, a relevant role has been reported for Omalizumab to resolve cardiovascular symptoms with or without anaphylaxis.[13]
When this approach is ineffective, a cytoreductive therapy with
cladribine or interferon α (IFN α) can be taken into consideration.[3]
Neurologic symptoms.
Depression, headache, cognitive impairment, and sleep disturbance are
common. H1-antihistamines showed to be effective on headaches but not
on other symptoms. Therefore, these patients should undergo a
psychiatric evaluation for a prompt start of tailored treatment.[4]
Osteoporosis.
Bone involvement leading to osteopenia, osteoporosis, and fragility
fractures are frequent in SM patients: the reported prevalence ranges
from 18 to 31%. It is important to note that an osteoporotic fracture
is not considered a sign of aggressive disease, as opposed to the
rarely encountered large, osteolytic bone lesions that can sometimes be
detected in patients with ASM or MCL and are classified as a C-finding.
The pathogenesis of osteoporosis has been attributed to the cytokines
and other mediators released from mast cells. Tumor necrosis factor
(TNF)-α, interleukin(IL)-1, and IL-6 promote osteoclast activity and
inhibit osteoblasts. Moreover, histamine has a stimulatory effect on
osteoclasts and their precursors.[14]
Since
there is a relative or absolute prevalence of bone reabsorption,
bisphosphonates are the first-line treatment option. Several molecules
are available orally (alendronate 70 mg a week, risedronate 35 mg a
week) and intravenously administered (pamidronate 90 mg a month;
zoledronate 5 mg every day 12-18 months or 4 mg a month in more severe
conditions). They positively affect vertebral bone mineral density
(BMD) and less on femoral neck BMD. Zoledronate showed the best
positive effect on both vertebral and femoral neck BMD.[15]
In
case of very severe osteoporosis and/or onset of new major fractures,
the use of IFN-α should be considered. IFN-α is able to decrease MC
burden and MC-related symptoms but may be poorly tolerated for flu-like
symptoms, bone pain, fever, cytopenias, depression, and hypothyroidism,
leading to poor compliance.[1]
It is
administered subcutaneously from a starting dose of 1-3 million units
(MU) three times per week to 3-5 MU 3-5 times per week.
Finally,
when bisphosphonates and IFN-α fail, a cytoreductive treatment with the
purine nucleoside analog 2-chlorodosxyadenosine (cladribine/2CdA) at a
dosage of 5 mg/m2 per 5 days every
4-8 weeks is the third-line treatment. Immunosuppression and
myelosuppression are important side effects that may lead to drug
discontinuation.[16]
Denosumab, a monoclonal
antibody directed against RANK-ligand(L), has been developed to treat
postmenopausal osteoporosis. RANKL, which is expressed by MCs, can
activate osteoclast by the RANK pathway. Denosumab, at the dosage of 60
mg subcutaneously every six months, showed to be effective in
increasing BMD at both vertebral and femoral neck sites after one year
of treatment. Denosumab could be used as a second-line in patients not
responding to bisphosphonates or not candidates to bisphosphonates
because of renal insufficiency: further studies in larger samples
are necessary to assess efficacy.[17]
Omalizumab.
Omalizumab, an anti-IgE humanized monoclonal antibody, is approved to
treat chronic spontaneous urticaria and extrinsic bronchial asthma when
standard treatments are not effective at maximum doses. At present, the
optimal dose and frequency of administration remain to be determined
(150 to 300 mg subcutaneously every two weeks to every four weeks).
The
largest trial included 55 patients with a mast cell disorder that
received Omalizumab. The diagnoses were ISM (29 patients), MC
activation syndrome (MCAS), and Cutaneous Mastocytosis (CM). A KIT
D816V mutation was found in 27 of 49 patients (particularly those with
ISM). The recommended starting dose was 150 mg subcutaneously every two
weeks. Omalizumab response was rapid, with a median time to first
response of 2 months and the best response after six months. It was
effective on all vasomotor symptoms, including those secondary to
anaphylaxis, and gastrointestinal and urinary symptoms, with a good
safety profile.[18]
In a smaller trial including
14 patients, the authors showed a significant improvement of vasomotor
symptoms and quality of life. However, the treatment was less effective
for gastrointestinal, musculoskeletal, and neuropsychiatric symptoms.[19]
In addition, the Schedule of drug administration (starting and
maintenance doses) varied among patients, considering initial symptoms,
clinical response, and treatment tolerance.
A recent review
showed that omalizumab treatment led to a complete resolution of
anaphylaxis episodes in 84% of the patients, while the efficacy on
respiratory, musculoskeletal, and neuropsychiatric symptoms was scarce.
The authors concluded that a randomized controlled trial is mandatory
to demonstrate the usefulness of Omalizumab in SM treatment.[13]
Cytoreductive Treatment of ISM and SSM
A cytoreductive treatment is not recommended for ISM and SSM unless anti-mediator therapy has failed.[20]
Midostaurin
is a multikinase inhibitor that is able to inhibit the kinase activity
of both wild-type and D816V mutated KIT. An open-label, non-randomized
phase 2 trial was conducted on 20 patients with ISM and severe mediator
symptoms not controlled with standard therapy. After 12 weeks, patients
showed a significant reduction in symptom-score and improved quality of
life. In addition, tryptase levels showed a significant decrease. All
patients stopped midostaurin after 24 weeks, and most of them showed a
relapse. Nausea, headache, and diarrhea were the most common side
effects. The effect on anaphylaxis-like symptoms was not studied
because of the insufficient number of subjects with cardiovascular
symptoms.[21]
Masitinib, an oral tyrosine kinase
inhibitor, was used in a randomized, double-blind, placebo-controlled
phase 3 trial. One hundred thirty-five adults patients with ISM and SSM
were enrolled in the study: 71 received masitinib and 64 placebo. The
dosage was 6 mg/kg per day in two doses. The primary endpoint was the
cumulative response in at least 1 of 4 severe baseline symptoms
(itching, flushing, depression, asthenia). After 24 weeks, masitinib
showed a significant cumulative response in the primary endpoint
compared to placebo (18.7% vs. 7.4%). The most frequent adverse events
in the active group were diarrhea, rash, and asthenia.[22]
Avapritinib
(BLU-285), a multikinase inhibitor, is a second-generation inhibitor of
KIT D816V. In a randomized, double-blind, placebo-controlled phase 2
trial (NCT03731260), avapritinib significantly improved mediator
symptoms compared to placebo, with a good safety profile. The dosage
ranged from 25 to 100 mg in a daily administration.
Cladribine
is a synthetic purine analog that inhibits DNA repair, blocks dividing
cells, and induces apoptosis in resting cells. It is effective in
reducing must cell burden in ASM and SM-AHN patients. Thirty-six
subjects with IM (6 CM, 28 ISM, and 2 SSM) were included in the study.
Each course of treatment was repeated with a 4 to 12-week interval for
a maximum of 9 courses at a dosage of 0.14 mg/kg/day from 1 to 5 days.
The treatment showed a significant improvement in flushing, itching,
neuropsychiatric and cardiovascular symptoms with a concomitant
reduction of tryptase levels. Common side-effects were
myelosuppression-related toxicity (47%) and infectious complications
(22%).[16]
Hymenoptera Venom Allergy and Mast Cell Activation Syndromes
There
is a frequent association between severe Hymenoptera venom allergy
(HVA) and elevated basal serum levels (>11.4 ng/mL). For example,
some authors found that 9 out of 137 (6.6%) patients with severe drug
or food allergy (6.6%) had a basal tryptase >11.4 ng/mL, and only
two (1.5%) were diagnosed with mastocytosis. On the other hand, 13.9%
of patients with HVA had elevated tryptase, and 11.1% had a clonal mast
cell disorder.[23]
American authors found a
mastocytosis prevalence of 10.1 per 100000 overall and 96.7 per 100000
among HVA patients. Nine out of 161 (5.6%) patients undergoing venom
immunotherapy (VIT) had basal tryptase >11.4 ng/mL, and 3 (1.8%) had
a clonal mast cell disorder.[24]
Typically, HVA
in mastocytosis patients is characterized by the absence of
urticaria/angioedema and the sudden onset of cardiovascular symptoms
leading to loss of consciousness. For this reason, these patients
should carry with them an emergency kit including two epinephrine
autoinjectors and should be trained in their use by the allergist.[23]
VIT
is mandatory since it is the only life-saving treatment for these
patients, and it should be prolonged long-life with a 3-4
month-interval, according to European and American guidelines.[25]
Mastocytosis patients are at higher risk of reactions during build-up
and maintenance phases, but some authors recently observed no adverse
events in 8 patients undergoing 12 ultra-rush VIT, both in the build-up
and maintenance phases. Nevertheless, because of severe reactions,
expert personnel should carry out an ultra-rush protocol with the
prompt availability of resuscitation equipment.[26,27]
In addition, some patients may experience recurrent anaphylaxis
following Hymenoptera sting and/or extremely invalidating intolerance
to VIT. In these cases, Omalizumab has been reported to induce
tolerance to VIT and reduce anaphylaxis episodes[13] successfully.
Treatment of Advanced Systemic Mastocytosis
The
term advanced systemic mastocytosis (advSM) identifies three different
diseases, namely aggressive SM (ASM), SM with an associated hematologic
neoplasm (SM-AHN), and mast cell leukemia (MCL). These subtypes are
characterized by mast cell-related organ damage, for which a
cytoreductive treatment is usually required, and a reduced survival.[1]
Considering
the protean clinical manifestations, the evaluation of aggressiveness
involves different body systems such as bone marrow, liver, spleen,
bones, and gastrointestinal tract, globally classified as C findings.
On the other hand, constitutional symptoms may be more invalidating in
some patients than the organ damage itself. The complexity of the
disease has led to the development of several response evaluation
criteria during the last 20 years, updated to include the grading
resolution of C findings and constitutional symptoms.[28-31]
Until
now, there is no curative option for patients with AdvSM. The available
therapeutic options include tyrosine-kinase inhibitors, interferonα,
and cladribine, with variable duration and extent of response (Table 2). Allogeneic stem cell transplantation is usually performed in younger selected patients.
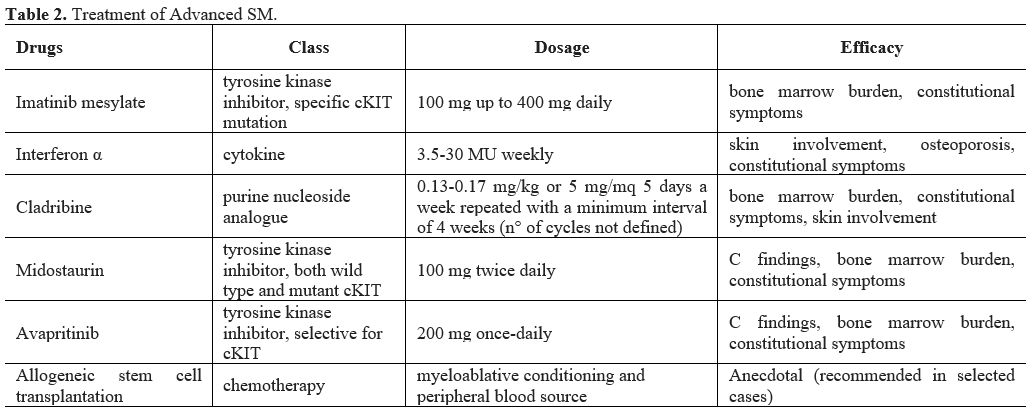 |
Table 2. Treatment of Advanced SM. |
Imatinib Mesylate
Imatinib mesylate is an in vitro inhibitor of several tyrosine kinases, particularly wild-type and specific mutant ckit.[32,33]
It was the first drug specifically approved by the FDA for adult
patients with SM lacking the cKIT D816V mutation or unknown cKIT
mutational status at 400 mg daily dosage. This indication came after a
report by Pardanani et al.,[34] in which 12 adults
with symptomatic SM were treated with imatinib mesylate 100 mg up to
400 mg daily. Three patients with eosinophilia (1 with ISS and 2 with
ASM, all cKIT D816V negative) obtained a complete remission, and 2 ASM
patients without eosinophilia obtained a bone marrow cytoreduction and
improvement of bone pain. However, three patients with ASM were
refractory to imatinib mesylate, irrespective of blood eosinophil
count; 2 patients with prevalent skin involvement had a progressive
decrease of symptoms, and two were not evaluable for response.
A Dutch open-label phase II study reported on the efficacy of imatinib mesylate among 14 patients, mostly with non-advSM.[35]
The authors found a reduction of hepatosplenomegaly and
skin/constitutional symptoms in almost half of the study population,
more pronounced in cKIT D816V negative but also detectable in cKIT
D816V positive patients. However, the concomitant use of steroids may
have contributed to the response.
Another American prospective
open-label phase II study recruited 11 patients with ISM and nine
patients with AdvSM for treatment with imatinib mesylate at 400 mg
daily.[36] During a median time on therapy of 9
months, 1 AdvSM patient and 6 ISM patients reported improvement of
symptoms; nevertheless, all patients interrupted the treatment for loss
of response. The authors concluded that imatinib mesylate might only
produce a significant clinical benefit in patients without cKIT D816V
mutation.
A monocentric retrospective study published in 2009
reported the outcome of 27 patients with ISM (30%) and AdvSM (70%)
treated with imatinib mesylate at a starting dose of 400 mg daily.[37]
Among 22 evaluable patients, only four responded (18% overall): 1 ISM
patient, 2 ASM patients, and 1 SM-AHN patient; the median duration of
response was 19.6 months for all patients (range 9-69 months).
More
recently, in 2017, a phase IV clinical trial tested the response to
imatinib mesylate of 10 patients with CM and SM with cKIT mutation
found outside the activation loop-coding region or wild-type cKIT.[38]
Four patients obtained a complete response in terms of MC burden in the
bone marrow, normalization of tryptase level, and resolution of
constitutional symptoms; 1 patient obtained a partial response
involving both MC burden and symptoms. All five unresponsive patients
were wild-type cKIT. The authors concluded that imatinib mesylate is
effective among SM patients with specific cKIT mutational status (i.e.,
mutation involving the extracellular and transmembrane regions) while
is less effective than previously reported in true wild type cKIT.
Interferon α
Historically, interferonα was the first treatment of SM, used by analogy with other myeloproliferative neoplasms,[39-41] sometimes in combination with steroids.
A
prospective multicentre phase II trial reported only 35% partial
response and 30% minor response among 13/20 evaluable patients with ISM
and ASM.[42] In this study, interferon α 3MU/m2
thrice weekly for six months improved both skin involvement and
constitutional symptoms while ineffective in organ involvement.
However, the authors also reported a discrete rate of withdrawal,
mainly because of worsening of cytopenia, and the reappearance of
systemic symptoms soon after the interruption of the drug, even for
patients who previously responded.
An Austrian study enrolled five patients treated with interferonα and steroids.[43]
The authors reported two complete resolutions of C findings, one
partial improvement of C findings, one stable disease, and one
progression to MCL.
A monocentric retrospective study published in
2009 reported the outcome of 47 patients with ISM (23%) and AdvSM (77%)
treated with interferonα at a median dosage of 15 MU weekly (range
3.5-30 MU weekly).[37] Among 40 evaluable patients,
the overall response rate was 53% for all categories, particularly 6
ISM patients, 6 ASM patients, and 9 SM-AHN patients; the median
duration of response was 12 months for all patients (range 1-67
months).
The main adverse events of treatment were
fatigue, cytopenia, depression, flu-like symptoms, and fever. In most
cases, symptoms are easily managed with dose reduction, but some cases
of drug interruption for severe cytopenia and/or depression were
reported.[42,43]
Cladribine
2-chlorodeoxyadenosine
(2-CdA) is a purine nucleoside analog used in different diseases of
hematopoietic origin, with significant activity against monocytes[44] that were thought to have a common progenitor with mast cells.[45] A
cases series of AdvSM patients refractory or intolerant to interferonα
reported the outcome of 4 patients after 4-6 cycles of 2-CdA (0.14
mg/kg 5 days a week) given every 1 to 6 months.[46]
The authors reported a significant improvement of systemic symptoms and
skin rash in three cases and persistence of response long after
treatment. In another series of 10 patients with ISM and AdvSM treated
with six courses of 2-CdA (0.13 mg/kg 5 days a week), the authors
concluded that three patients with ASM obtained a response after a
median time of 6 months.[47] Moreover, skin
involvement was reduced at least 50% in all affected patients, and bone
marrow involvement was significantly reduced in 8 cases; constitutional
symptoms were markedly decreased. A
monocentric retrospective study published in 2009 reported the outcome
of 26 patients with ISM (38%) and AdvSM (62%) treated with 2-CdA (0.14
mg/kg 5 days a week) given every 1 to 3 months.[37]
Among 22 evaluable patients, the overall response rate was 55% for all
categories: five ISM patients, one ASM patient, and six SM-AHN
patients; the median duration of response was 11 months for all
patients (range 3-74 months).A
multicenter French study recruited 68 patients with CM (9%), ISM (44%)
and AdvSM (47%) treated with a median of 3 courses (range 1-9) of 2-CdA
(0.13-0.17 mg/kg or 5 mg/mq 5 days a week) given every 1 to 3 months.[16]
The authors reported an overall response rate of 72%, with the highest
response rate in cutaneous/indolent form (100% and 89%, respectively)
compared to ASM and SM-AHN (43% and 59%, respectively). Although no
complete response was reported, the median duration of response was 3.7
years, with no significant difference between ISM and AdvSM. The
main adverse events of treatment were grade 3-4 myelosuppression and
infection, which may lead to dose reduction and treatment delay. Herpes
reactivation can be managed with antiviral prophylaxis.[16,47]
Dasatinib
Dasatinib
is a tyrosine kinase inhibitor that exerts a potent action on the
mutant D816V cKIT in vitro. On this basis, Verstovsek et al. conducted
an open-label phase 2 study on 18 ISM and 15 AdvSM patients treated
with dasatinib 140 mg daily.[48] The authors reported
an overall response rate of 33%: only two patients obtained a complete
response, lasting for 5 and 16 months, and both were negative for D816V
cKIT mutation; the other nine patients achieved an improvement in
constitutional symptoms. The authors concluded that dasatinib is
ineffective in treating patients with SM carrying D816V cKIT mutation.
Nilotinib
Nilotinib
is a tyrosine kinase inhibitor that is also active against the KIT in
vitro. Following preliminary results, Hochhaus et al. conducted a
multicenter phase-2 registration trial on 61 patients with advSM (69%)
and ISM (31%) treated with nilotinib 400 mg twice a day.[49]
Unfortunately, evaluable responses were available only in the ASM
group: the authors reported a minor response in 8/37 patients, while no
complete response was documented.
Midostaurin
Midostaurin
is a potent multi-target tyrosine kinase inhibitor, active on wild-type
and mutant cKIT D816V. Following the preliminary efficacy report, an
international multicenter single-group open-label phase-2 study
recruited 116 patients with AdvSM for treatment with midostaurin 100 mg
twice daily.[50] The authors reported an overall
response rate of 46% and a median duration of treatment of 11.4 months
(range 0.3-51.5). According to WHO categories, the response rates were
75%, 58%, and 50% for patients with ASM, SM-AHN, and MCL. Responses
included improved organ function (reduction of bone marrow burden,
spleen volume, and normalization of liver enzymes), reduction or
interruption of transfusion dependence, improvement of constitutional
symptoms and quality of life, and recovery of weight loss. However, the
median duration of response was not reached in patients with ASM (95%
CI, 24.1 months to not estimated) and with MCL (95% CI, 3.6 months to
not estimated), while it was 12.7 months (95% CI, 7.4 to 31.4) in
patients with SM-AHN. Similarly, the median overall survival was not
reached (95% CI, 28.7 months to not estimated) in patients with ASM,
while it was 20.7 months (95% CI, 16.0 to 44.4) and 9.4 months (95% CI,
7.5 to not estimated) in patients with SM-AHN and MCL, respectively.
Overall, median progression-free survival was 14.1 months: in ASM
patients, it was 28.7 months, 11.0 months higher than SM-AHN, and 11.3
than MCL patients. Based on this study, in 2017 FDA approved midostaurin for the treatment of AdvSM, regardless of cKIT D816V mutation status. Another
multicenter phase-2 trial reported on the long-term outcome of 26
patients with AdvSM treated with midostaurin 100 mg twice daily for up
to 12 cycles and beyond in case of response.[51]
During the 12-cycle period, the overall response rate was 69%, with a
median time to response of 25.5 days (range 4–56) and a median time to
best response of 56 days (range 25–229). Five patients reported only
stable disease, and three patients progressed. The best rate of
response was reported in patients with SM-AHN (76%) and MCL (67%),
rather than ASM (33%). After the 12-cycle period, the overall response
rate did not change. Median overall survival was not reached for 3 ASM,
while it was 40 months (95% CI, 24.2–55.9) for the 17 SM-AHN patients
and 18.5 months (95% CI, 0–62.2) for MCL patients. Overall, the median
PFS was 41.0 months (4.4–77.6). A Polish study reported data on the real-world efficacy of midostaurin on 13 patients with AdvSM.[52]
After a median duration of treatment of 9 months (range 1-21), a
clinical benefit was detectable in 77% of patients, and half of the
patients with measurable organ damage obtained a response. After a
median follow-up of 19 months, the authors reported seven patients with
ongoing therapy and three patients died of progressive disease. The
main adverse events of treatment were nausea and vomiting, diarrhea,
increased transaminase, and cytopenia. Gastrointestinal symptoms
usually improve after the first months of treatment. Cytopenia and
abnormal liver function can be managed with drug interruption and dose
reduction, according to the toxicity grading.[50-52]
Brentuximab Vedotin
Brentuximab
vedotin (BV) is a chimeric immunoglobulin G1, specific to human CD30,
covalently attached to the microtubule-disrupting agent monomethyl
auristatin E (MMAE) used mostly in lymphoproliferative diseases
expressing surface CD30. After in vitro study on CD30+ human mast cell
lines, a phase 2, open-label study was performed, with the primary
objective of evaluating the antitumor activity of BV in patients with
CD30-positive non-lymphoid malignancies.[53] Two
patients with ISM and two patients with ASM were included and treated
with BV at a dosage of 1.8 mg/kg or 2.4 mg/kg every three weeks. The
authors reported one major response, one improvement of constitutional
symptoms, and two disease progression.Another
phase 2 open-label, single-group was conducted to determine the
efficacy and safety of BV 1.8 mg/kg every three weeks among patients
with AdvSM, and at least 20% of surface CD30 expression, assessed by
flow cytometry.[54] After a median follow-up of 722
days (range 18-1246 days) and a median number of five cycles (range 1-8
cycles), no significant and/or durable response was observed among ten
recruited patients, assessed by reduction of tumor burden and
constitutional symptoms. The authors concluded that BV has no clinical
activity in this setting of patients.
Avapritinib
Avapritinib (BLU-285) is a selective cKIT D816V inhibitor that showed promising results in phase 1 clinical trial.[55]
The phase-1 trial enrolled 53 patients with AdvSM for treatment
with avapritinib: among 32 evaluable patients, the overall response
rate was 76% after a median follow-up of 27.3 months. At this time, the
median overall survival has not been reached in this population. The phase-2 registration trial reported the outcome of 32 patients treated with avapritinib 200 mg daily.[56]
The overall response rate was 75%, specifically 100% for ASM, 81% for
SM-AHN, and 25% for MCL. The median time to response was two months,
and the median time to best response was 5.6 months; at a median
follow-up of 10.4 months, all responses persisted, and median overall
survival was not reached. Based on these data, the FDA approved avapritinib for Adv SM treatment in June 2021.The
main adverse events of treatment were periorbital and peripheral edema,
fatigue, gastrointestinal symptoms, cytopenia, and cognitive
impairment.[55,56]
Allogeneic Transplantation
The
role of allogeneic stem cell transplantation is still not defined in
the treatment of disease: debulk strategy, the timing of the procedure,
choice of best conditioning regimen and donor source, and possible
maintenance therapy post-transplant are still matter of debate in the
clinical practice.[57,58] Allogeneic
stem cell transplantation has been performed with various outcomes in
mast cell disease patients associated with another hematological
neoplasm. Some authors reported the disappearance of leukemia-related
mast cells clone after allogeneic transplant,[59] while others showed neoplastic mast cell persistence despite complete remission of concomitant hematological neoplasm.[60]
Moreover, evidence of the graft-versus-mast-cells effect implies an
immunological mechanism underlying the clearance of neoplastic
infiltration.[61,62] Literature
data about conditioning regimens and donor sources are scarce. Nakamura
et al. reported the outcome of 3 patients with MCL and SM-AHN
conditioned with a non-myeloablative regimen (cyclophosphamide and
fludarabine).[63] Engraftment was reached in all 3
cases, and no transplant-related mortality was observed, but all
patients relapsed despite a transient graft-versus-mast cell effect
after immunosuppression withdrawal. More recently, a retrospective
multicenter study reported the outcome of 57 patients with AdvSM
transplanted in the United States and Europe: the overall response was
about 70% for all categories.[57] Considering
survival, the OS and PFS differ among the three categories.
Particularly, considering ASM, SM-AHN, and MCL, OS at three years was
43%, 74%, and 17%, respectively, while PFS at three years was 43%, 63%,
and 17%. Excluding the patients with MCL, which had the worse
prognosis, risk factors for reduced survival were diagnosis of ASM and
a reduced-intensity conditioning regimen. All three patients who
received a transplant from cord blood and HLA-haploidentical relative
and all patients with MCL who received RIC died.As
a general recommendation, myeloablative conditioning and peripheral
blood source should be considered in younger people with aggressive
clinical course.
Conclusions
Mastocytosis
is a complex disease for which a multidisciplinary approach is
mandatory for a comprehensive evaluation and choice of therapy. In most
cases, patients might need a personalized treatment with a specific
combination of different drugs, ranging from antihistamines and
bisphosphonates to TKI and chemotherapy. In addition, tolerance of
treatment, disease symptoms control, and adverse events should be
frequently evaluated and carefully balanced in these patients.
References
- Pardanani A. Systemic mastocytosis in adults: 2021
Update on diagnosis, risk stratification and management. Am J Hematol
2021; 96: 508-525. https://doi.org/10.1002/ajh.26118 PMid:33524167
- Pardanani
A. How I treat patients with indolent and smoldering mastocytosis (rare
conditions but difficult to manage). Blood 2013; 121 3085-3094. https://doi.org/10.1182/blood-2013-01-453183 PMid:23426950
- Castells
M, Butterfield J. Mast cell activation syndrome and mastocytosis:
initial treatment options and long-term management. J Allergy Clin
Immunol Parct 2019; 7: 1097-1106. https://doi.org/10.1016/j.jaip.2019.02.002 PMid:30961835
- Nurmatov
UB, Rhatigan E, Simons FER, Sheikh A. H1-antihistamines for primary
mast cell activation syndromes: a systematic review. Allergy 2015; 70:
1052-1061. https://doi.org/10.1111/all.12672 PMid:26095756
- Butterfield
JH, Singh RJ. Divergent PGD2 and leukotriene C4 metabolite excretion
following aspirin therapy: Ten patients with systemic mastocytosis.
Prostaglandins Other Lipid Mediat. 2021 Aug;155:106563. doi:
10.1016/j.prostaglandins.2021.106563. Epub 2021 May 21. https://doi.org/10.1016/j.prostaglandins.2021.106563 PMid:34029712
- Scherber RM, Borate U. How we diagnose and treat systemic mastocytosis in adults. Br J Haematol 2018; 180: 11-23. https://doi.org/10.1111/bjh.14967 PMid:29048112
- Mackey
S, Pride HB, Tyler WB. Diffuse cutaneous mastocytosis. Treatment with
oral psoralen plus UV-A. Arch Dermatol 1996; 132: 1429-1430. https://doi.org/10.1001/archderm.1996.03890360013002 PMid:8961869
- Kurosawa
M, Amano H, Kanbe N, et al. Heterogeneity of mast cells in mastocytosis
and inhibitory effect of ketotifen and ranitidine on indolent systemic
mastocytosis. J Allergy Clin Immunol 1997; 100: S25-32. https://doi.org/10.1016/S0091-6749(97)70001-0
- Hirschowitz
BI, Groarke JF. Effect of cimetidine on gastric hypersecretion and
diarrhea in systemic mastocytosis. Ann Int Med 1979; 90: 769-771. https://doi.org/10.7326/0003-4819-90-5-769 PMid:373561
- Horan
RF, Sheffer AL, Austen KF. Cromlyn sodium in the management of systemic
mastocytosis. J Allergy Clin Immunol 1990; 85: 852-855. https://doi.org/10.1016/0091-6749(90)90067-E
- Schuch A, Brockow K. Mastocytosis and anaphylaxis. Immunol Allergy Clin N Am 2017; 37: 153-164. https://doi.org/10.1016/j.iac.2016.08.017 PMid:27886904
- Tanno LK, Alvarez-Perea A, Pouessel G. Therapeutic approach of anaphylaxis. Curr Opin Allergy Clin Immunol 2019; 19: 393-401. https://doi.org/10.1097/ACI.0000000000000539 PMid:31058676
- Jendoubi
F, Gaudenzio N, Gallini A, et al. Omalizumab in the treatment of adult
patients with mastocytosis: a systematic review. Clin Exp Allergy 2020;
50: 654-661. https://doi.org/10.1111/cea.13592 PMid:32107810
- Rossini
M, Zanotti R, Orsolini G, et al. Prevalence, pathogenesis, and
treatment options for mastocytosis-related osteoporosis. Osteoporos Int
2016; 27: 2411-2421. https://doi.org/10.1007/s00198-016-3539-1 PMid:26892042
- Rossini
M, Zanotti R, Viapiana O, et al. Zoledronic acid in osteoporosis
secondary to mastocytosis. Am J Med. 2014; 127: 1127.e1-1127.e4. https://doi.org/10.1016/j.amjmed.2014.06.015 PMid:24954632
- Barete
S, Lortholary O, Damaj G, et al. Long-term efficacy and safety of
cladribine (2-CdA) in adult patients with mastocytosis. Blood
2015;126:1009-1016. https://doi.org/10.1182/blood-2014-12-614743 PMid:26002962
- Orsolini
G, Gavioli I, Tripi G, et al. Denosumab for the treatment of
mastocytosis-related osteoporosis: a case series. Calcif Tissue Int
2017; 100: 595-598. https://doi.org/10.1007/s00223-017-0241-z PMid:28229176
- Lemal
R, Fouquet G, Terriou L, et al. Omalizumab therapy form mast
cell-mediator symptoms in patients with ISM, CM, MMAS and MCAS. J
Allergy Clin Immunol Pract 2019; 7: 2387-2395. https://doi.org/10.1016/j.jaip.2019.03.039 PMid:30954641
- Broesby-Olsen
S, Vestergaard H, Mortz CG, Jensen B, Havelund T, Hermann AP,
Siebenhaar F, Møller MB, Kristensen TK, Bindslev-Jensen C; on behalf of
Mastocytosis Centre Odense University Hospital (MastOUH). Omalizumab
prevents anaphylaxis and improves symptoms in systemic
mastocytosis: efficacy and safety observations. Allergy 2018; 73:
230-238. https://doi.org/10.1111/all.13237 PMid:28662309
- Fletcher L, Borate U. Novel approaches for systemic mastocytosis. Curr Opin Hematol 2019; 26; 112-18. https://doi.org/10.1097/MOH.0000000000000486 PMid:30694839
- van
Anrooij B, Oude Elberink JNG, Span LFR, et al. Midostaurin in patients
with indolent systemic mastocytosis: an open-label phase 2 trial. J
Allergy Clin Immunol 2018; 142: 1006-1008. https://doi.org/10.1016/j.jaci.2018.06.003 PMid:29890238
- Lortholary
O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of
severely symptomatic indolent systemic mastocytosis: a randomized,
placebo-controlled, phase 3 study. Lancet 2017; 389; 612-20. https://doi.org/10.1016/S0140-6736(16)31403-9
- Bonadonna
P, Bonifacio M, Lombardo C, Zanotti R. Hymenoptera allergy and mast
cell activation syndromes. Curr Allergy Asthma Rep 2016; 16: 5. https://doi.org/10.1007/s11882-015-0582-5 PMid:26714690
- Schuler
4th CF, Volertas S, Khokhar D, et al. Prevalence of mastocytosis and
hymenoptera venom allergy in the United States. J Allergy Clin Immunol.
2021 Apr 22:S0091-6749(21)00652-7
- Golden
DBK. Insect sting allergy: new guidelines form the European and USA
consensus groups: algorithms and recommendations. Curr Opin Allergy
Clin Immunol. 2019 Oct;19(5):456-461 https://doi.org/10.1097/ACI.0000000000000570 PMid:31335358
- González
de Olano D, Alvarez-Twose I, Esteban-López MI, et al. Safety and
effectiveness of immunotherapy in patients with indolent systemic
mastocytosis presenting with Hymenoptera venom anaphylaxis. J allergy
Clin immunol 2008; 121: 519-526. https://doi.org/10.1016/j.jaci.2007.11.010 PMid:18177694
- Gruzelle
V, Ramassamy M, Bulai Livideanu C, Didier A, Mailhol C, Guilleminault
L. Safety of ultra-rush protocols for hymenoptera venom immunotherapy
in systemic mastocytosis Allergy 2018; 73: 2260-2263. https://doi.org/10.1111/all.13557 PMid:29984458
- Valent
P., Akin C., Sperr W.R., Escribano L., Arock M., Horny H.P., Bennett
J.M., Metcalfe D.D. Aggressive systemic mastocytosis and related mast
cell disorders: Current treatment options and proposed response
criteria. Leuk. Res. 2003;27:635-641. https://doi.org/10.1016/S0145-2126(02)00168-6
- Valent
P., Akin C., Escribano L., Födinger M., Hartmann K., Brockow K.,
Castells M., Sperr W.R., Kluin-Nelemans H.C., Hamdy N.A., et al.
Standards and standardization in mastocytosis: Consensus statements on
diagnostics, treatment recommendations and response criteria. Eur. J.
Clin. Investig. 2007;37:435-453. https://doi.org/10.1111/j.1365-2362.2007.01807.x PMid:17537151
- Pardanani
A, Tefferi A. A critical reappraisal of treatment response criteria in
systemic mastocytosis and a proposal for revisions. Eur. J. Haematol.
2010;84:371-378. https://doi.org/10.1111/j.1600-0609.2010.01407.x PMid:20059531
- Gotlib
J, Pardanani A, Akin C, et al. International Working
Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT)
& European Competence Network on Mastocytosis (ECNM) consensus
response criteria in advanced systemic mastocytosis. Blood.
2013;121(13):2393-2401. https://doi.org/10.1182/blood-2012-09-458521 PMid:23325841 PMCid:PMC3612852
- Heinrich
MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of
c-kit receptor tyrosine kinase activity by STI 571, a selective
tyrosine kinase inhibitor. Blood 2000; 96: 925-32. https://doi.org/10.1182/blood.V96.3.925 PMid:10910906
- Ma
Y, Zeng S, Metcalfe DD, et al. The c-KIT mutation causing human
mastocytosis is resistant to STI571 and other KIT kinase inhibitors;
kinases with enzymatic site mutations show different inhibitor
sensitivity profiles than wild-type kinases and those with
regulatory-type mutations. Blood 2002; 99: 1741-44. https://doi.org/10.1182/blood.V99.5.1741 PMid:11861291
- Pardanani
A, Elliott M, Li CY, Baxter EJ, Cross NCP, Tefferi A. Imatinib for
systemic mast-cell disease. Lancet 2003;362(9383):535-6 Lancet 2003 https://doi.org/10.1016/S0140-6736(03)14115-3
- Droogendijk
HJ, Kluin-Nelemans HJ, van Doormaal JJ, Oranje AP, van de Loosdrecht
AA, van Daele PL. et al. Imatinib mesylate in the treatment of systemic
mastocytosis: a phase II trial. Cancer. 2006 Jul 15;107(2):345-51 https://doi.org/10.1002/cncr.21996 PMid:16779792
- Vega-Ruiz
A et al. Phase II study of imatinib mesylate as therapy for patients
with systemic mastocytosis. Leuk Res 2009;33(11):1481-4 https://doi.org/10.1016/j.leukres.2008.12.020 PMid:19193436 PMCid:PMC4184059
- Lim
KH, Pardanani A, Butterfield JH, Li CY, Tefferi A. Cytoreductive
therapy in 108 adults with systemic mastocytosis: Outcome analysis and
response prediction during treatment with interferon-alpha,
hydroxyurea, imatinib mesylate or 2-chlorodeoxyadenosine. Am J Hematol
2009;84(12):790-4. https://doi.org/10.1002/ajh.21561 PMid:19890907
- Alvarez-Twose
I, Matito A, Morgado JM, et al. Imatinib in systemic mastocytosis: a
phase IV clinical trial in patients lacking exon 17 KIT mutations and
review of the literature. Oncotarget 2016;8(40):68950-68963 https://doi.org/10.18632/oncotarget.10711 PMid:28978170 PMCid:PMC5620310
- Kluin-Nelemans
HC, Jansen JH, Breukelman H, Wolthers BG, Kluin PM, Kroon HM, Willemze
R. Response to interferon alfa-2b in a patient with systemic
mastocytosis. N Engl J Med 1992;326(9):619-23. https://doi.org/10.1056/NEJM199202273260907 PMid:1370856
- Worobec
AS, Kirshenbaum AS, Schwartz LB, Metcalfe DD. Treatment of three
patients with systemic mastocytosis with interferon alpha-2b. Leuk
Lymphoma 1996;22(5-6):501-8. https://doi.org/10.3109/10428199609054789 PMid:8882964
- Butterfield JH. Response of severe systemic mastocytosis to interferon alpha. Br J Dermatol 1998;138(3):489-9 https://doi.org/10.1046/j.1365-2133.1998.02131.x PMid:9580806
- Casassus
P, Caillat-Vigneron N, Martin A, et al. Treatment of adult systemic
mastocytosis with interferon-alpha: results of a multicentre phase II
trial on 20 patients. Br J Haematol. 2002 Dec;119(4):1090-7 https://doi.org/10.1046/j.1365-2141.2002.03944.x PMid:12472593
- Hauswirth
AW, Simonitsch-Klupp I, Uffmann M, Koller E, Sperr WR, Lechner K,
Valent P. Response to therapy with interferon alpha-2b and prednisolone
in aggressive systemic mastocytosis: report of five cases and review of
the literature. Leuk Res. 2004; 28:249-257 https://doi.org/10.1016/S0145-2126(03)00259-5
- Carrera
CJ, Terai C, Lotz M, Curd JG, Piro LD, Beutler E, Carson DA. Potent
toxicity of 2-chlorodeoxyadenosine toward human monocytes in vitro and
in vivo. A novel approach to immunosuppressive therapy. J Clin Invest
1990;86(5):1480-8 https://doi.org/10.1172/JCI114865 PMid:1700795 PMCid:PMC296893
- Kirshenbaum
AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD. Demonstration
that human mast cells arise from a progenitor cell population that is
CD34(+), c-kit(+), and expresses aminopeptidase N (CD13). Blood
1999;94(7):2333-42 https://doi.org/10.1182/blood.V94.7.2333.419k30_2333_2342 PMid:10498605
- Pardanani
A, Hoffbrand AV, Butterfield JH, Tefferi A. Treatment of systemic mast
cell disease with 2-chlorodeoxyadenosine. Leuk Res 2004 ;28(2):127-31. https://doi.org/10.1016/S0145-2126(03)00185-1
- Kluin-Nelemans HC, Oldhoff JM, Van Doormaal JJ, et al. Cladribine therapy for systemic mastocytosis. Blood 2003 ;102(13):4270-6 https://doi.org/10.1182/blood-2003-05-1699 PMid:12933573
- Verstovsek
S, Tefferi A, Cortes J, et al. Phase II study of dasatinib in
Philadelphia chromosome-negative acute and chronic myeloid diseases,
including systemic mastocytosis. Clin Cancer Res 2008 ;14(12):3906-15 https://doi.org/10.1158/1078-0432.CCR-08-0366 PMid:18559612 PMCid:PMC5018899
- Hochhaus
A, Baccarani M, Giles FJ, et al. Nilotinib in patients with systemic
mastocytosis: analysis of the phase 2, open-label, single-arm nilotinib
registration study. J Cancer Res Clin Oncol ;141(11):2047-60 https://doi.org/10.1007/s00432-015-1988-0 PMid:26002753 PMCid:PMC4768228
- Gotlib
J, Kluin-Nelemans HC, George TI, et al. Efficacy and Safety of
Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med.
2016;374(26):2530-41 https://doi.org/10.1056/NEJMoa1513098 PMid:27355533
- DeAngelo
DJ, George TI, Linder A, et al. Efficacy and safety of midostaurin in
patients with advanced systemic mastocytosis: 10-year median follow-up
of a phase II trial. Leukemia 2018;32(2):470-478 https://doi.org/10.1038/leu.2017.234 PMid:28744009
- Szudy-Szczyrek
A, Bachanek-Mitura O, Gromek T, et al. Real-World Efficacy of
Midostaurin in Aggressive Systemic Mastocytosis. J Clin Med 2021
;10(5):1109 https://doi.org/10.3390/jcm10051109 PMid:33799933 PMCid:PMC7961806
- Borate
U, Mehta A, Reddy V, Tsai M, Josephson N, Schnadig I. Treatment of
CD30-positive systemic mastocytosis with brentuximab vedotin. Leuk Res.
2016;44:25-31 https://doi.org/10.1016/j.leukres.2016.02.010 PMid:26994848
- Gotlib
J, Baird JH, George TI, et al. A phase 2 study of brentuximab vedotin
in patients with CD30-positive advanced systemic mastocytosis. Blood
Adv 2019;3(15):2264-2271 https://doi.org/10.1182/bloodadvances.2019000152 PMid:31350306 PMCid:PMC6693006
- Deininger
MW, Gotlib J, Robinson WA, et al. Avapritinib (BLU-285), a selective
KIT inhibitor, is associated with high response rate and tolerable
safety profile in advanced systemic mastocytosis: results of a phase 1
study [abstract]. HemaSphere. 2018;2(S1). Abstract PF612
- DeAngelo
DJ, Reiter A, Radia D, et al CT023 - PATHFINDER: Interim analysis of
avapritinib (ava) in patients (pts) with advanced systemic mastocytosis
(AdvSM) Cancer Res 2021 vol 81 is 13 https://doi.org/10.1158/1538-7445.AM2021-CT023
- Ustun
C, Reiter A, Scott BL, et al. Hematopoietic stem-cell transplantation
for advanced systemic mastocytosis. J Clin Oncol. 2014 Oct
10;32(29):3264-74 https://doi.org/10.1200/JCO.2014.55.2018 PMid:25154823 PMCid:PMC4876356
- Ustun
C, Gotlib J, Popat U, et al. Consensus Opinion on Allogeneic
Hematopoietic Cell Transplantation in Advanced Systemic Mastocytosis.
Biol Blood Marrow Transplant. 2016 Aug;22(8):1348-1356 https://doi.org/10.1016/j.bbmt.2016.04.018 PMid:27131865
- Sperr
WR, Drach J, Hauswirth AW, et al: Myelomastocytic leukemia: Evidence
for the origin of mast cells from the leukemic clone and eradication by
allogeneic stem cell transplantation. Clin Cancer Res 2005 11:6787-6792
https://doi.org/10.1158/1078-0432.CCR-05-1064 PMid:16203765
- Rønnov-Jessen
D, Løvgreen Nielsen P, Horn T: Persistence of systemic mastocytosis
after allogeneic bone marrow transplantation in spite of complete
remission of the associated myelodysplastic syndrome. Bone Marrow
Transplant 1991 8:413-415
- Spyridonidis
A, Thomas AK, Bertz H, et al: Evidence for a graft-versus-mast-cell
effect after allogeneic bone marrow transplantation. Bone Marrow
Transplant 2004 34:515-519. https://doi.org/10.1038/sj.bmt.1704627 PMid:15273711
- Gromke
T, Elmaagacli AH, Ditschkowski M, et al: Delayed graft-versus-mast-cell
effect on systemic mastocytosis with associated clonal haematological
nonmast cell lineage disease after allogeneic transplantation. Bone
Marrow Transplant 2013 48:732-733 https://doi.org/10.1038/bmt.2012.198 PMid:23085825
- Nakamura
R, Chakrabarti S, Akin C, et al. A pilot study of nonmyeloablative
allogeneic hematopoietic stem cell transplant for advanced systemic
mastocytosis. Bone Marrow Transplant 2006 37:353-358 https://doi.org/10.1038/sj.bmt.1705245 PMid:16400343
[TOP]