Ugo Testa, Germana Castelli and Elvira Pelosi
Department of Oncology, Istituto Superiore di Sanità, Rome, Italy.
Correspondence to: Ugo
Testa, Department of Oncology, Istituto Superiore di Sanità, Viale
Regina Elena 299, 00161. Rome, Italy.
Published: September 1, 2022
Received: July 3, 2022
Accepted: August 18, 2022
Mediterr J Hematol Infect Dis 2022, 14(1): e2022069 DOI
10.4084/MJHID.2022.069
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Hematopoietic
stem cells (HSCs) ensure the coordinated and balanced production of all
hematopoietic cell types throughout life. Aging is associated with a
gradual decline of the self-renewal and regenerative potential of HSCs
and with the development of clonal hematopoiesis. Clonal hematopoiesis
of indeterminate potential (CHIP) defines the clonal expansion of
genetically variant hematopoietic cells bearing one or more gene
mutations and/or structural variants (such as copy number alterations).
CHIP increases exponentially with age and is associated with cancers,
including hematologic neoplasia, cardiovascular and other diseases. The
presence of CHIP consistently increases the risk of hematologic
malignancy, particularly in individuals who have CHIP in association
with peripheral blood cytopenia.
|
Hematopoiesis, Hematopoietic Stem Cells and Aging
Hematopoietic
stem cells (HSCs) are the blood-forming stem cells that possess the
property of both self-renewing and of differentiation generating all
hematopoietic elements; the biological activity of HSCs is essential
during all life to maintain hematopoiesis and to promote hematopoietic
cell regeneration in stress conditions or after transplantation. HSCs
are phenotypically and functionally heterogeneous.
Fetal Hematopoiesis.
The hematopoietic system is generated through a complex developmental
process starting from the early stages of embryonic development. In
mammals, the first transient waves of blood cell generation occur at
the level of the extra-embryonic yolk sac; two waves of yolk-sac
hematopoiesis have been described: a primitive wave of hematopoiesis
leads to the production of primitive nucleated erythrocytes and
primitive macrophages; the second wave of hematopoiesis is
characterized by the generation of erythron-myeloid progenitors (EMPs)
and lymphoid progenitors that transiently migrate and seed into the
fetal liver.[1]
Generation of HSC.
HSCs are originated from a peculiar population of endothelial cells
(hemogenic endothelium) located at the level of the dorsal aorta or the
aorta-gonad-mesonephros (AGM) region around 30-42 days post-conception.[1]
Following their budding into blood vessels, HSCs undergo a migration
process to the fetal liver, where the stem cell pool considerably
expands.[1] The final step of the developmental
generation of the hematopoietic system is represented by the migration
of HSCs to the bone marrow, where these cells further expand and
differentiate, orchestrating the whole hematopoiesis.[1]
In
human embryonic tissues, the hematogenous potential is present inside
the embryo, while hematopoietic cells originating in the yolk sac do
not contribute to the generation of HSCs sustaining definitive
hematopoiesis.[2]
However, the yolk sac
contributes to the generation of macrophagic cells that are maintained
in adult life. Studies in murine hematopoiesis have shown three waves
of macrophage cell development: (i) A primitive wave of macrophage
generation related to primitive hematopoiesis occurring at the level of
the yolk sac (in this phase, in addition to macrophages, are also
generated primitive erythroid and megakaryocytic cells); these
primitive macrophages are responsible for the generation of microglia
in adult mouse brain. (ii) The second wave of macrophage production is
related to erythron-myeloid (EMP) progenitors responsible through their
differentiation for the generation of tissue-resident macrophages.
(iii) The third wave of macrophage generation is mediated by the
differentiation of HSCs formed in the AGM region of the embryo and
colonizing the fetal liver; these macrophages represent a population of
long-lived macrophagic cells persisting throughout life. In mouse, most
adult tissue macrophages resident in the liver, brain, epidermis, and
lung originates from yolk sac EMLs distinct from HSCs.[3]
Studies carried out in human embryonic yolk sac cells support the yolk
sac origin of a part of macrophages on day 23-25 post-conception, a
population of early yolk sac-derived primitive macrophages was
observed; on day 39-42 post-conception, erythron-myeloid progenitors
are present in the yolk sac, generating Mac1 macrophages: these
macrophages have a distinct identity compared to HSC-derived
macrophages, as evidenced by their expression of CDH5 and HBE1.[4] Analysis of human embryonic heads showed that most microglia cells originated from primitive yolk sac-derived macrophages.[4]
Importantly, HSC-independent macrophages maintain a peculiar
transcriptomic and epigenetic identity in adult life compared to their
HSC-derived counterparts.[4] Single-cell RNA
sequencing studies have led to the identification of three macrophage
subsets: one of these subsets detected both in mouse and in humans is
characterized by TIMD4, LYVE1 and/or FOLR2 (TFL+) expression: these macrophages are maintained through self-renewal with minimal monocyte input].[5] Atkins et al. reported a human pluripotent stem cell (PSC) in vitro differentiation model for yolk sac hematopoiesis: activin A, BMP4 and FGF2 signaling drives PSC differentiation to KDR+-CD235a/b+
mesoderm cells that generate yolk sac-type primitive erythroid cells
and macrophages via hemogenic endothelial cells and these cells, in
turn, generate erythroid-myeloid progenitors with multipotent
differentiation capacities.[6] This study supports the
hypothesis that the development of hematopoiesis is mediated through
the formation of hemogenic endothelium that in the yolk sac primes a
primitive wave of hematopoiesis associated with transient multipotency,
and in the AGM region, a definitive wave of hematopoiesis, associated
with sustained multipotency and long-term self-renewal capacity (HSC
wave).
Recent studies carried out in zebrafish embryos suggest
that both hemogenic endothelium and aortic endothelium originated from
a common angioblast precursor; this angioblast precursor generates
arterial ECs and HECs: these cells display a spatial separation in the
dorsal aorta where most ECs are in the floor and most HECs in the roof.[7]
The specification of ECs or HECs from the precursor angioblasts is
modulated by ETV2 levels through differential regulation of Fli1a,
Notch, and Sclβ.[7] In addition, enforced RUNX1 expression in ECs promotes the transition to HECs.[7]
The
process through which HE generates hematopoietic stem/progenitor cells
is an endothelial to hematopoietic transition. Intra-aortic
hematopoietic clusters (IAHCs) contain immature HSCs, HSCs, and
committed progenitors.[8] The single-cell
transcriptomic analysis of HE cells, of cells undergoing EHT
transition, and whole IAHCs isolated from mouse embryo aortas allowed
identifying transcription factor networks activated during the HE
transitions to IAHCs.[8]
Studies in mouse and human embryos have led to a partial characterization of HE and IAHCs.[9-11]
One of the studies in mice showed that the transition from ECs to
pre-HECs is characterized at molecular level by increased accessibility
of chromatin regions enriched for SOX, FOX, GATA, and SMAD binding
motifs.[9] The transition from pre-HE to HE is associated with reduced RUNX1 enhancer accessibility.[9] Two populations of IAHCs are generated: an initial wave of lympho-myeloid biased progenitors, followed by precursors of HSCs.[9] The second study in mice was based on a single-cell transcriptomic analysis of the dorsal aorta from embryonic day 8 to E11.[10]
At E10, two types of endothelial cells were observed (EC and HEC) and
HSC-competent HECs; the sequential analysis showed that primitive
vascular endothelial progenitors at E8 first undergo an EC arterial
fate choice, followed by a hemogenic (HEC) fate conversion.[10]
The study on human embryos showed that arterial endothelial cells with
hemogenic potential in wk4 human embryos were characterized by
upregulation of RUNX1, MYB, and ANGPT1 expression; endothelial cells
expressing CD44 were particularly enriched in hemogenic potential.[11] HE cells primed to hematopoietic transition were characterized by transient overexpression of EMCN, PROCR, and RUNXT1.[11]
The
placenta represents, in addition to the yolk sac, another site of
extra-embryonic hematopoiesis. In humans, at weeks 5-6 of development,
the human placenta contains CD34+ cells and erythron-myeloid
progenitors as assayed by the common in vitro clonogenic assays.[12]
At the stage of 5-7 weeks of gestation, the human placenta is a site
for terminal maturation (enucleation) of primitive erythrocytes,
characterized by the synthesis of embryonic hemoglobin.[13]
Despite the early presence of CD34+ cells in the human placenta, HSCs
are detected in this organ only after week 9 of gestation.[14] Repopulating HSCs were detected in the human placenta from 15 to 24 week gestation but were absent at term.[15]
Calvanese
et al. have recently performed a single-cell transcriptome analysis of
human hematopoietic tissues from the first trimester to birth, and
through this analysis, they provide an overview of the whole
development process occurring during embryonic and fetal life.[16] HSC origin in human embryos was tracked to hemogenic endothelial cells characterized by the positivity of ALDH1A1 and KCNK[17]
markers; these cells were regulated from a subset of endothelial cells
identified as pre-hemogenic IL33+ALDH1A1+ endothelial cells; this
process occurred at the level of intra-aortic hematopoietic clusters.[16] The single-cell RNA sequence of CD34+ and/or CD31+
cells isolated from the AGM region of 4.5-5 weeks old human embryos
allowed to define of a molecular signature that characterized HSCs and
included the co-expression of RUNX1, HOXA9, MLLT3, MECOM, HLF and SPINK2
genes.[16] AGM HSCs also possessed a distinct
endothelial signature, characterized by the expression of genes such as
PROCR and EMCN. Clustered cells expressing this RNA expression
signature were observed in AGM, placenta, yolk-sac, umbilical and
vitelline vessels, head, and heart.[16]
Interestingly,
AGM HSCs also possessed a distinct endothelial signature, characterized
by the expression of genes such as PROCR and EMCN.[16]
The analysis of this RNA signature in fetal livers of different ages
showed that the HSC transition to the liver occurs around six weeks of
gestation; furthermore, an extended analysis of gene expression profile
suggested that fetal liver HSCs undergo a maturational process after
eight weeks, progressively exhibiting features like those observed in
adult HSCs with the acquisition of the HSC maturity markers CD133 and
HLA-DR.[16] The comparative analysis of HSCs
populating different sites suggested that extra-embryonic HSCs are
positioned one step downstream from the most immature AGM HSCs and
could colonize the liver to initiate multilineage hematopoiesis.[16]
The
migration of HSCs from the AGM region to the fetal liver is a
fundamental event in the development of hematopoietic tissue because,
in the hepatic environment, the migrated HSCs undergo an intensive
expansion process to generate the HSC pool required to sustain
hematopoiesis for the lifetime of an individual. The fetal liver
remains the main site of hematopoiesis until it is replaced before
birth by the bone marrow. Fetal liver HSCs exhibit several properties
different from adult bone-marrow stem cells, related to higher cycling,
self-renewing activity, and a higher engraftment/repopulating activity
of fetal liver compared to cord blood and bone marrow.[17]
Fetal HSCs concomitantly display two functional properties, such as
high proliferating and repopulating activity, while in adult HSCs,
proliferation and repopulating activity are two inversely related
functional properties.[18,19] Studies in murine fetal
liver HSCs suggest that these cells possess the unique property to
tolerate high proliferation without reducing their multipotential
differentiation capacities through a mechanism seemingly related to
increased DNA damage response observed in fetal HSCs compared to
postnatal HSCs.[20] Gene expression studies have shown that a purified cell population enriched in long-term repopulating HSCs (GPI-80+-enriched
fraction) purified from fetal liver exhibited an enhanced expression of
some genes related to aryl hydrocarbon receptor (AHR) family and of RGCC, LMNA and ID genes.[21] The most expressed gene of the AHR family, TIPARP, is a negative regulator of AHR activity and AHR inhibitors favor HSC expansion.[21] LMNA gene
encodes the nuclear lamina protein lamin A/C and its expression is
preferentially observed in repopulating HSCs and declines with aging.[21] ID and RGCC
genes are related to cell-cycle control and their expression of fetal
HSCs may contribute to the increased proliferation and engraftment
capacities of fetal HSCs.[21]
Fetal liver HSCs
migrate and seed the bone marrow, where these cells, in the presence of
a different microenvironment, generate a program of definitive
hematopoiesis, promoting an extensive myeloid diversification with the
production of neutrophils, eosinophils, and basophils and dendritic
cell subsets; furthermore, in fetal bone marrow, B lymphoid elements
undergo a consistent expansion, starting a process of generation of the
adaptive immune repertoire, subsequently completed in post-natal and
adult bone marrow.[22]
Fetal and adult
hematopoietic stem cells display different proliferation rates (higher
in fetal compared to adult HSCs), gene expression profiles, and lineage
differentiation biases. The molecular mechanisms responsible for these
developmental-related changes in HSCs are largely unknown. However, a
recent study provided evidence that the transition from proliferating
fetal HSCs to quiescent adult HSCs is mainly related to intrinsic
mechanisms, gradual events independent of the microenvironment,
occurring in a stochastic manner and, at least in part, mediated by the
type I interferon.[23]
HSC self-renewal.
HSCs have the unique property of replicating to create two daughter
identical cells and to differentiate to generate a progeny of
hematopoietic cells, and through these essential biological functions,
they are able to both maintain and restore blood cell production.
However, adult HSCs are characterized by a very low replication rate.
In humans, the analysis of the changing ratio with the age of
maternal/paternal X-chromosome phenotypes in blood cells from females
allowed to define the replication rate of human HSCs, estimated on
average of one replication every 40 weeks, with a range comprised
between 25 to 50 weeks.[24]
Both intrinsic
mechanisms mediated by molecular regulators and extrinsic mechanisms
mediated by environmental signals regulate the quiescence of HSCs;
these mechanisms include transcription factors, cell-cycle regulators,
microenvironmental mediators, and epigenetic factors. All these
mechanisms imply a peculiar regulation of cell metabolism of HSCs, and
the transition from quiescence to activation is accompanied by marked
changes in cell metabolism (protein synthesis, oxidative
phosphorylation, glycolysis, autophagy). Recent studies have identified
some important regulators of HSC quiescence/proliferation.
Cell-cycle
components are important regulators of quiescence/proliferation of
HSCs. The HSC pool is heterogeneous concerning the repopulating
capacities, and HSCs can be subdivided into long-, intermediate- and
short-term (LT, IT, and ST) HSCs. Both LT-HSCs and ST-HSCs are
quiescent but differ in the timing of exit from quiescence that is
longer for LT-HSCs compared to ST-HSCs, and this difference seems to be
related to the level of cyclin-dependent kinase 6 (CDK6) that is higher
in ST-HSCs than in LT-HSCs.[25]
The study of
transcriptional signatures of human quiescent LF-HSCs showed some
remarkable differences compared to those observed in activated ST-HSCs:
in fact, the Act/HSPC (activated/hematopoietic stem progenitor cells)
signature is observed in activated ST-HSCs and is characterized by the
activation of CCCTC-binding factor (CTCF) binding sites; silencing of
CTCF expression derepressed expression of stemness genes and maintained
the long-term repopulating activity of quiescent HSCs.[26]
These observations suggest that chromatin interactions mediated by CTCF
control the transition of HSC from quiescence to an activated
condition.[26]
Other studies have supported a major role of lysosomes in the control of HSC quiescence.[27-28]
These organelles are not simply catabolic degradation structures but
are also major signaling centers for complex molecular assembly. A
first study showed a connection between lysosomal activity and
metabolic activity of HSCs.[27] Quiescent,
highly-repopulating HSCs are characterized by low mitochondrial
membrane potential (MMP), this potential being higher in primed,
activated HSCs.[27] Cycling, primed HSCs are
characterized by high glycolysis, and inhibition of glycolytic activity
in these cells induces an enhancement of their repopulating capacity.[27]
Quiescent HSCs are characterized by the presence of large, scarcely
active lysosomes. Inhibition of lysosomal activation in HSCs suppresses
glucose uptake and further stimulates their repopulating activity.[27]
In the second study, Gracia-Prat et al. explored the peculiar
mechanisms of lysosomal activity observed in HSCs; particularly, they
examined how transcription factor EB (TFEB) and MYC regulate the
catabolic and anabolic processes required for HSC quiescence or
activation.[18] Particularly, TFEB-mediated induction
of the endolysosomal pathway triggers membrane receptor degradation,
limiting HSC metabolic activation and mitogenic activation, promoting
stem cell quiescence and self-renewal; in contrast, MYC promotes
biosynthetic processes and inhibits lysosomal catabolic functions, thus
driving HSC activation.[28]
Some transcription
factors are selectively expressed at the level of HSCs and play a key
role in the maintenance of the self-renewal capacity of these cells.
Thus, Hepatic Leukemia Factor (HLF) is highly expressed in normal HSCs
and multipotent progenitors (MPP) and is rapidly lost during the
differentiation of these cells.[29] HLF-deficient
mice are viable with normal hematopoietic parameters, including a
normal HSC pool. However, when these mice were challenged through
transplantation showed an impaired capacity to reconstitute
hematopoiesis and were gradually exhausted after transplantation.[30]
In mouse embryos, HLF expression is limited to intraembryonic HSCs
(intra-aortic and fetal liver) but not to extra-embryonic HSCs (yolk
sac).[31]
HLF expression marks human HSCs at all stages of hematopoietic
development, from intra-aortic embryonic hematopoiesis to cord blood
and bone marrow.[32]
MLLT3 is a transcription
factor whose expression is highly enriched in fetal, neonatal, and
adult human HSCs and is gradually decreased in culture, determining a
loss of HSC activity.[33] Enforced expression of
MLLT3 in human HSCs reduces this decrease in stem activity during in
vitro culture and potentiates about tenfold the expansion of
repopulating HSCs.[33] Importantly, fusion proteins
involving MLLT3 have the property to transform normal HPCs into
leukemic stem cells. Another factor exerting an essential role in the
control of HSC self-renewal is the RNA-binding protein Musashi 2
(MSI2); this factor plays a key role in the expression of master
regulators of HSCs through a post-transcriptional mechanism:
particularly, MSI2 expression promotes HSCs and HPCs proliferation
through downregulation of aryl hydrocarbon receptor (AHR) signaling.[34]
The expression of MSI2 in the HSCs is promoted by two transcription
factors, PLAG1 and USF2, which are able to bind to the MSI2 gene
promoter resulting in increased gene transcription.[35]
In line with these findings, an AHR small molecule antagonist,
StemReginin (SR1) promotes ex vivo expansion of transplantable human
HSCs/HPCs: a phase I/II clinical trial using SR1-expanded cord blood
stem/progenitor cells for transplantation showed faster neutrophil
engraftment compared to unmanipulated stem/progenitor cells, but not an
improvement in hematopoietic recovery.[36] A small molecule screen allowed to identify pyrimidoindole derivatives (UM171) as stimulators of cord blood HSC expansion in vitro.
Short-term expansion of cord blood HSCs/HPCs using UM171 is a safe and
feasible strategy and is under active clinical evaluation in the
treatment of patients with hematological malignancies lacking a
suitable HLA-matched bone marrow donor.[37]
Importantly, the UM171 expansion markedly improved the usability of CB
units stocked in CB banks, allowing the use of smaller CB units for
transplantation purposes.[38]
HSC differentiation.
Hematopoiesis is a finely regulated process of cell differentiation
through which HSCs generate blood elements of all hematopoietic
lineages belonging to myeloid and lymphoid lineages. Historically, two
theories have been proposed to explain the process of HSC commitment:
an instructive model and a stochastic model.[39-41]
Following the instructive model, the HSC differentiation choices are
driven by external signals mediated by cytokines. According to the
stochastic model, the commitment of HSCs is promoted by spontaneous,
stochastic variations of cell phenotypes that are selected through
selective signals/mechanisms mediated by cytokines.[39-41]
A remarkable difference between the two models is related to the level
of cell heterogeneity that is expected to be high in the stochastic
model and low in the instructive model; the stochastic model suggests
the existence of cell-to-cell variability, particularly at the early
steps of hematopoietic differentiation before the occurrence of
selective processes.
The hematopoietic system represents a highly
complex biological system of cell differentiation, leading to the
controlled generation of different hematopoietic blood cell types
through the differentiation of small populations of HSCs and
Hemopoietic Progenitor Cells.[39-41] Historically,
the study of hematopoietic cell differentiation was promoted by the
isolation of single stem/progenitor cells grown in vitro
using colony assays or transplantation into myeloablated mice. These
studies allowed to define HSCs and HPCs with various differentiation
potentials into a cell differentiation hierarchy with HSCs at the apex
and mature cell types at the bottom; between these two extremes, there
are many defined intermediate stages, the first one being represented
by the bifurcation of HSCs into myeloid and lymphoid branches, through
the generation of a common myeloid progenitor (CMP) and a common
lymphoid progenitor (CLP); the subsequent steps are represented by the
generation of unilineage hematopoietic progenitors, generating the
first undifferentiated precursors of the various blood lineages
entering into the maturation compartment.[39-41]
During the years 2005 to 2015, this model was integrated through new
acquisitions that have led to considering the HSC compartment more
heterogeneous both in terms of self-renewal and differentiation
capacities, the presence of a multipotent progenitor LMPP (Lymphoid
Myeloid Pluripotent Progenitor) linking the myeloid and lymphoid
lineages below the HSC stage. From 2016 onwards, a continuum model of
hematopoietic differentiation was proposed, suggesting that
hematopoietic lineage commitment is more reliably represented by a
continuous process of differentiation trajectories rather than by
stepwise differentiation series of distinct hematopoietic progenitor
cell populations.[39-41] The development of new
technologies of single-cell transcriptomics and proteomics, as well as
lineage tracing and functional studies, have led to the important
conclusion that there is a continuum of the lineage commitment from
HSCs up to mature blood elements, with most of the lineage choices
being promoted at the level of HSCs and MPPs.[39-41]
The
development of fluorescently labeled HSCs from transgenic donor mice
allowed analyzing their differentiation capacities along five different
hematopoietic lineages (erythroid, megakaryocytic, neutrophilic,
monocytic, B-lymphoid, and T-lymphoid cells). This assay identified a
class of myeloid-repopulating progenitor cells able to generate a cell
progeny composed of platelet (MkRP), platelet-erythrocyte (MERPs), or
platelet-erythrocyte-neutrophil-monocyte lineages (CMRPs). These
repopulating progenitors may be originated through direct
differentiation of HSCs through asymmetric cell divisions generating
one multipotent stem daughter cell and one lineage committed
repopulating daughter progenitor.[42] These
repopulating progenitors display the capacity of repopulating in vivo
part of the hematopoietic system but cannot be serially transplanted,
thus indicating that they do not possess self-renewal capacity.[43,44]
Single-cell
and HSC transplantation cell tracking experiments supported a
consistent differentiative heterogeneity of hSCs with the evidence
about the existence of some myeloid-biased HSCs and some HSCs adopting
a fate towards effective and stable replenishment of a
megakaryocyte/platelet lineage tree but no other cell lineages.[44]
These findings were confirmed by Rodriguez-Fraticelli and coworkers,
who used transposon tagging to clonally trace the fates of progenitors
and stem cells in native hematopoiesis; this analysis showed the
existence of some long-term HSCs are a source of
megakaryocyte-restricted progenitors, suggesting that in mice the
megakaryocyte lineage id the predominant fate of long-term HSCs.[45] Finally, Upadhaya et al. have used a system for in vivo
genetic labeling of HSCs, combined with high-dimensional single-cell
analysis to characterize the kinetics of HSC differentiation under
native hematopoiesis; this study showed early emergence of
megakaryopoiesis, the subsequent divergence of erythroid and myeloid
development from lymphopoiesis.[46]
Two studies
have combined lineage tracing and single-cell RNA sequencing to obtain
simultaneous evaluations of clonal history and cell identity in murine
hematopoiesis.[47,48] This method elucidates how
single HSCs and their corresponding progeny develop through the
continuous differentiation steps shown by single cell transcriptomics.
The results of the study of Weinreb et al. showed that in the
hematopoietic differentiation process, different sequences of molecular
events might lead to the same differentiation terminal event; a notable
example is given by monocytes. Furthermore, sister cell experiments
provided evidence that cells with very similar gene expression profiles
can be committed to different cell fates, thus suggesting that
transcriptional networks alone are insufficient to determine the
potential of the hematopoietic cell towards different fates.[47]
The second study, performed by Pei et al. using a similar
methodological approach, showed that HSCs are heterogeneous, with
differentiation-inactive, multilineage, and lineage-restricted HSC
clones corresponding to different regions of the transcriptional
landscape of hematopoiesis.[48]
Adult hematopoiesis.
Studies in human hematopoietic cells support the revised model of
hematopoietic differentiation based on the observation that HSC and
multipotent progenitors progressively acquire lineage biases along
various differentiation fates. Through an integrative analysis of
transcriptomic, flow cytometry, and functional data at a single-cell
level, Velten et al. explored the early steps of hematopoietic cell
differentiation at HSC and HPC stages: this analysis supported the
existence of a continuum of low-primed undifferentiated HSCs and HPCs;
in this context, the separation of megakaryocytic/erythroid and
lympho-myeloid represented the main routes of lineage specification.[49]
According to these findings, a model of human hematopoietic
differentiation was proposed based on a continuum and transient state
of lineage commitment at the stem/progenitor cell compartment level.
Buenrostro et al. have used another approach to explore a regulatory
landscape of human hematopoietic differentiation through single-cell
epigenomic analysis based on an assay for Transpase Accessible
Chromatin with high single-cell RNA sequencing on ten phenotypically
defined HSC and HPC subsets; this analysis showed the existence of an
association of changes in chromatin accessibility with changes in
transcription factor expression during differentiation.[50] Particularly,
the transcription factor-chromatin accessibility variability in HSCs
follows megakaryocytic-erythroid/lymphoid pathways and shows the
existence of two granulo-monocytic subsets: the more primitive and
least-primed subset of HSCs and HPCs is characterized by low cell
cycling activity, low RNA content, low gene expression, low cellular
respiration and expression of HOX motif; the stem/progenitor cells
primed along cell differentiation display an increased cell cycling
activity, increased gene expression and gradient of expression of
transcription factor regulators ID3, CEBP and GATA1 toward lymphoid,
myeloid and erythroid differentiation, respectively.[50]
Additional
studies strongly supported the continuum nature of human hematopoiesis.
Thus, Psaila et al. have performed a single-cell analysis of human
megakaryocyte-erythroid progenitors isolated from cord blood, showing a
consistent differentiation capacity of these cells: pre-MEPs
predominantly display erythroid-myeloid differentiation but with
residual myeloid potential; MEPs were strongly biased to erythroid
differentiation; Mk-MEPs primarily showed megakaryocytic
differentiation capacities.[51] In addition,
Karamitros et al. performed a single-cell analysis of lympho-myeloid
progenitors present in human cord blood, indicating that
lymphoid-primed multipotential progenitors (LMPPs), granulo-monocyte
progenitors (GMPs) and multi-lymphoid progenitors (MLPs) are
functionally unipotent, bipotent and multipotent and transcriptionally
heterogeneous.[52] Single-cell analysis of human
hematopoietic progenitors further supported the continuum
differentiation model of human hematopoiesis.[53]
These
studies have supported the view that: (i) lineage commitment occurs at
early stages of hematopoietic differentiation from primed HSCs; (ii)
hematopoietic progenitor cells are highly heterogeneous and classical
erythroid-megakaryocytic, and granulo-monocytic progenitors englobe
oligopotent progenitor cells; (iii) the two main branches of
hematopoietic differentiation involve a GATA2-positive branch of
erythroid, megakaryocytic and eosinophilic/basophilic/mast cell
progenitors and a GAAT2-negative branch of lympho-myeloid progenitors,
including progenitors of neutrophils, monocytes, and dendritic cells.
Single-cell
transcriptomic and proteomic studies, coupled with flow cytometry
analysis, have allowed defining some immunophenotypic profiles
associated with differentiation properties of human stem/progenitor
cells. Human hematopoiesis is mostly sustained by CD34-positive cells,
a cell surface marker identifying the large majority of HSCs and HPCs.
Using a combination of cell surface markers, Notta and co-workers
distinguished a subset of CD34+CD38-CD90+CD49f+ cells enriched in functional HSCs and a subset of CD34+CD38-CD90-CD49f- cells enriched in MPPs.[54] The combination of single-cell transcriptomic studies and xenotransplantation assays allowed to identify CD34+CD38-CD45RA-EPCR+
cells as a cell subset highly enriched in multipotent HSCs: 1/3 of
these cells display functional properties of repopulating HSCs;
furthermore, these cells are slow cycling and exhibit a low metabolic
profile.[55] EPCR+ cells are at the apex of a HSC/HPC hierarchy: CD34+CD38-CD90+ cells display a lower stem repopulating capacity, estimated in the order of 1/119 cells; CD34+EPCR+ cells can generate CD34+EPCR- cells but not the contrary; CD34+EPCR- cells can generate MPPs and more committed progenitor cells.[55] Unicellular transcriptomic studies showed that CD34+EPCR+ cells display a multipotent/stem profile with a moderately myeloid biased phenotype.[55] Importantly, CD34+EPCR+ cells resulted in being relatively homogeneous, as expected for cells that can be located at the apex of hematopoiesis.
The
techniques that simultaneously measure mRNA and surface protein
expression in single cells allowed to define of cytometry assays
carefully reflecting single-cell RNA sequencing-based molecular data at
the level of various hematopoietic differentiation stages.[56]
Aksoz et al. have explored the individual transcriptome profile of
human bone marrow hematopoietic cells highly enriched in HSCs (CD34+CD38-CD90+CD45RA-)
and showed that these cells displayed a consistent heterogeneity: HSCs
with multilineage signatures correlated with high cellular output
signatures, whereas platelet bias and low-cellular-output signatures
correlated at the single-cell level.[57]
One of
the fundamental biologic properties of HSCs consists in their capacity
to undergo symmetrically (with the generation of two HSCs or two HPCs)
or asymmetric (with the generation of one HSC and one HPC) divisions;
through this unique biologic property, HSCs can adapt to the
regenerative need of hematopoietic tissue. Asymmetric cell divisions
imply the unequal repartition of cellular components during cell
division, a condition that determines a different cell fate of daughter
cells. Recent studies have suggested that asymmetric cell divisions are
determined by an asymmetric reorganization of the cytoskeleton during
cell division, determining a condition of cellular polarization with
consequent asymmetric distribution of cell fate determinants. Studies
carried out in murine HSCs have shown that the asymmetric distribution
of the cellular degradative machinery (including lysosomes,
autophagosomes, mitophagosomes, and the NUMB-can protein) is associated
with the activation/differentiation in that the daughter cells
receiving this machinery maintain a stem cell condition.[58]
The asymmetric distribution of lysosomes plays a relevant role in the
mechanism of cell fate determination of human HSCs. In fact, Loeffler
et al. have shown that human HSCs, undergoing asymmetric divisions,
receive more lysosomes at the level of daughter cells, maintaining
their stemness condition. In contrast, daughter cells receiving fewer
lysosomes are more prone to undergo cell differentiation.[59]
Interestingly, in addition to lysosomes, active mitochondria can also
be asymmetrically partitioned: daughter cells receiving more lysosomes
tend to receive fewer active mitochondria during an asymmetric HSC
division.[59]
The model of continuum
hematopoietic differentiation implies a possible involvement of HSCs
and MPPs in the homeostatic maintenance of hematopoiesis. Initial
studies performed in mice have supported the view that unperturbed
hematopoiesis is mainly maintained by MPPs and committed progenitor
cells but not by HSCs.[60-61] However, recent studies
have challenged this view, providing evidence through different in vivo
labeling systems that murine adult HSCs considerably contribute to
steady-state, unperturbed hematopoiesis.[62-64]
Particularly, Chapple et al. showed that adult murine HSCs contribute
robustly to steady-state hematopoiesis, with a major efflux toward the
myeloid lineages compared to lymphoid lineages;[63]
Sawai et al. showed that murine HSCs give a major contribution to all
blood lineages, including myeloid cells and lymphocytes, except tissue
macrophages and B1a lymphoid cells;[62] Sawen et al.
provided evidence that HSCs contribute to native hematopoiesis, but the
HSC contribution to multilineage hematopoiesis declines with increasing
age.[64]
Role of HSCs in hematopoietic reconstitution after bone marrow transplantation.
Recent studies have explored the contribution of HSCs to hematopoietic
reconstitution after bone marrow transplantation. These studies took
advantage of the clonal tracking studies carried out in gene
therapy-treated patients exploiting vector integration sites (ISs) as
molecular markers for monitoring and assessing the dynamics of
hematopoietic reconstitution induced by infusion of bone marrow cells
genetically manipulated.[65-66] The analysis of ISs
at the level of different blood elements after transplantation allowed
to define the kinetics of blood cell generation from individual HSCs.
Studies carried out in murine systems have shown that individual ISs
are, in the majority of instances, present in either myeloid or
lymphoid blood cells and only in a few cases are shared in both these
cell types.[67] Lu et al. have used a
high-sensitivity quantitative cloning tracking technology to explore
HSC commitment after transplantation in the absence of conditioning and
after conditioning with irradiation or with anti-c-kit antibody
treatment.[68] Under conditions of unperturbed
hematopoiesis, donor HSCs homogeneously contribute to the various
stages of hematopoiesis, thus suggesting that HSC lineage commitment
develops with an equal contribution from each clone.[68]
At variance of unconditioned mice, in irradiated mice, a small fraction
of engrafted HSC clones constantly expanded faster than other clones
during differentiation and generated most neutrophils and
B-lymphocytes; it was estimated that in both irradiated and c-kit
conditioned mice, about 50% of total neutrophil and B-lymphoid
production is generated from the differentiation these few HSC dominant
clones. [68] The conditioning regimens (such as the
irradiation dose) and the transplantation conditions (such as the
number of helper cells used in the transplantation procedure)
consistently induce HSC lineage bias; lineage bias is originated from
dominant differentiation events occurring at distinct lineage
commitment steps.[68]
Studies in non-human
primates have confirmed the results observed in mice. A quantitative
method for the assessment of the self-renewal and differentiation
patterns of lentivirally-labeled macaque HSCs allowed to show that: (i)
individual HSC clones may display stable myeloid or lymphoid bias for
many years; (ii) output of individual HSCs and HPCs was stable for many
years, with very limited evidence of clonal succession.[69]
In
humans, the study of patients treated with genetically manipulated
HSCs/HPCs allowed the unique opportunity to explore the dynamics of
human hematopoietic reconstitution at the clonal level, exploiting the
capacity to identify the differentiated progeny at clonal level through
the analysis of the insertion of the therapeutic vector in a unique
genomic site.[65] Furthermore, the longitudinal study
of individual clones of hematopoietic cells for many years after
transplantation of genetically modified HSCs/HPCs allowed to define
peculiar patterns of clonal dynamics during early and steady-state
reconstitution phases: in the initial phase of engraftment, the
generation of myeloid cells is ensured by committed myeloid
progenitors, such as CMPs and GMPs; after this phase of engraftment
there is the early phase of reconstitution that is ensured during the
first 18 months post-transplantation by short term-repopulating HSCs
and MPPs and from 18 to 24 months post-transplantation by long-term
repopulating HSCs; the phase of steady-state hematopoiesis, occurring
after 24 months post-transplantation, is ensured by long-term
repopulating HSCs.[65-66] These studies also showed that lymphoid-biased
stem/progenitor cells may be capable of long-term survival and can be
maintained independently of their generation from HSCs.[66] Another study
based on the analysis of patients undergoing HSC/HPC gene therapy for
Wiscott-Aldrich Syndrome or beta-hemoglobinopathies provided evidence
that HSCs/HPCs can be classified into three groups according to their
clonal lineage outputs, reflecting stable, distinct differentiation
programs: myeloid-dominant, lymphoid-dominant and balanced.[70]
Bone Marrow Transplantation Derived Observations.
The observations made during the last 20 years on bone marrow
transplantation also support the existence of a decrease in HSC
function associated with aging. In 2001, a retrospective analysis of a
large cohort of patients who have undergone bone marrow transplantation
(BMT) and analyzed various donor-related parameters that could affect
overall survival (OS), disease-free survival (DFS), acute and chronic
graft-versus-host disease (GVHD), engraftment and relapse. Among the
various donor parameters evaluated, age was the only donor trait
significantly associated with OS and DFS for both HLA-matched and
HLA-mismatched transplants: the use of younger donors lowers the
incidence of GVHD and improves survival after BMT.[71]
The same authors confirmed these findings through the analysis of
11,039 BMTs from unrelated donors; after adjusting for patient disease
and transplantation characteristics, survival was better for
transplantations of grafts from young donors: for every 10-year
increment in donor age, there was a 5.5% in the hazard ratio for
overall mortality.[72] The study of a cohort of 889 patients who have
undergone haploidentical BMT showed that increasing donor age by decade
was associated with poorer OS. In addition, worse progression-free
survival and a higher frequency of GVHD; these less-favorable results
with older donors were related to worse non-relapse mortality.[73]
When
bone marrow cells are infused in patients for transplantation, only
5-30% of HSCs get home to the bone marrow, while the remainder is lost
and distributed in the lung, spleen, and liver. The relatively low
homing/engraftment of infused HSCs/HPCs is more pronounced in clinical
studies carried out using gene-edited stem/progenitor cells, a
phenomenon related to the gene editing procedure or the use in these
studies of purified populations of HSCs/HPCs deprived of T helper
cells.[74] To bypass this limitation, some clinical studies have used the
intra-bone administration of genetically-corrected autologous
HSCs/HPCs, a procedure that accelerates the kinetics of hematopoietic
recovery post-transplantation.[75] Transplantation studies carried out in
immunodeficient mice showed that intra-bone marrow transplantation of
HSCs and HPCs, but not of LT-HSCs: the higher engraftment of HPCs
compared to HSCs seems to be due to a higher expression of the CXCR4
receptor on HPCs compared to HSCs.[76] The removal of HPC and the
transplantation of an HSC-enriched cell population intra-bone improved
the engraftment HSCs.[76] Induction of a higher expression of CXCR4 on
HSCs improves homing and engraftment of these cells.[76]
Aging of Hematopoietic Stem Cells
Aging
is associated with a decline and alterations of mature blood cells and
there is evidence that a combination of intrinsic and extrinsic
mechanisms is responsible for these changes. Growing evidence suggests
that changes occurring at the level of HSCs are, in large part,
responsible for the aging of the hematopoietic system.
At the
level of intrinsic mechanisms, several studies have characterized the
changes occurring at the level of the HSC compartment in mice and in
humans.
The studies carried in murine HSCs have shown that: (i)
the number of HSCs increases with age, but the competitive repopulating
activity of these cells declines, thus suggesting a decrease of HSC
biologic function associated with age; (ii) aging mouse displays a
decrease in lymphopoiesis, associated with an increase in myelopoiesis,
changes that are at least in part related to a prevalence of myeloid
biased HSCs in elderly mice.[77-79] A fundamental study by Ganuza et al. used a non-invasive in vivo
color-labeling system to evaluate the changing clonal complexity of
steady-state hematopoiesis during murine lifespan.[80] Steady-state
hematopoiesis is characterized by a mechanism of clonal instability in
which pools of HSCs increase and decrease their contribution to
hematopoiesis during life.[80] The clonal complexity of hematopoiesis consistently decreases with age, particularly at the HSC and MPP cell compartments.[80]
Aging was associated with a consistent increase in the functional
heterogeneity of HSCs and with a reduction in their repopulating
activity.[80] Serial transplantations exerted marked
effects on hematopoiesis, as evidenced by: (i) a marked reduction of
clonal diversity; (ii) an increase of clonal instability; (iii) an
increase of mutational burden, a phenomenon much more evident in aged
bone marrow than in young bone marrow.[80]
Studies carried out in human bone marrow showed an increase of cells with an HSC immunophenotype (CD34+CD38-CD90+CD45RA-) among CD34+ cells associated with aging.[81] Immunophenotypic evaluation of HPC subsets showed a significant decrease of CLPs in elderly bone marrow.[81] Elderly HSCs displayed a reduced capacity to generate a lymphoid progeny and an increased myeloid-differentiation capacity.[81] Thus, aged human HSCs are less quiescent and exhibit myeloid-biased differentiation potential compared to young HSCs.[81]
At the gene expression level, elderly HSCs transcriptionally
up-regulate genes associated with cell cycle, myeloid lineage
specification, and myeloid malignancies.[81] Other studies confirmed that aged human BM displays a reduced content of CLPs, associated with increased frequencies of MEPs.[82] Kuranda et al. confirmed these results showing an increased frequency of CD34+CD38-
cells in the elderly bone marrow; however, xenotransplantation
experiments in NOD/SCID mice showed that the number of repopulating
HSCs does not change with aging.[83] Elderly bone marrow HSCs showed a reduced myeloid reconstitution.[83]
Studies
in murine HSCs have explored the cycling activity of HSCs. In this
context, a study by Kowalzyk et al. provided evidence of a consistent
decrease of the G1/S phase cells among old HSCs compared to young
HSCs (7% vs. 22%, respectively); this finding was interpreted as
evidence that aged HSCs traverse through G1 faster.[84]
Ethinyl deoxyuridine (EdU) is a chemical compound used for fast and sensitive detection of DNA synthesis.[85]
Kovtonyuk et al., using this technique, measured the cycling status and
the compartment sizes of HSCs, HPCs, and granulocytes in mice of four
different ages: 3-week, 2-month, 1-year, and 2-year-old mice.[86]
The compartment size gradually increased with age from 3 wk old mice to
2-year-old mice; in contrast, the cycling activity of HSCs decreased
progressively and significantly with age.[86] Thus, an increase of HSC in dormancy is responsible for the increased size of the HSC pool in aging.[86]
Cdc42
is a small Rho GTPase present in two activation states: an active
guanosine-triphosphate (GTP)-bound state and an inactive guanosine
diphosphate (GDP)-bound state. Cdc42 expression is altered in aging
HSCs, and this event causes a loss of polarity of these cells.[87]
In fact, Florian et al. showed that Cdc42 expression is high in aging
HSCs and correlates with a loss of polarity in aged HSCs.[87]
Experiments with pharmacologic inhibitors of Cdc42 restored the
polarization of aged HSCs and the levels and spatial distribution of
histone H4 lysine 16 acetylation.[87] The loss of polarization induces
several effects on aged HSCs, such as preferential self-renewing
symmetric divisions, resulting in the degeneration of daughter HSCs
with reduced regenerative potential and lymphoid differentiation
capacities.[88] A recent study showed that also the
aging of human HSCs is associated with changes in Cdc42 activity; Amoah
and coworkers showed that: (i) the number of aged HSCs increased, and
these cells display a delayed response in vitro
to cytokine stimulation; (ii) Cdc42 activity in aged human HSCs is
increased and correlates with an increased number of HSCs; (iii) the
frequency of HSCs exhibiting a polarization for Cdc42 and tubulin
decreases with aging; (iv) treatment of aged human HSCs with casin, a
Cdc42 inhibitor, restores cell repolarization and rapid response to
cytokines.[89]
DNA damage is universally
considered one of the fundamental mechanisms driving tissue aging: DNA
damage affects most aspects of the aging phenotype.[90]
Proliferating progenitor cells are dependent on reliable homologous
recombination (HR) pathway for DNA reparation, while quiescent HSCs use
a different mechanism of DNA reparation called the error-prone
nonhomologous end joining (NHEJ) repair pathway.[91]
This mechanism exposes quiescent stem cells to the risk of genomic
rearrangements that can persist and contribute to developing
hematopoietic abnormalities.[91] The condition of HSC
quiescence and the concomitant decrease of DNA repair and response
pathways are conditions that favor DNA damage accumulation in HSCs
during aging.[92] Furthermore, cycling old HSCs
display increased levels of replication stress activity, associated
with cell cycle defects and chromosome gaps and breaks: this condition
determines a functional decline of aged HSCs.[93]
In
murine HSCs, the induction of the quiescence state in response to
conditions that model physiological stress, such as chronic blood loss
or infection, provoked the gradual decrease of normal HSC activity.[94] This observation suggests a link between physiological stress and DNA damage in normal HSCs.[94]
5-hydroxymethylcytosine
binding, cell-specific (HMCES), is a gene enabling single-stranded DNA
binding activity, involved in cellular response to DNA damage stimulus
and protein-DNA covalent cross-linking. HMCES represents a guardian of
genome integrity and long-term self-renewal capacity of HSCs during the
stress response, such as response to myeloablation and transplantation.[95]
The
presence and the level of expression of DNA damage repair mechanisms is
a major determinant of radiosensitivity of hematopoietic cells. In
addition, marked differences in radiation sensitivity exist between the
lymphoid and myeloid cells, with lymphoid cells being significantly
more sensitive than cells of the myeloid lineage; in the myeloid
lineage, monocytes/macrophages are the most radio-resistant cell types.[96]
Irradiated
human HSCs/HPCs, but not committed HPCs rapidly undergo apoptosis
through an ATM-dependent process. This apoptotic process is inhibited
by interaction with bone marrow stromal cells.[97] HSCs/HPCs showed reduced NHEJ processes compared to committed HPCs.[97]
The interaction with stroma does not affect the level of NHEJ activity.
10% of HSCs/HPCs surviving to irradiation display clonal chromosomal
aberrations.[97]
The Discovery of Clonal Hematopoiesis of Indeterminate Potential (CHIP)
During
their lifespan, cells divide and may accumulate somatic mutations: most
of these mutations are neutral; however, a minority of mutations may
increase cellular fitness and confer a growth advantage, resulting in
clone expansion. This phenomenon was observed in many normal tissues
and increases with age.[98] A median somatic mutation frequency of 2.8x10-7
per bp was estimated using human dermal fibroblasts; this frequency of
mutational events is higher than that observed in germline tissues,
calculated in the order of 10-8 per bp.[99]
Whole exome sequencing studies were used to quantify and track somatic mutations in normal hematopoietic cells.[100,101]
These studies were based on isolating single stem/progenitor cells by
fluorescence-activated cell sorting and expanding these cells in vitro to generate clonal populations of hematopoietic cells sufficient to permit extensive whole genome sequencing.[100,101]
According to the results obtained, it was estimated in one study
carried out on a single subject a mutational accumulation rate of 11.7
mutations per year[100] and in the other study,
carried out on seven healthy donors ranging in age from 0 (umbilical
cord blood) to 63 years, a constant accumulation rate of 14 mutations
per year.[101] Importantly, blood mutations occurred in a characteristic trinucleotide context.[101]
The analysis of 11 healthy subjects of different ages showed a positive
correlation between the number of base substitutions in hematopoietic
stem/progenitor cells and the age of the donors, with an accumulation
of 14.6 base substitutions per year of life.[102]
The analysis of the mutational spectra of normal stem/progenitor cells
showed two predominant mutational signatures: hematopoietic
stem/progenitor signature (previously identified as a signature
predominantly observed in normal stem/progenitor cells) and single base
substitution signature 5.[103] Most point mutations
consist of 1 bp deletion of a C or a T and 1 bp insertion of a T, a
process commonly ascribed to polymerase slippage during replication of
the replicated DNA strand.[104] In addition to mutational events, chromosomal studies have shown some recurring aging-related alterations.
A
notable example is loss of the Y chromosome, detected in 43% of men
older than 70, while sub chromosomal rearrangements have been observed
in 2-3% of older individuals.[105] The complex
cellular and molecular mechanisms orchestrating and regulating cell
division represent a major source of mutational events and chromosomal
abnormalities generation].[106] Furthermore, genome integrity is also
compromised by other molecular mechanisms, such as defective DNA-repair
mechanisms.[106] Endogenous and exogenous mutagens favor the generation of the genetic alterations ineluctably associated with aging.[106]
Among
the various forms of tissutal clonal expansion, clonal hematopoiesis
was intensively investigated. The term clonal hematopoiesis of
indeterminate significance (CHIP) was introduced to describe
individuals with a hematologic malignancy-associated somatic mutation
in peripheral blood or bone marrow cells but without any other
diagnostic criteria for a hematologic malignancy.[107]
CHIP must be distinguished from myelodysplastic syndromes (MDS) by the
absence of cytopenias and the diagnostic morphologic criteria for
dysplasia that define MDS and can be considered analogous to monoclonal
gammopathy of undetermined significance and monoclonal B-lymphocytosis.[107]
Initial
studies aiming to measure the ratios of X-inactivation if females led
to the identification of age-associated skewing (AAS) in blood cells,
particularly at the level of the myeloid compartment; a potential cause
of AAS is the acquisition of somatic mutations inducing a growth
advantage, with consequent clonal hematopoiesis: in line with this
hypothesis, Busque et al. reported the occurrence of TET2 and DNMT3A mutations in one of three individuals with AAS; the subsequent exploration of 179 older women with AAS showed the presence of TET2 mutations (nonsense, missense and frameshift mutations) in 5.6% of these subjects.[108]
Three
pivotal studies reported in 2014 the main biological features
associated with CHIP: CHIP-associated mutations are rare in individuals
younger than 40 years of age but rose significantly with age,
particularly after 60 years; the risk of leukemic progression is
influenced by VAF, the number and the type of mutant genes; the
majority of mutations were observed at the level of three genes, DNMT3A, TET2 and ASXL1;
the presence of CHIP was associated with an increased risk of
hematologic malignancies, an increase in all-cause mortality and in the
risk of coronary heart disease.[109-111]
As it
will be discussed, CHIP is associated with aging and is
over-represented in some diseases irrespective of age, where it may
contribute to disease outcomes and all-cause mortality.
Age-associated CHIP, gene mutations. In 2014, three studies reported the age-related accumulation of mutations in leukemia-related genes (DNMT3A, TET2, ASXL1)
using NGS with variant allele frequency >0.02 (2%), due to the error
rate. Benign mutant clones were rarely detected in individuals younger
than 60 years but in 10-20% of individuals older than 60-70 years.[109-111]
Based on these studies, CHIP was operatively defined as the presence of
somatic mutations otherwise detected in hematologic malignancies in
subjects' blood without any evidence of a morphologically defined
hematologic malignancy, with a VAF of 2% or greater.[107] The most frequent mutations occurred at the level of three genes, DNMT3A, TET2 and ASXL1 followed by less frequent mutations of TP53, JAK2, SF3B1, SRSF2, GNB1 and CBL.[109-111]
McKerrell et al. confirmed these findings by performing an ultra-deep
targeted analysis of 4,219 individuals and reporting clonal
hematopoiesis in 0.8% of individuals under 60 years, rising to 19.5 in
those ≥90 years.[112] DNMT3A-R882 mutations were the most frequent; the mutations involving spliceosome genes SF3B1 and SRSF2 were
detected only in individuals aged >70 years.[112] Another study based
on ultra-deep targeted sequencing involved the analysis of 2530
hematologically normal individuals with an age range from 55 to 101
years. 13.7 % of subjects displayed CHIP, ranging from <10% in the
55-59 year group to >40% in the 90-101 year group.[113] Some remarkable differences were observed between DNMT3A and TET2 mutations: TET2 mutations
were more age-dependent, associated with a modest neutropenic effect,
familial aggregation and chronic obstructive pulmonary disease; DNMT3A mutations do not have an impact on hematologic parameters.[113]
Young
et al. explored the occurrence of CHIP by targeted error-corrected
sequencing, which enables the detection of clonal mutations as rare as
0.0003 VAF; they observed that CHIP could be detected using this very
sensitive technique in nearly 95% of 50-70 years old individuals.[114]
Furthermore, the exploration of clonal variants in purified T
lymphocytes, B lymphocytes and myeloid cells purified from the blood of
13 individuals showed the presence in 10/13 cases of the same clonal
single nucleotide variant in both lymphoid and myeloid elements, thus
supporting the cellular origin of CHIP at the level of hematopoietic
stem/progenitor cells.[114]
Arends et al.
performed a detailed analysis of clonal hematopoiesis in 437 elderly
individuals in different blood cell populations (Table 1).
VAFs of the main CHIP-mutated genes were significantly higher in
monocytes, granulocytes, and NK-cells compared to B and T-cells.[115]
Importantly, in all cases analyzed, CHIP mutations were detected at the
level of the CD34+CD38- HSCs and at the level of myeloid-committed
progenitors.[115]
Zink et al. explored
the occurrence of CHIP in a population of 11,262 Icelanders by
whole-genome sequencing. They observed a frequency of 12.5% in the
whole population, variable with age and reaching a frequency of 23%,
32%, and 52% in the age groups of 65-75, 75-85 and 85-110 years,
respectively (Table 1).[116]
In this population, the presence of CHIP was associated with higher
death rates and an increased risk of hematological malignancy.[116]
Another
recent study of targeted error-corrected NGS confirmed the presence of
CHIP in the large majority of individuals ≥80 years: 57% in the 80-82
yr group and 72% in the ≥86 yr group.[20] In this old
population of individuals, only the presence of multiple mutations was
associated with an increased risk of death and of developing
hematological malignancy.[117]
Rossi et al.
reported the analysis of CHIP in 1794 individuals aged ≥80 years:
somatic mutations were observed in about one-third of these subjects[118] (Table 1). Somatic mutations were observed in about one-third of individuals aged >80 and were associated with reduced survival.[118] Most variants occurred in 3 genes: DNMT3A, TET2, and ASXL1; a significant prevalence of TET2 and ASXL1 mutations after the age of 90 years was observed; mutations in JAK2
and splicing genes, multiple mutations, and variant allele frequency
≥0.096 had predictive value for increased probability of developing
myeloid neoplasms.[118]
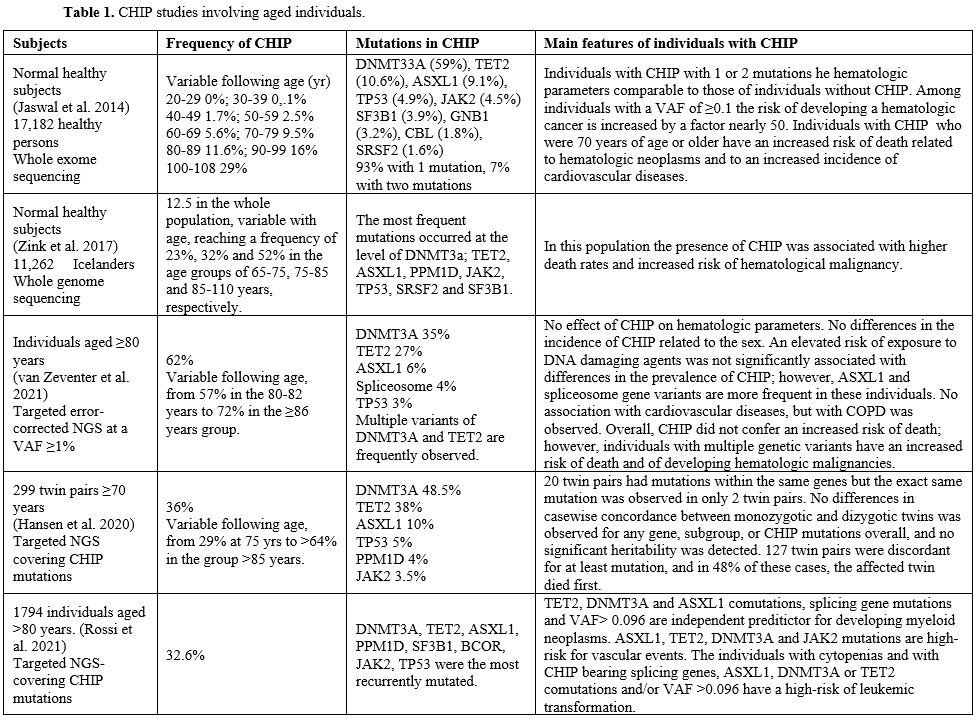 |
Table
1. CHIP studies involving aged individuals. |
Kar
et al. reported the data of CH in a large cohort of 200,453 UK Biobank
participants aged between 38-72 years analyzed by whole-exome
sequencing.[119] DNMT3A, TET2 and ASXL1 genes were the most commonly mutated genes in CHIP, followed by DNA damage response genes PPM1D, TP53, ATM and splicing factor genes SRSF2 and SF3B1 and JAK2 and GNB1 genes.[119] However, the age-related rise in prevalence differed for the different driver genes: compared to DNMT3A mutations, mutations in ATM were detected three years before, while ASXL1, PPM1D, SRSF2, SF3B1 mutations were observed 1-3 years later.[119]
Fabre
et al. reported the longitudinal dynamics of CHIP clones over a median
of 13 years; 92.4% of these clones expanded at a stable exponential
rate over observation: different mutations exhibited different growth
rates.[120] Thus, DNMT3A and TP53 grow with an average annual growth rate of about 5%; clones bearing mutations in TET2, ASXL1, PPM1D and SF3B1 displayed a growth rate of about 10%; fast-growing clones with SRSF2, PTPN11 and U2AF1 mutations expand with a growth rate of 15-20%/yr.[120] The individual growth variability of fast-growing clones, such as those associated with SRSF2-P95H mutant or U2AF1, was low compared to the consistent individual variability of slow-growing clones, such as those related to DNMT3A mutations.[120] The reconstruction of the dynamics of development of the different mutant clones showed that: DNMT3A-mutant
clones preferentially expand early in life and exhibit a slower growth
in old age, whereas splicing gene mutations expand later in life, and TET2-mutant clones grow and expand at all ages.[120]
The
compendium of all genes driving clonal hematopoiesis is far from
complete and recent studies have identified new drivers. Pich et al.
have adopted a reverse somatic calling approach, exploring the analysis
of whole-genome and whole-exome blood/tumor paired samples of two large
cohorts of cancer patients.[121] Using this
approach, they identified more than 60 genes showing signs of positive
selection in clonal hematopoiesis and thus with the properties of
clonal hematopoiesis drivers.[121] Beauchamp and
coworkers reported analysis of sequencing data from 84,683 individuals
and identified novel drivers of clonal hematopoiesis in the
5-methylcytosine reader ZBTB33 (0.18%) and in YLPM1 (0.07%), SRCAP (0.06%) and ZNF318 (0.12%).[122] Functional studies in mouse models suggested that mutated ZBTB33 induces the expansion of hematopoietic stem cells.[122]
Growing
evidence indicates that elderly individuals have evidence of CHIPs even
in the absence of known driver mutations. Thus, Zink and coworkers, in
their screening of 11262 Icelanders, provided proof of clonal
hematopoiesis with and without driver mutations: using a comprehensive
genome sequencing approach, they detected a much higher proportion of
individuals with CHIP compared to the frequency observed using a
detection approach based on the analysis restricted to the 18 genes
including all high-impact mutations observed in hematopoietic tissue.[123]
Poon et al. have developed a strategy to decipher the genome-wide rate
of positive selection based on the analysis of the VAF distribution of
synonymous mutations: most synonymous mutations reach high VAF due to
genetic hitchhiking, a phenomenon implying the co-occurrence of
synonymous mutations in association with positively selected driver
mutations which might be undetected.[123] Thus, the
high number of VAF synonymous variants provides information about the
genome-wide rate of driver mutations. The application of this framework
to data from the physiological blood of normal individuals showed that
a large part of mutations driving clonal expansions is located outside
of canonical cancer driver genes.[123] Mitchell et al. have evaluated the clonal dynamics of hematopoiesis during the human lifespan;[124]
this study was based on the analysis by whole-exome sequencing of
colonies grown from single stem/progenitor cells, enabling
comprehensive identification of somatic mutations and reconstruction of
lineage relationships between cells, similarly to the study previously
performed by Lee Six et al..[100] HSC/Progenitors
accumulated 17 mutations/year after birth and lost 30bp/year of
telomere length. Hematopoiesis in adults aged <65 years was largely
polyclonal, with consistent clonal diversity and with a population of
20,000-200,000 stem/progenitor cells contributing to blood cell
production; in individuals aged >75 was oligoclonal, with 12-18
independent clones globally contributing to 30-60% of total
hematopoiesis and each contributing to 1-34% of blood production.[124]
Most clones start their expansion before the subjects reach 40 years of
age. However, only 22% of these clones had known driver mutations;
genome-wide selection analysis estimated that 3% to 8% of mutations
were drivers.[124] Thus, this study raised several
fundamental observations on age-related clonal hematopoiesis, showing
that: the prevalence of clones with more than 1% of VAF is virtually
universal over the age of 70 years; (ii) the number of expanded clones
per individual is 10-20; (iii) the fraction of overall hematopoiesis
sustained by mutant clones in 30-60%; (iv) clonal expansions are
generated from mutations occurring decades earlier.[124]
Abascal
and coworkers explored the somatic mutation landscape at
single-molecule resolution in individuals of different ages observing
that differentiated blood granulocytes displayed remarkably similar
mutation loads and signatures compared to their corresponding
stem/progenitor cells, although mature granulocytes had undergone
considerably more cell divisions.[125] This observation suggests that mutational events occurring in hematopoietic stem cells may be independent of cell division.[125] Similar comments have been made for the colon and other tissues.[125]
Although
the development of clonal hematopoiesis is an age-related event, the
generation of somatic mutations in hematopoietic cells occurs even in
fetal hematopoietic stem/progenitor cells. Using an error-corrected
sequencing approach enabling the detection of variants with a VAF as
low as 0.01%, Wong et al. reported the presence of clonal hematopoiesis
in 18.2% of cord blood samples, with a VAF ranging from 0.2% to 0.6%.[126]
Hasaart et al. explored the occurrence of mutations in normal and Down
syndrome human fetal hematopoiesis and observed that: in fetal liver
hematopoietic stem/progenitor cells, there is an accumulation of about
100 base substitutions which is about two times and 5.8 times higher
than in cord and in post-infant hematopoietic stem/progenitor cells,
respectively.[127] Most of these mutations are located in introns, a minority in exons, and none are classified as drivers.[127]
Interestingly, fetal stem/progenitor cells displayed a higher relative
contribution of single base substitution signature 1 compared to
post-infant stem/progenitor cells.[127] Campbell et
al. have explored the accumulation of somatic mutations in fetal
hematopoietic stem/progenitor cells to investigate the dynamics of
human prenatal development and the origins of primitive and definitive
hematopoiesis, showing that fetal progenitors acquire tens of somatic
mutations by 18 weeks after conception.[128]
The
heritability of CHIP was explored in a large group of 299 twin pairs
≥70 years: 20 twin pairs had CHIP mutations within the same genes, but
the same mutation was observed in only two twin pairs; furthermore, no
difference in casewise concordance between monozygotic and dizygotic
twins was observed for any gene, subgroup, or CHIP mutations overall,
and no significant heritability was detected.[129]
One hundred twenty-seven twin pairs were discordant for at least one
mutation, and in 48% of these cases, the affected twin died first.[129]
The
CHIP study was largely confined to the analysis of somatic variants
involving a subset of genes recurrently mutated in myeloid
malignancies. However, recently, Niroula et al. hypothesized that
clonal hematopoiesis can also be detected in the lymphoid lineage and
could represent a condition of increased risk for developing lymphoid
malignancies. Thus, they defined a list of 235 genes recurrently
mutated in lymphoid malignancies and examined somatic variants in these
lymphoid driver genes using wide-exome sequencing data from 46,706
individuals aged 40-70 years with no previous diagnosis of hematologic
malignancy in the UK Biobank resource: 1.3% individuals carried
variants in one of these lymphoid driver genes and were referred as
lymphoid CHIP (L-CHIP).[130] L-CHIP increased with
age as well as myeloid-CHIP (M-CHIP); at variance with M-CHIP, L-CHIP
variants were distributed along a wide number of genes whose frequency
of occurrence was similar, such as DUSP22, FAT1, KMT2D, SYNE1, ATM, KMT2C, PCLO, PEN, ARID1A, NEB, MGA.[33]
Importantly, L-CHIP was associated in individuals screened in the UK
Biobank with an increased incidence of lymphoid malignancies,
particularly evident among individuals with larger clones; importantly,
the incidence of lymphoid malignancies was much lower among individuals
without CHIP or with M-CHIP; finally, only one individual with L-CHIP
developed a myeloid malignancy.[130] In addition,
some rare individuals displayed both L-CHIP and M-CHIP: these
individuals had a higher frequency of myeloid than lymphoid
malignancies.[130]
Clonal hematopoiesis, when
measured with a VAF sensitivity of >0.02, is clearly increasing with
age; this age-related effect is due to a cell-autonomous mechanism
linked to an increase in HSC self-renewal and positive selection.[131] In fact, the mutations in epigenetic regulators, such as DNMT3A and TET2
,provide an advantage by increasing self-renewal of stem/progenitor
cells and thus favoring their expansion, while the mutations in genes
involved in DNA damage response may increase cell survival.[131]
CHIP
detection in mice could provide an important animal model to explore
the mechanisms and physio-pathological consequences of clonal
hematopoiesis. To this end, Chin et al. screened for the most common
CHIP mutations in 4-month-old wild-type C57BL/6j mice, the most
extensively used mouse strain for hematologic studies.[132]
Hematopoietic clones with non-synonymous mutations in CHIP genes were
only detected in 2% of mice at 24 months. However, in transplanted
mice, the CHIP clones expanded: the detection of the same mutations in
multiple recipients of the same donor supported the view that CHIP
mutations could be acquired early in life.[132] In conclusion, the aged mice cannot provide a suitable model to study human clonal hematopoiesis.
Interestingly,
a recent study showed an experimental approach based on CRISPR/Cas9
technology to develop a simple model of clonal hematopoiesis.[133] Site-specific mutations were introduced in specific sites of ASXL1, DNMT3A, and TET2 in CD34+
progenitors derived from umbilical cord blood. The biological effects
induced by these genetic modifications were assayed in short-term and
long-term cultures, evaluating changes in self-renewal and cell
differentiation; TET2, but not DNMT3A and ASXL1
mutations induced enhanced self-renewal in short-term cultures; all the
three mutants and particularly the combined three mutants elicited a
clear increase of self-renewal, as evidenced by long-term culture
experiments.[133] In addition, the analysis of
clonal expansion after long-term culture showed a mutation-specific
impact on stem/progenitor cells.[133]
The study
of long-term survivors of allogeneic stem cell transplantation grafted
with CHIP-positive donors offers the unique opportunity to explore
the expansion of CHIP clones during hematopoietic reconstitution.
Boettcher et al. have studied 5 of these patients exhibiting
donor-engrafted CHIP: 4/5 cases displayed increased CHIP clones’ size
in recipients compared with donors, as measured by VAF; CHIP mutations
were constantly found in the myeloid lineage, but with variable
penetrance in the B and T lymphoid lineages; telomere shortening was
observed in granulocytes, supporting a proliferative activity of
hematopoietic stem cells.[134] Wong et al. have made
similar observations in a group of patients transplanted with younger
unrelated donors: some rare clonal mutations engrafted in recipients
and persisted over time.[135] Other studies have evaluated whether the presence of CHIP-related mutations after either autologous[136] or allogeneic matched sibling transplantation[137]
influences transplant outcomes. Both studies concluded that the
presence of CHIP did not affect transplant outcomes, including the time
to hematopoiesis recovery, relapse incidence, transplant-related
mortality, and progression-free and overall survival.[136,137]
However, the risk of acute graft versus host disease was higher in
allogeneic CHIP-positive donors compared to CHIP-negative donors.[136,137]
In
conclusion, these studies showed that engraftment, repopulation, and
long-term survival with donor CHIP are possible; after transplantation,
survival of hematopoietic cells with donor CHIP is only modest and
associated with a low clonal expansion in recipients. However, it
cannot be excluded that some CHIP-related variants could be associated
with increased clonal fitness. It is important to note that one of the
five patients reported in the study of Boettcher et al.[134]
developed a myelodysplastic syndrome. Particularly, one donor-recipient
pair developed MDS, diagnosed 18 and 21 years posttransplant,
respectively, and in both cases, derived from a shared founding clone.[134]
Nevejan et al. reported four subjects of CHIP-positive donor
cell-derived hematologic neoplasms (DCHN) in 263 HLA identical sibling
transplantation: the recipient patients, but not the donors, developed
myeloid neoplasia 17-20 years posttransplant; a higher VAF of the
pathogenic variant was observed in recipients compared to the donors; a
variable presence of driver variants in CHIP was observed.[138]
These observations show that malignant progression of donor-engrafted
clonal hematopoiesis in sibling recipients may occur many years after
stem cell transplantation.[138]
Chromosomal alterations in clonal hematopoiesis.
In addition to variants related to point mutations, other studies have
defined the presence of chromosomal alterations observed in clonal
hematopoietic cells. This condition, called age-related mosaic
chromosomal alterations (mCAs), and observed in DNA derived from blood
elements and related to clonal structural somatic alterations
(deletions, duplications, or copy number neutral loss of
heterozygosity), present in a more or less small fraction of peripheral
leukocytes, can indicate the existence of a condition of clonal
hematopoiesis. Two pivotal studies in 2012, based on the analysis of
DNA purified from peripheral blood of normal individuals, reported a
frequency of chromosomal mosaic abnormalities (duplications, deletions,
copy-neutral loss of heterozygosity) increasing with age: <0.5%
before 50 years and rising to 2-3% at >70 years.[139,140]
Recent
studies have characterized mCAs in British and Japanese normal
populations. The characterization of the British population was carried
out on the basis of DNA analysis of 151,202 UK Biobank participants;
4.94% of individuals displayed mCAs: most detected mCAs (70%) were
present at inferred fractions <5%.[141] The
detected mCAs were classified as loss or gain or copy-number neutral
loss of heterozygosity. The most common events were the loss of
chromosome X in females and the loss of chromosome Y in males; commonly
deleted regions <1 MB involve tumor suppressor genes; focal
deletions most frequently involved DNMT3A, TET2,
and 13q14; gains on chromosome 15 were much more frequent in elderly
males, while 16p11.2 deletions and 10q terminal deletions were more
frequent in females.[141] mCAs displayed
associations with germline variants on the same chromosomes. CNN-LOH
events on chromosome 1p are strongly associated with rare risk
haplotypes at the level of the MPL proto-oncogene at 1p34.1; CNN-LOH events on chromosome 11q are associated with a rare risk haplotype surrounding the ATM gene at 11q22.3; CNN-LOH and loss events at chromosome 15q are associated with a rare inherited 70-kb deletion spanning the TM2D3 and TARSL2 genes at 15q26.3.[141]
Terao et al. have reported the results of a study based on the analysis
of mosaic chromosomal alterations in a population of 179,417
participants in the Japan BioBank.[142] This
analysis detected a frequency of 15.5% of individuals with mCAs;
detectable mosaicism reached 40.7% in men and 31.5% in women over 90-y,
thus suggesting that mCAs are almost inevitable in elderly individuals.[142]
This study highlighted the existence of several remarkable differences
between Japanese and European individual in the frequency of some mCAs:
mosaic deletion of the TRA
locus on chromosome 14q, indicating clonal expansion at the level of
the T cell lineage, were common in the Japanese individuals but rare in
the European individuals (82% vs. 11%) and deletions at the IGH and IGL immunoglobulin
loci, indicating clonal expansion at the level of the B cell lineage,
were common in the European but rare in Japanese individuals (39% vs.
5% on chromosome 14 and 58% vs. 2% on chromosome 22); the three mCAs,
chromosome 12 gain, 13q loss, and 13q CN-LOH, events commonly observed
in CLL and in individuals who later develop CLL, are less frequent in
Japanese individuals than in European individuals, in line with the
lower incidence of CLL in the former ones compared to the latter ones.[142]
Zekavat
et al. reported the results of a very large study based on the analysis
of mCAs from 768,762 individuals from 5 biobanks without hematological
cancer at the time of DNA acquisition.[143] These
authors explored the association of mCAs with hematological traits
providing evidence that autosomal mCAs and loss of chromosomes X and Y
are significantly associated with altered hematologic features.[143]
Particularly, autosomal mCAs are related to elevated lymphocyte and
white blood cell leukocyte counts; loss of chromosome Y with increased
white blood cell leukocyte counts, neutrophil, monocyte, and platelet
counts; loss of chromosome X with increased white blood cell leukocyte
counts, lymphocyte, monocyte, and neutrophil counts.[143]
Autosomal mCAs were more associated with an increased risk of
hematological cancer (particularly lymphoid leukemia) than the loss of
chromosome Y.[143] Loss of chromosome X was associated with an increased risk of CLL, lymphoid leukemia, and AML development.[143]
As
mentioned above, the loss of the Y chromosome is the most common
chromosome abnormality in the hematopoietic lineage of aging men.
Peripheral leukocytes often exhibit mosaic loss of chromosome Y in
aging men, with an estimated frequency of >40% in the UK BioBank.[144] The origin of the loss of the Y chromosome may be explained by chromosome mis-segregation events during mitosis.[144]
A recent study provided evidence that 75% of men with loss of
chromosome Y also carried mutations in genes typically associated with
CHIP.[145] The study of the mutational profile of
the monocytes of 26 individuals with loss of chromosome Y showed
frequent CHIP pathogenic variants in TET2, DNMT3A, SF3B1, ASXL1 and TP53 genes; furthermore, BCOR, ZRSF2, BCORL1, FBWW7, FLT3 and GATA2 gene resulted also to be frequently mutated.[145] Another recent study showed that patients with chromosome Y loss in their bone marrow cells often have mutations of DNMT3A, TET2 and ASXL1 genes.[146]
The analysis of bone marrow cells of elderly subjects with loss of Y
chromosome showed that individuals with ≥75% of metaphases with LOY
have a greater likelihood of having myeloid-associated mutations and a
higher risk of developing myeloid neoplasia.[146]
Interestingly, a recent study reported the results of the study of the
association of mLOY with alterations in blood cell counts in a large
cohort (206,353 UK males) of UK BioBank men: associations between mLOY
and reduced erythrocyte count, elevated platelet count, elevated
leukocyte count (particularly for neutrophils and monocytes and less
for lymphocytes); these associations were independent of the effects of
aging and smoking.[147]
Some recent studies
have provided a fundamental contribution to the study and to the
understanding of clonal hematopoiesis through the combined study of
single-nucleotide variants and copy number alterations.[130,148]
Saiki et have investigated the occurrence of CHIP-related gene
mutations by targeted NGS and CNAs by gene array in 11,234 Japanese
individuals (in large part aged ≥60 years) without hematological
malignancies from the BioBank Japan cohort, including 672 individuals
with subsequent development of hematological malignancy and have
studied the effects of these genetic alterations on hematological
phenotypes, including the development of hematological malignancies.[148]
27.3% of these individuals exhibited CHIP-related mutations: 20.6% with
one mutation, 5.2% with two mutations and 1.5% with at least three
mutations; the most frequent alterations were DNMT3A (13.5%), TET2 (9.5%), ASXL1 (2.2%) and PPM1D (1.4%).[28]
CNAs, including CNN-LOH or uniparental disomy (UPD), were detected in
20.1% of cases, of which 3.7% exhibited multiple CNAs; 14qUPD, +21q,
del(20q), and +15q were frequent CNAs, whereas del(20q), 16pUPD and
17pUPD were associated with the largest mean clone size.[148]
The evaluation of the combined occurrence of CNAs and CHIP mutations
showed a frequency of 40% in individuals ≥60 years and 56% of cases who
developed hematological malignancies.[28] 16% of all individuals displaying clonal hematopoiesis have both types of genetic lesions: mutations in TP53, TET2, JAK2, SF3B1 and U2AF1
were more frequently accompanied by concomitant CNAs; the maximum clone
size in clonal hematopoiesis-positive cases correlated with the total
number of CHIP mutations and CNAs.[148]
Interestingly, in some cases, multiple co-occurring lesions were
estimated to be present in the same large clone. In other cases, the
mutations and CNAs affect the same genetic loci.[148] In addition to age, other factors affected CHIP mutations and CNAs: ASXL1, PPM1D, TP53, splicing factors and CNAs, including +15,del(20q), +21 and 14qUPD correlated with male gender and smoking.[148] Furthermore, individuals with high platelet counts have a higher frequency of JAK2 mutations and 9pUPD, while individuals with cytopenia of any type display frequent U2AF1 mutations and del(20q).[148]
Both CHIP mutations and CNAs were significantly associated with higher
mortality deriving from hematological malignancies;
hematological-related mortality in clonal hematopoiesis-positive
individuals was more attributable to myeloid than to lymphoid
neoplasms; the highest risk of hematological mortality was related to U2AF1, EZH2, RUNX1, SRSF2 and TP53 mutations and +1q CNA.[148]
Importantly, the presence of both CHIP mutations and CNAs was
associated with increased hematological-related mortality compared with
that of CHIP mutations or CNAs alone; this combined effect seems to be
largely related to the total number of alterations rather than by the
type of alterations.[148] The results of this study
underscore the importance of measuring both lesions for an accurate
evaluation of the risk of developing a hematological malignancy in
individuals with clonal hematopoiesis.
Niroula et al. have
analyzed CHIP mutations in 55,383 individuals and autosomal mCAs in
420,969 individuals with no history of hematologic malignancies in the
UK BioBank and Mass General Brigham BioBank.[130] As
discussed above, this study distinguished myeloid and lymphoid somatic
gene mutations at the CHIP level; furthermore, this study used a
similar strategy to differentiate myeloid and lymphoid mCAs. Thus, to
categorize mCAs, in a group of individuals with a hematologic
malignancy, mCAs have been defined as M-mCA (0.38% in the whole
population) or L-mCA (0.83%) according to their differential prevalence
in myeloid or in lymphoid malignancies; mCAs common to both
malignancies were defined as ambiguous (A-mCA, 0.32%); a significant
number of mCAs (2%) cannot be classified.[130] The
presence of M-mCA increased the risk of myeloid malignancies, L-mCA
increased the risk of lymphoid malignancies, and A-mCA increased the
risk of both lymphoid and myeloid malignancies.[130]
Frequencies of myeloid malignancies, such as acute myeloid leukemia,
myelodysplastic syndrome, and myeloproliferative neoplasms, are higher
among individuals with M-CHIP and M-mCA; L-CHIP and L-mCA are
associated with increased risk of chronic lymphocytic leukemia (CLL)
and small lymphocytic lymphoma (SLL) and also with follicular lymphoma
and diffuse large B-cell lymphoma (DLBCL); the annual rate of myeloid
malignancies increased from 0.02%-0.03% among individuals with no
clonal hematopoiesis to 0.17% and 0.82% among individuals with M-CHIP
and M-mCA, respectively and the annual rate of lymphoid malignancies
increased from 0.01% among individuals without clonal hematopoiesis to
0.22% and 0.6% among individuals with L-CHIP and L-mCA, respectively.[130]
Some individuals exhibited concomitant M-CHIP and M-mCA and others
concomitant L-CHIP and L-mCA; these individuals showed an increased
tendency to develop myeloid and lymphoid malignancies, respectively,
compared to those with a single type of genetic alteration.[130]
Interestingly, in a significant proportion of cases M-CHIP and L-CHIP,
mCAs (mostly CNN-LOH) overlapping the mutated genes were present,
resulting in biallelic variants in specific driver genes.[130]
Analysis of standard hematological parameters showed that patients with
both abnormal myeloid cell parameters and M-CHIP/M-mCA alterations and
with both abnormal lymphoid cell parameters and L-CHIP/L-mCA
alterations exhibit a particularly increased risk of developing myeloid
and lymphoid malignancies, respectively.[130]
482,378
individuals from the UK biobank were investigated for the coexistence
of different types of clonal hematopoiesis: LOX, LOY, autosomal mCAs
(gains, losses, and copy neutral LOH), CHIP, and MPN
(myeloproliferative neoplasia).[149] Positive
phenotypic associations between autosomal mCAs with LOY, LOX, CHIP, and
MPN were observed; CHIP was positively associated with MPN but
negatively associated with LOY; individuals with higher cellular
fractions with autosomal mCA events have a greater probability of also
having LOX, CHIP and MPN; higher autosomal mCA cellular fractions were
inversely associated with LOY; individuals with higher VAF of CHIP have
more chances to have detectable autosomal mCAs.[149]
Furthermore, 6.4% of individuals with CHIP and 5.3% of individuals with
autosomal mCAs displayed both CH types: these individuals display a
peculiar pattern of co-occurrence of either CHIP mutations or mCAs;
some of these individuals show co-localization of mutational and CA
events at the level of TET2, DNMT3A and JAK2:
the analysis of the VAF of CHIP mutations and the cellular distribution
of mCAs suggests that the acquisition of the CHIP mutation preceded the
acquisition of autosomal mCAs.[149] Genome-wide
studies suggested genetic correlations between LOY and LOX and between
LOY and MPN, suggesting the existence of shared biologic mechanisms
promoting or predisposing clonal expansion development.[149]
The
presence of an unexplained alteration in a hematological trait must be
explored at the molecular level for its possible association with
clonal hematopoiesis. Van Zeventer et al. examined a cohort of 144,676
adults for the presence of monocytosis (defined as monocytes ≥1x109/L and ≥10%), not associated with known pathological conditions.[150]
Among individuals ≥60 years, the prevalence of monocytosis was 0.8% and
increased with age; these older individuals with monocytosis displayed
more frequently CHIP than age-matched individuals without monocytosis
(50.9% vs. 35.5%, respectively).[150] Monocytosis
has been associated with more frequent multiple gene mutations and
mutations in spliceosome genes but not of isolated mutated DNMT3A, TET2 and ASXL1.[150] Myeloid malignancies developed in 4 individuals with monocytosis, all displaying CHIP.[150]
Germline
Risk of Clonal Hematopoiesis and Rare Inherited and Not-Inherited
Diseases Associated with Clonal Hematopoiesis Expansion
Studies
carried out in the last years have provided evidence that inherited
factors shape the incidence of clonal hematopoiesis.[151]
The simultaneous analysis of germline and somatic genetic variation on
a population scale has shown some heritable germline susceptibility to
CHIP, mCA and mLOY.[151] The most remarkable
finding emerging from these studies is that the DNA damage response and
telomere maintenance pathway genes are commonly implicated in genetic
association with clonal hematopoiesis subtypes.[151]
Bick et al. performed the widest population screening of the inherited
causes of clonal hematopoiesis in a population of 97,691 individuals
analyzed by whole-genome sequencing; they identified 4,229 individuals
with CHIP.[152] Three main germline genetic determinants of CHIP were identified: germline genetic variants at the TERT locus were identified as predisposing to clonal hematopoiesis; one locus on chromosome 3 in an intergenic region spanning KPNA4 and TRIM59; one locus on chromosome 4 near TET2.[152]
Recent
studies have evidenced a consistent contribution of clonal
hematopoiesis in the genesis of several rare inherited or not-inherited
conditions associated with frequent myeloid neoplasms.
Erdheim
Chester Disease (ECD) is a rare histiocytic neoplasm classified as a
macrophage dendritic neoplasm in the 2016 hematopoietic and lymphoid
tumors classification. About 50% of ECD patients display a BRAFV600E
mutation, and the rest usually have other activating mutations in the
MAPK pathway. As a result, about 10% of ECD patients develop myeloid
neoplasms. A recent study by Cohen Aubart et al. reported the targeted
NGS of bone marrow cells of 120 ECD patients: mutations associated with
CHIP were identified in 42.5% of patients; the most frequent mutations
were TET2 (22%), ASXL1 (9%) and DNMT3A (8%).[153]
In addition, 15% of these patients developed myeloid neoplasms: 31% of
ECD patients with CHIP developed a myeloid neoplasia, compared to 3%
among those without CHIP; 89% of patients who developed a myeloid
neoplasia have CHIP, compared to 34% among those who did not develop
myeloid neoplasia.[153]
DNMT3A overgrowth
syndrome (DOS), also known as Tatton-Brown Rahman Syndrome (DOS), is
one of the overgrowth syndromes caused by constitutional mutations in
genes encoding epigenetic regulators and characterized by complex
phenotypes. DOS patients had de novo heterozygous germline mutations in DNMT3A,
missense, frameshift, and nonsense mutations, all involving the
functional domain of DNMT3A protein. The clinical features of DOS
include overgrowth and intellectual disability. The analysis of the 200
known DOS patients worldwide showed that 8 of these patients developed
hematological malignancies.[154] The study of DNA methylation in peripheral blood cells of DOS patients showed that heterozygous DNMT3A mutations induce a focal hypomethylation phenotype, most severe with the dominant negative DNMT3AR882H.[155]
A very recent study reported a detailed analysis of the hematological
phenotype of individuals with DNMT3A overgrowth syndrome, characterized
by the presence of nonanemic RBC macrocytosis, a relative decrease in
peripheral blood lymphocytes (with an increase of the CD4/CD8 ratio),
and monocytes and an increase in neutrophils; this hematological
phenotype was recapitulated in murine models of DNMT3A overgrowth
syndrome.[156]
Shwachman-Diamond Syndrome (SDS)
is a genetic disorder (ribosomopathy) associated with a high risk of
developing myeloid neoplasia early in life; this disorder is due to a
biallelic mutation in the SBDS
gene encoding the SBDS protein required for the formation of the
translationally active 80S ribosome. Recent studies have clarified the
mechanisms responsible for the frequent generation of myeloid neoplasms
in these patients. Thus, Xia et al. have explored clonal
hematopoiesis's occurrence in different types of congenital
neutropenia, including SDS.[157] CHIPs were observed in 59% of SDS cases; mutations of TP53
were observed in 48% of SDS CHIPs, while mutations of this gene were
undetectable in the CHIPs of controls or of cyclic neutropenia or
severe congenital neutropenia patients.[157] TP53 mutations were present in a low fraction of bone marrow cells, and their presence correlates with the age of patients.[157]
Kennedy et al. performed a detailed molecular characterization study of
a large cohort of 110 SDS patients. The analysis of bone marrow cells
of 86 patients without diagnosis of myeloid neoplasia provided evidence
of CHIP in 72% of cases: the majority of these CHIP-positive patients
displayed more than one mutation; recurrent mutations in EIF6 (59%), TP53 (40%), PRPF8 (10.8%), CSNK1A1 (7.2%) were observed.[158] It was proposed that germline SBDS mutations induce a fitness constraint that determines the selection of somatic clones through two distinct mechanisms: EIF6 mutations
induce EIF6 inactivation with consequent amelioration of the ribosome
defect of SDS with no effects on leukemic transformation; TP53 mutations
drive a maladaptive pathway with leukemic potential through
inactivation of tumor suppressor checkpoints without correcting the
ribosome defect.[158] Leukemia development in these patients was associated with acquiring biallelic TP53 alterations.[158]
Furutani and coworkers reported the study of 153 subjects with SDS and
explored the hematologic phenotype of these patients, particularly for
that concerns the hematologic complications: (i) absolute neutrophil
counts were positively associated with age; (ii) platelet counts and
bone marrow cellularity were negatively associated with age; (iii)
severe marrow failure necessitating transplantation was observed in 8
patients; (iv) 17% of patients developed a malignancy (16 MDS and 10
AML) at a median age of 12.3 years; (v) 1 patient developed a lymphoid
malignancy.[159]
Sterile Alpha Motif Domain 9 (SMAD9) and its paralogue SAMD9-like (SAMD9L)
are two genes located on chromosome 7q21, encoding two cytoplasmic
proteins. The function of these two genes seemingly originated from a
common ancestral gene duplication, remains enigmatic. Germline SAMD9 and SAMD9L
cause a variety of multisystem syndromes with a propensity for
cytopenia, bone marrow failure, and an elevated risk of early-onset
myeloid neoplasms, particularly myelodysplastic syndromes.[160-162] Germline SAMD9 and SAMD9L
mutations predispose to myelodysplastic syndromes. Schwartz, in an
extensive study of evaluation of the genomic landscape of pediatric
MDSs, showed that germline variants in SAMD9 or SAMD9L were present in
17% of primary MDS patients; these variants were lost in the tumor
mechanism by a rescue mechanism involving either chromosomal deletions
though monosomy 7 or copy number neutral loss of heterozygosity
(CN-LOH).[163]
Another recent study further supported the existence of rescue mechanisms essential for the leukemic transformation of SAMD9 or SAMD9L
germline mutated cells; in fact, it was shown that 61% of these
patients undergo somatic genetic rescue, resulting in clonal
hematopoiesis involving both maladaptive (monosomy 7) and adaptive
(isodisomy 7q) mechanisms.[164] Bone marrow single-cell DNA sequencing showed multiple competing somatic genetic rescue events in individual patients.[164]
A
recent study showed that SMAD9 and SMAD9L are multifunctional proteins
that determine alterations in cell cycle, cell proliferation, and
protein translation in hematopoietic stem/progenitor cells; mutant SAMD9 or SAMD9L induce the generation of a cellular environment that causes DNA damage repair defects.[165]
Clonal Hematopoiesis in Cancer
Clonal
hematopoiesis increases with age and is a condition that predisposes to
hematologic malignancies. In addition, many studies have shown that
clonal hematopoiesis occurs with higher frequency in individuals with
lymphoid and solid tumors and increases following exposure to genotoxic
stress. Furthermore, clonal hematopoiesis may represent a major
determinant for the risk of therapy-related neoplasms (t-MN),
comprising therapy-related acute myeloid leukemia and myelodysplastic
syndrome, as late complications of cytotoxic therapy, chemotherapy,
and/or radiotherapy, used in the treatment of both malignant and
non-malignant diseases.
The terrorist attack on the World Trade
Center (WTC) in 1993 generated a unique environmental exposure to
aerosolized dust, gases, and potential carcinogens in a small
population of individuals; it represented a peculiar opportunity to
explore the effect of potential environmental carcinogens in clonal
hematopoiesis development.[166] Deep targeted
sequencing of blood samples showed a significantly higher proportion of
individuals with CHIP among WTC-exposed first responders compared to
non-WTC-exposed firefighters after controlling for age, sex, and
race/ethnicity (10% vs. 6.7%, respectively).[166] CHIP mutations predominantly affected TET2 and DNMT3A.[166]
In
2017, Coombs et al. reported the analysis of paired tumor and blood
samples derived from 8,810 individuals with non-hematological
malignancies by deep-coverage, targeted NGS; in these patients, clonal
hematopoiesis was identified in 25% of cases and was associated with
increased age, tobacco smoking and prior radiotherapy.[167]
In these patients, clonal hematopoiesis was associated with increased
white blood cell counts, increased absolute monocyte and neutrophil
counts, increased mean corpuscular volume, and decreased platelet
count.[167] The most frequently mutated genes at the level of clonal hematopoiesis were DNMT3A, TET2, PPM1D, ASXL1, ATM and TP53; TP53 and PPM1D mutations were significantly associated with prior exposure to cancer chemotherapy.[167] Clonal hematopoiesis in these patients was associated with an increased incidence of subsequent hematologic cancers.[167]
These findings were confirmed in a larger analysis involving 17,469
cancer patients: clonal hematopoiesis-associated mutations were
observed in 26.5% of these patients, mostly involving DNMT3A, TET2 and PPM1D; clonal hematopoiesis was most frequent among skin cancer and non-small cell lung cancer patients.[168]
Clonal
hematopoiesis mutations can be tentatively identified in unpaired NGS
assays of tumor samples of solid tumors, but these mutations putatively
related to clonal hematopoiesis must be confirmed by paired blood
sequencing.[169,170]
Gao and coworkers reported
the results of a combined analysis of mCAa and CHIP mutations in a
cohort of 32,442 cancer patients: the incidence of mCAs was low,
increasing with age from under 1% in individuals under 50 years to more
than 3% among individuals more than 80 years old; mCAs were most
frequently observed among cancer patients with soft tissue sarcoma,
thyroid and lung cancer; mCA was positively associated with external
beam radiation therapy but not with cytotoxic treatment.[171]
63% of clonal hematopoiesis cases with mCAs co-occurred with at least
one gene mutation, particularly with high mutation number and VAF.[47]
CHIP mutations were observed in 30% of these cancer patients. mCAs
displayed a peculiar pattern of co-occurrence with gene mutations,
showing mechanisms of cooperation between chromosomal alterations and
gene mutations through cis (biallelic inactivation) and trans (functional cooperation) mechanisms.[171]
These patients exhibited clonal hematopoiesis with composite genotypes,
a subgroup associated with a high risk of developing a hematologic
malignancy.[171]
Other studies have evaluated the
effects of anticancer treatments (chemotherapy, radiation therapy,
targeted therapy, and immunotherapy) on clonal hematopoiesis.
Furthermore, it was investigated a possible contribution of clonal
hematopoiesis to the generation of therapy-related myeloid neoplasms
(t-MN), a condition that occurs after treating primary malignancies
using chemotherapy and/or radiation therapy or after immunosuppression
for solid organ transplant or autoimmune disease.[172,173]
Two recent studies have explored the evolution of hematopoietic cells
under cancer therapy, exploring the mutational footprints induced by
cancer therapies.[174] Cells of AMLs secondary to
treatment with platinum-based drugs show the mutational footprint
typical of these drugs. These mutations also appeared in non-malignant
cells at the drug exposure time. Furthermore, platinum drugs produce a
mutagenic effect of the same magnitude in both normal and tumoral
cells.[175] In contrast, no trace of the
5-fluorouracil mutational signature is found in AMLs secondary to
exposure to 5-FU, suggesting that cells establishing the leukemic
process could be quiescent during treatment (in fact, 5-FU-induced
mutations appear only in cells that are nor quiescent during drug
exposure).[175] In a second study, using wide genome
sequencing therapy-related myeloid neoplasias (tMN) samples from
patients exposed to various anti-cancer drugs and pre-therapy samples,
have characterized the mutational impact of chemotherapy and used
chemotherapy-induced signatures (CIS) as a temporal barcode to
reconstitute tumor evolution.[176] tMNs evolving
without CISs display features similar to those observed in de novo
AMLs. In contrast, tMNs with CIS, such as those occurring in platinum-
or melphalan-treated patients, are hypermutated and more frequently
associated with complex genomes, including chromothripsis and copy
number alterations.[176] Interestingly the procedure
of treatment involving high-dose melphalan and ASCT allows clonal
hematopoiesis to escape chemotherapy exposure and to be reinfused to
expand to malignancy; these patients can develop tMN bearing no
evidence of melphalan-induced mutations.[176]
TMNs
are molecularly heterogeneous and are represented by tMDSs and tAMLs;
the majority of tMNs display chromosomal abnormalities associated with
MDS or KMT2A rearrangements.[177]
At molecular level, the majority (about 80%) of these tMNs display an
abnormal karyotype, while a minority (about 20%) is associated with a
normal karyotype; TMNs with normal karyotype compared to those with
abnormal karyotype are enriched for mutations in TET2, NPM1, ASXL1, SRSF2, RUNX1 and STAG2, many genes frequently mutated in CHIP.[177]
Clonal hematopoiesis in lymphoma.
A consistent number of studies explored the occurrence of CHIP in
patients with various types of lymphoma or myeloma. Wong et al.
investigated 119 patients with a history of malignancy (lymphoma and
myeloma): 81 received previous chemotherapy with or without radiation,
and they were explored by error-corrected NGS for 46 genes associated
with clonal hematopoiesis.[178] 28.4% of patients
exposed to cytotoxic therapy displayed clonal hematopoiesis possessing
at least one mutation with VAF ≥2% and 82.7% with VAF ≥0.1%; the
incidence of clonal hematopoiesis, as well as the number of genetic
variants, is higher in patients receiving cytotoxic therapy compared to
those who not received it and to the healthy controls.[178]
The analysis of the CHIP mutational spectrum showed several remarkable
differences between patients who received cytotoxic therapy and those
not receiving these treatments. While the frequency of DNMT3A and TET2 mutations was similar in these two groups, TP53 and PPM1D
mutations displayed a markedly higher frequency in patients who
received cytotoxic therapy compared to those who did not receive it.[178]
In addition, 73% of patients with clonal hematopoiesis after cytotoxic
therapy displayed multiple variants; 59% had multiple variants in the
same gene, 31% had multiple variants in DNA damage response genes, and
26% had three or more variants.[178] In these patients, DNMT3A, PPM1D and TP53 mutations are present in both myeloid and lymphoid elements, suggesting their origin from hematopoietic stem cells.[178]
In a set of 40 of these patients undergoing autologous stem cell
transplantation, it was evaluated the effect of transplantation on
clonal hematopoiesis expansion; two gene variants, DNMT3A and PPM1D, were mostly modulated by transplantation: of 51 DNMT3A variants, 33% significantly increased ≥ 2-fold in VAF after transplantation, while 6% decreased; of 23 PPM1D variants, only about 9% increase in VAF after transplantation, while 30% decreased.[178] Finally, the leukemogenic potential of expanded TP53
mutant hematopoietic clones was higher than that of clones bearing
mutations at the level of other DNA damage response genes, such as PPM1D.[178]
Husby
et al. explored 565 lymphoma patients undergoing autologous stem cell
transplantation in the context of transplant centers in Denmark: 25% of
these patients displayed at least one CHIP mutation; overall, patients
with CHIP did not reveal an inferior survival; however, those with
mutations in DNA repair genes, such as PPM1D, TP53, RAD21 and BRCC3 showed a reduced overall survival.[179]
Gibson et al.[180]
explored the occurrence of clonal hematopoiesis mutations in the blood
of 401 lymphoma patients (Hodgkin and non-Hodgkin lymphoma) undergoing
autologous stem cell transplantation (ASCT): about 30% of these
patients displayed in peripheral blood clonal hematopoiesis-associated
mutations; 69% of these patients showed 1 mutation and 31% ≥ 2
mutations.[180] In these patients, mutations in PPM1D and TP53 are much more frequent compared to the values reported for aged individuals: PPM1D and TP53 mutations accounted for 32% and 11% of all the clonal hematopoiesis-related mutations observed in these patients.[180]
Importantly, patients with CHIP, particularly those with CHIP bearing
PPM1D exhibited a shorter overall survival than those without CHIP.[180]
In another study, Eskelund et al. evaluated a homogeneous cohort of 149
mantle cell lymphoma (MCL) patients undergoing ASCT and achieving a
condition of minimal residual disease negativity (MRD-):
CHIP clones consistently expanded during chemotherapy and ASCT and
stabilized after the end of therapy; no clinical impact of CHIP in this
cohort of good-prognosis patients was observed; 98% of CHIP mutations
were already detectable before exposure to any chemotherapy.[181]
Few
studies have explored CHIP in Hodgkin lymphoma (HL) patients. Husby et
al. reported the occurrence of CHIP in 14% of HL patients undergoing
ASCT.[179] Venanzi explored the presence of CHIP in 40 HL patients and observed positivity in 12.5% of cases.[182]
Massive genome sequencing of tumor tissue showed that only one of the
five CHIP-positive patients, mutant clonal hematopoiesis seeded the
neoplastic clone.[182]
Saini et al. explored
the occurrence of clonal hematopoiesis among 114 patients with large
B-cell lymphoma treated with anti-CD19 CART cells; 36.8% of these
patients displayed CHIP most frequently mutated in PPM1D and TP53 genes.[183]
CHIP-positive patients had a more frequent incidence of immune-effector
cell-associated neurotoxicity syndrome (ICANS) than CHIP-negative
patients (45% vs. 25%) Higher incidence of ICANS and cytokine release
syndrome were observed among patients with CHIP associated with DNMT3A, TET2 and ASXL1 gene mutations.[183]
In addition, the cumulative 24-h incidence of tMN was significantly
higher in CHIP-positive than in CHIP-negative patients (19% vs. 4.2%,
respectively).
The presence of CHIP in lymphoma patients may also
be associated with drug-associated toxicity. Thus, TET2-clonal
hematopoiesis was observed in 8.2% of patients with lymphoma;
importantly. In addition, TET2-CH was associated with
anthracycline-induced cardiotoxicity.[184]
The exploration of CHIP
in patients with angioimmunoblastic T-cell lymphoma (AITL) is
particularly interesting in that these tumors harbor frequent mutations
in the epigenetic modifiers TET2, DNMT3A and IDH2 and in the small GTPase RHOA.[185]
In a cohort of 25 patients with AITL, it was found that 15/22 displayed
CHIP: in these patients, identical mutations were detected in the
neoplastic T cells and in myeloid compartments, including DNMT3A and TET2
mutations; four of these patients developed myeloid neoplasia, all of
which shared CHIP-type mutations with their AITL tumor counterpart.[185] These observations indicate that CHIP is prevalent in AITL patients and can evolve to AITL and MNs.[185]
Blastic
plasmacytoid dendritic cell neoplasm (BPDCN) is an example of another
lymphoma condition associated with prevalent CHIP incidence. BPDCN is
an aggressive tumor with features of cutaneous lymphoma and/or
leukemia; 20% of these patients displayed MDS or chronic myelomonocytic
leukemia with a shared clonal origin.[186] NGS on bone marrow cells,
skin, or sorted cells provided evidence of mutations in BPDCN (100% of
cases) and in bone marrow hematopoietic cells (65% of cases), with
similar high frequencies of TET2 (58% vs. 60%) and ASXL1 (40%
vs. 33%) mutations; karyotypic abnormalities were frequently observed
in BPDCN (66%) but only very rarely in BM hematopoietic cells (2%).[186] These observations support a high prevalence of bone marrow clonal hematopoiesis in BPDCN.[186]
Clonal hematopoiesis in multiple myeloma.
Several studies have evaluated CHIP occurrence in multiple myeloma (MM)
patients. However, only two studies have characterized large sets of MM
patients. One of these two studies was based on the analysis of the
hematopoietic stem cell products of 629 MM patients treated with
autologous stem cell transplantation at Dana Farber Cancer Institute:
using deep targeted sequencing (VAF≥1%) observed a CHIP prevalence
corresponding to 21.6%.[187] Recurrently mutated genes in these patients were DNMT3a, TET2, TP53, ASXL1 and PPM1D.[187]
In patients not receiving immunomodulatory drug maintenance (IMDM), the
presence of CHIP is associated with decreased overall survival due to
an increase in MM progression; in patients receiving IMDM, drug
treatment improved overall survival and progression-free survival,
regardless of CHIP status.[187] The second study was
based on the analysis of MM patients enrolled in PETHEMA and
GEM2012MENOS65 trials. At diagnosis, about 10% of MM displayed
MDS-associated phenotypic alterations (MDS-PA) and 11.6% monocytic
MDS-PA.[188] Bulk and single-cell sequencing studies of CHIP-associated genes at the level of CD34+ progenitors showed the occurrence of CHIP in 50% of cases with MDS-PA and 22% in those without MDS-PA; TET2 and NRAS were the most recurrently mutated genes.[188]
MDS-PA at diagnosis predicted an increased risk of hematopoietic
toxicity and was independently associated with shorter PFS and OS.[188]
Waldenstrom
macroglobulinemia (WM) is an indolent NHL characterized by
immunoglobulin M-secreting lymphoplasmacytic cells and mutations at the
level of genes involved in B-cell signaling. A recent study showed that
CHIP was identified in 14% of patients with IgM monoclonal gammopathy
of undetermined significance (MGUS), smoldering WM (SWM), or WM; the
most recurrent mutations involved DNMT3A, TET2, and ASXL1 genes.[189] In addition, patients with CHIP had an increased risk of progression from MGUS to SWM to WM but not worse overall survival.[189]
Clonal hematopoiesis in solid tumors.
Several studies have characterized in patients with solid tumors: (i)
the association between the presence of some mutant genes in CHIP and
the response to chemotherapy; (ii) the influence of various
chemotherapy treatments on the mutations observed at the level of CHIP
and of t-MNs.
PPM1D
mutations were observed in pMNCs of patients with solid cancers, such
as breast, colon, ovarian, and lung cancer. Thus, Zajkowicz et al.
reported frameshift PPM1D mutations
in 0.92% of non-small cell lung cancer patients; all the positive
patients had squamous lung cancer (1.5% of positive patients).[190] Pharoah et al. reported PPM1D
mutations in 0.37% of ovarian cancer patients at the level of
lymphocyte DNA: all positive cases corresponded to patients analyzed
after chemotherapy treatment.[191] Studies carried out in ovarian cancer patients showed that PPM1D
mutations detected in peripheral blood mononuclear cells were
significantly associated with prior chemotherapy and, in patients
treated with chemotherapy, with older age.[192] Interestingly, in these patients, TP53 mutations were not related to previous chemotherapy and age.[192]
Truncating mutations in the terminal exon of PPM1D induce a chemoresistance phenotype that results in the selective expansion of PPM1D-mutant hematopoietic cells in the presence of chemotherapy; this finding explains the clonal expansion of PPM1D-mutant cells.[193]
A
recent study explored a very large set of cancer patients (24,439
individuals) and observed CHIP in 30% of these patients: 68% of these
patients had one mutation in CHIP, and 32% had two or more mutations;
the most frequently mutated genes were the epigenetic regulators DNMT3A and TET2 and the genes involved in DNA Damage Response (DDR) pathway, including PPM1D, TP53 and CHEK2; 90% of the mutations observed in CHIP were classified as driver myeloid mutations.[194]
The spectrum of gene mutations observed in CHIP was similar in
different cancer types, with the exception of DDR gene mutations,
particularly of the PPM1D gene, which were enriched in ovarian and endometrial cancers.[194] The presence of specific gene mutations was associated with some pathogenic events: (i) mutations of the spliceosome genes SRSF2 and SF3B1 were less frequent than other CH mutations and are associated with age; (ii) CHIP mutations in the DDR genes TP53, PPM1D and CHEK2 were strongly associated with prior oncologic therapy (mutations in PPM1D
were mainly associated with previous exposure to platinum and
radionuclide therapy, but also with topoisomerase II inhibitors and
taxanes; mutations in TP53 were associated with previous platinum, radiation therapy, and taxanes; mutations in CHEK2 were associated with platinum and topoisomerase II inhibitors); (iii) CHIP mutations in ASXL1 gene were strongly associated with smoking.[194]
Furthermore, the fitness associated with mutations in epigenetic
regulators or splicing regulators was not markedly modulated by
oncologic therapy.[194] The environmental factors
most strongly associated with the development of CHIP myeloid driver
mutations are represented by radiation therapy, platinum (mostly
carboplatin) chemotherapy and exposure to topoisomerase II inhibitors.[194]
The characterization of the clonal dynamics of evolution of CHIP
mutations in 525 cancer patients in a median lapse time of 23 months
provided evidence that 62% remained stable, 28% increased, and 10%
decreased in clonal size; the growth rate was most pronounced for CHIP
mutations in DDR genes.[194] The incidence of CHIP
far exceeds that of t-AML, and the main determinants of the risk of a
CHIP transforming into therapy-related myeloid neoplasia are related to
the type of CHIP mutations (mostly TP53 and spliceosome genes SRSF2, U2AF1 and SF3B1 mutations), the number of CHIP mutations and clonal size.[56] As discussed above, TP53 is one of the mutated genes frequently involved in t-AML: the analysis of 34 t-MN seemingly evolving from CHIP displayed TP53 mutations in 44% of cases; 73% of theseTP53-mutant t-MNs displayed pre-tMN TP53 mutations; 73% of TP53-mutated
t-MNs showed complex karyotype alterations, an event acquired at the
level of neoplastic transformation, but absent in pre-neoplastic CHIPs.[194]
The
incidence of t-MN is variable in different cancer types and treatment
regimens; t-MN usually develops 3-10 years after exposure to
chemotherapy and/or radiation treatment; t-MN occurs more frequently in
patients who received alkylating agents or topoisomerase II inhibitors
and less often in those who received taxanes or antimetabolites;
high-dose chemotherapy followed by autologous stem cell transplantation
increases the risk of developing t-MN.[195-197]
Several
recent studies support the view that CHIPs may have a relationship with
t-MN development, at least in a significant proportion of patients.
Thus. Wong et al. explored 22 cancer patients who developed t-MN by
error-corrected NGS whole genome sequencing; for 7 of these patients
displaying clonal TP53
mutations, peripheral blood or bone marrow samples were available 3-8
years before the development of t-MN: four of these seven patients
showed biallelic TP53 mutations in peripheral blood or bone marrow years before chemotherapy treatment.[60] In addition, studies in chimeric mice supported a competitive advantage of TRP53-mutated HSCs over TRP53-WT HSCs.[198]
Takahashi
and coworkers have explored the possible association between clonal
hematopoiesis and the risk of t-MNs; they initially explored a group of
14 cancer patients developing t-MN and found that 71% displayed
preleukemic mutations in their peripheral blood samples.[199]
In a control group of cancer patients, CHIP was found in 31 cases:
these CHIP-positive patients exhibited a higher frequency of t-MN
development after a five-year follow-up than the rest of the
CHIP-negative patients (30% vs. 7%).[199] Finally,
in another cohort of lymphoma patients treated with CHOP chemotherapy,
7% developed t-MN and 80% of them displayed CHIP.[199]
In line with these observations, Gillis et al. reported that cancer
patients (that were 70 years or older at diagnosis and were treated
with chemotherapy) with t-MN were more likely to have CHIP than those
who do not develop t-MN (62% vs. 27%, respectively); the mutational
spectrum of patients with t-MN compared to those without t-MN differed
for a higher prevalence of TP53 mutations.[200]
Other
recent studies explored the possible role of clonal hematopoiesis in
t-MN observed in gynecological cancer patients. For example, Kwan et
al. showed that 2.1% of ovarian cancer patients enrolled in the ARIEL2
and RIEL3 studies based on the administration of rucaparib, a PARP1
inhibitor, developed t-MN.[201] Furthermore, the
frequency of homologous recombination repair gene mutations in the
tumor was associated with an increased prevalence of t-MN; the
frequency of pre-existing CHIP containing TP53
gene mutations with a VAF of 1% or greater was significantly higher in
PBMNs from cases with t-MN compared to those without t-MN (45% vs.
13.6%).[201] The longitudinal analysis showed that pre-existing TP53-mutated CHIPs underwent an expansion in patients who developed t-MN.[201]
Khalife-Hachem
performed a molecular analysis of 77 cancer patients with gynecologic
and breast cancer both developing t-MN involving the molecular
profiling of myeloid neoplasia at diagnosis of t-MN and CHIP in the
peripheral blood or bone marrow at the time of diagnosis of primary
cancer.[202] The molecular characterization of t-MNs
provided evidence that these leukemias can be classified into three
different subgroups, according to Lindsley et al.:[203] TP53/PPM1D subgroup, MDS-like subgroup (defined by the presence of mutations such as SRSF2, SF3B1, U2AF1, ASXL1, EXH2, BCOR, STAG1) and de-novo/pan AML subgroup. CHIP was detected in 66% of these patients.[202] The patients with TP53 or PPM1D
mutations had more treatment lines and more complex karyotypes (7.5
months median OS); the patients with MDS-like features were older and
with more gene mutations (14.5 months OS); the patients with
de-novo/pan-AML mutations were younger with more balanced chromosomal
translocations (25.2 months OS).[203]
Targeted
sequencing of known cancer genes showed that clonal hematopoiesis
occurred in about 4% of children after cytotoxic treatments.[194]
t-MNs represent a major cause of premature death in childhood cancer
survivors and affect about 7-11% of children undergoing treatment for
neuroblastoma or sarcoma. Coorens et al. reported the analysis of two
children affected by neuroblastoma and developing t-MN after cytotoxic
treatment of their tumors, including autologous stem cell
transplantation; in both these patients, there was evidence of CHIP
mutations present in peripheral blood/bone marrow at the time of
diagnosis of primary cancer.[204]
The molecular mechanisms through which mutant TP53
mutations promote hematopoietic stem and progenitor cell expansion are
largely unknown. However, a recent study provided evidence that mutant TP53 induces a growth, and competitive advantage for HSC/HPCs, as shown by transplantation and post-radiation recovery studies.[205] These effects are mediated, at least in part, through interaction between mutant TP53
and EZH2, thus increasing the expression of genes involved in the
self-renewal of HSCs.[205] These observations suggest that EZH2 may
represent a therapeutic target for preventing CHIP progression.[205]
Clonal Hematopoiesis and Acute Myeloid Leukemia Development
Initial
studies have shown that aged subjects with clonal hematopoiesis
mutations have an increased risk of developing hematologic
malignancies, including AML.[4-6] Two studies have explored the possible
association between CHIP and AML development.Abelson
et al. have used an experimental strategy to distinguish individuals at
high risk of developing AML from those with a low risk of developing
AML based on the deep sequencing of genes recurrently mutated in AML in
the peripheral blood of 95 individuals who later developed AML (pre-AML
group), compared with 414 individuals who do not develop AML (control
group). Several remarkable differences were observed between these two
groups of individuals. For example, (i) CHIP with driver
mutations was observed in 73.4% of pre-AML individuals, compared to
36.7% of controls; (ii) 39% of pre-AML individuals harbored a driver
mutation with VAF >10%, compared to 4% of controls; (iii) the median
number of CHIP driver mutations per individual increased with age and
was higher in the pre-AML group compared to the control group; (iv)
analysis of the distribution of VAF supported the existence of larger
size clones among pre-AML individuals compared to control individuals;
(v) the proportion of pre-AML harboring driver mutations such as DNMT3A, TET2, SRSF2, ASXL1, TP53, U2AF1, JAK2, RUNX1, IDH2 was higher in pre-AML cases compared to controls.[206] Importantly, mutations in certain genes, such as mutations of TP53 and the splicing factor U2AF1, were associated with the highest risk of AML development, while mutations of DNMT3A and TET2 were associated with a lower risk of leukemia development.[206]
Interestingly, the comparative, longitudinal analysis of the
hematological parameters of the two groups of individuals showed that
those who developed AML had significantly higher red cell distribution
width (RDW) than the control group; high RDW preceded AML development
by several years.[206] Furthermore, the presence of two or more CHIP
mutations per individual and CHIP mutations with VAF ≥ 9% were also
associated with a high risk of AML development.A
similar approach was adopted by Desai et al.; they explored 188 women
developing AML and 212 controls not developing AML during a follow-up
of about ten years for the presence of CHIP mutations in their
peripheral blood.[207] The group developing AML was explored for CHIP mutations when healthy, years before AML development.[207] Individuals of the pre-AML group displayed a higher number of CHIP mutations than the control group.[207] The most common mutations observed in CHIP with a VAF above 1% were DNMT3A (36.7% in pre-leukemia vs 18.8% in controls), TET2 (25% vs 5.5%), TP53 (11.2% vs 0%), SRSF2 (6.9% vs 0%), IDH2 (6.4% vs 0%), SF3B1 (5.9% vs 1.1%), JAK2 (5.3% vs 0.6%) and ASXL1 (3.% vs 3.3%); collectively, spliceosome genes (SF3B1, SRSF2, U2AF1) were detected in 13.8% of pre-leukemia individuals compared to 1.1% of controls.[207]
The mutational complexity was higher in pre-leukemia (46.8% of cases
with co-mutations) compared to controls (5.5% of cases with
co-mutations). Some of these gene mutations showed greater specificity
and penetrance for leukemia development, such as TP53, IDH1/IDH2, and RUNX1/PHF6, in that 100% of individuals with these mutations developed leukemia.[207] Multivariate analysis showed that some mutations conferred a high risk of AML development: TP53, IDH1, IDH2, sf3b1, SRSF2, U2AF1, TET2, and DNMT3A, in decreasing order of penetrance.[207] The presence at baseline of these CHIP mutations shortened the time to AML presentation.[207] The mutations of TP53 and IDH genes at any VAF were associated with an increased AML risk; DNMT3A, TET2, and spliceosome genes conferred a higher risk of AML development when present at high VAF (i.e., >10%).[207]Young
et al. have explored with a similar approach a pre-leukemia group of 35
cases and a group of 69 controls using error-corrected NGS to assess
somatic mutational profile at VAF ≥ 0.1%.[208] They
observed AML-associated mutations at the level of CHIP in 97% of all
participants; Individuals with mutations ≥ 1% VAF displayed a
significantly increased risk of AML development, as well as individuals
with higher-frequency clones and those with DNMT3A R882H/C mutations.[208] Watson
et al. have analyzed the literature data on blood sequencing from about
50,000 individuals and have reached the conclusion that positive
selection and not drift, is the major force driving and shaping clonal
hematopoiesis; this analysis led the authors to quantify the fitness
advantage of specific genetic driver variants and their capacity to
confer a higher risk of developing AML: thus, using this approach, 20
high-risk variants were identified.[209]
Incorporating specific gene variants and their VAF into predictive
algorithms may provide an important contribution to predicting the risk
of developing AML and thus identifying individuals with a high-risk
condition of leukemic development.[209]As discussed above, the presence of TP53
gene alterations in CHIPs is the somatic genetic alteration that
induces the highest risk of leukemic transformation of these cells.
About 10-15% of AMLs exhibit TP53 alterations, either like mutations either deletions, or a combination of both alterations.[210] Several observations support the view that TP53-mutated AML forms a peculiar group of AMLs with typical cellular and molecular properties.[210] TP53
aberrations in AML include gene mutations, mostly involving the DNA
binding domain of the gene, and deletions of different sizes implying
the TP53 locus at the level of chromosome 17p13. Functional studies on missense TP53
mutant variants commonly observed in AML indicate loss-of-function
effects and induction of effects comparable to those observed with
complete TP53 inactivation; these findings have suggested a dominant-negative effect as the primary force of selection of TP53 mutations in myeloid malignancies.[211] In addition to somatic TP53 mutations, Tp53 germline mutations are observed in a minority of AML patients and are more frequent in t-AML.[212] The prognostic impact of different TP53 mutations is heterogeneous; in fact, Stengel et al. have explored a large cohort of TP53-mutated AMLs: TP53 mutations were detected in 13% of cases (mutation-only 7%; mutation + deletion 5%; deletion - only 1%); all patients with TP53 mutations alone or in association with TP53 deletions, but not cases with TP53 deletions-only, were associated with a poor prognosis and reduced overall survival.[213] Recent studies have addressed the problem of the heterogeneity of VAF of TP53 mutations observed in AMLs. Prochazka and coworkers characterized TP53 mutations
in 98 AML patients and reported a VAF >40% in 62.2% of cases, VAF
20-40% in 19.4% of cases, and VAF <20% in 18.4% of cases; AMLs with
subclonal TP53 mutations (VAF <20%) displayed fewer complex karyotypes and chromosomal losses than AMLs with clonal TP53 mutations.[214] All three mutant TP53 groups showed similarly reduced PFS and OS compared to TP53-WT AMLs.[214] Another study confirmed a worse prognosis of TP53-mutated AMLs was observed irrespective of the allele burden, including cases with VAF <20%.[215] Transplantation studies of AML samples with clonal TP53
mutations into immunodeficient mice provided evidence that these
mutations characterize pre-leukemic stem cells in AML; this conclusion
was supported by the observation that TP53 mutations were detected both in myeloid and lymphoid cell compartments, including T lymphocytes.[216] Interestingly, AML specimens with subclonal TP53 mutations also displayed multilineage engraftment potential in transplantation studies in immunodeficient (NOD/SCID) mice.[217] P53 pathway is deregulated in AML more frequently than predicted based on TP53
mutational frequency. In fact, loss of p53 function may also originate
through the aberrant expression of a protein that acts as a
physiological regulator of p53 stability and function. One of these
mechanisms is related to the overexpression of MDM2, a negative
regulator of TP53, overexpressed in a part of AMLs with TP53-WT; these AMLs were characterized by the absence of p21 expression and by a negative prognosis.[218]
Other studies have shown that overexpression in AML also of MDMX (also
known as MDM4) and the pharmacologic inhibition of both MDM2 and MDM4
resulted in inhibition of cell proliferation and induction of apoptosis
of primary AML cells.[219] Absent
or reduced protein levels of p53 are observed in 50% of AMLs. P53
haploinsufficiency or loss plays a key role in the development of AMLs
with FLT3-ITD mutation.[220]
Stem Cell Transplantation, Clonal Hematopoiesis and the Risk of Leukemic Transformation
The
study of the expansion and potential leukemic evolution of CHIPs
offered a tool to understand better the biology of clonal hematopoiesis
and the risk of leukemic progression. In fact, cell-extrinsic stress
associated with the post-HSCT reconstitution of autologous (auto-SCT)
or donor-derived (allo-SCT) hematopoiesis accelerates the timing of
events underlying CHIP evolution. In this context, particularly
informative was the study performed by Ortmann et al. on 81 patients
with solid tumors or lymphoid diseases undergoing auto SCT.[221]
These patients were studied for CHIP-associated mutations at diagnosis,
at stem cell collection for transplantation, and at first follow-up PB
analysis post-transplantation.[221] 22% of these
patients displayed CHIP; 16 of the 28 CHIP mutations found in the
post-transplantation analysis were tracked back to the graft: for 15/16
of these mutations, the clone size increased after transplantation.[221]
This finding may be interpreted assuming that CHIP mutations conferred
a reconstitution advantage to mutated HSCs in the setting of auto-SCT.[221] Soerensen
et al. explored, in a cohort of 1130 cancer patients undergoing
auto-SCT, the CHIP mutational profile of 36 patients with non-myeloid
malignant disease developing t-MNs following auto-SCT (case subjects).[222]
These patients were compared to an equal number of patients undergoing
auto-SCT and not developing t-MNs (controls). Two remarkable
differences distinguished these two groups of patients: (a) case
subjects were poorer mobilizers of CD34+
cells at leukapheresis compared to controls, thus suggesting that they
have a reduced bone marrow function; (b) while case patients and
controls display a comparable CHIP mutational frequency at the level of
DNMT3A and TET2 genes, case patients exhibited a markedly higher frequency of low VAFs TP53, ASXL1, ZRSR2, SRSF2 and SF3B1 mutations.[222]
In these patients, t-MNs could directly originate from these CHIPs
through a process of both clonal expansion and acquisition of new
mutations driven by stresses induced by HSC reconstituting activity.[222]
Similar conclusions were reached by Berger et al. in a group of
lymphoma patients developing t-MNs after auto-SCT, suggesting that
these leukemias developing after auto-SCT originate from HSCs bearing
pre-t-MN mutations that are present years before disease onset; in
fact, in 70% of these patients CHIPs were identified.[223] Furthermore, 3/7 patients CHIP-positive developing t-MN possess TP53 mutations at low VAF, exhibiting a marked increase of their VAF in t-MN.[85]
However, sequencing studies of CHIP and corresponding t-MNs do not
support, in all cases, a direct origin of leukemias from the evolution
of CHIPs.[223] The
study of patients who underwent allo-SCT from donors with CHIP offers a
unique opportunity to explore the mechanisms of leukemic evolution of
clonal hematopoiesis. Frick et al. explored the role of donor clonal
hematopoiesis in allo-SCT; the analysis of 500 stem cell donors' blood
samples showed clonal hematopoiesis in 16% of donors.[224]
The presence of donor CHIP did not affect thrombocyte engraftment time
but induced slightly faster leukocyte engraftment; 2/82 recipients of
donors with CHIP developed cell malignancies compared to 0/426 in
allo-SCT from donors without CHIP.[224] One of the two patients developing donor cell leukemia had a donor CBLC mutation
with a VAF of 8%, which rapidly increased after transplantation and
remained stable until leukemia diagnosis; sequencing of the DCL sample
showed acquisition of TP53 R175H mutation.[224] The other patient developing DCL exhibited the presence of an ASXL1 and a DNMT3A mutation with a VAF between 2% and 3%.[224]Recently,
Gibson et al. reported the results of targeted error-corrected
sequencing on samples of 1,727 donors for allo-SCT and evaluated the
effect of donor clonal hematopoiesis on transplantation outcomes.
Clonal hematopoiesis was detected in 22.5% of donors; the presence of DNMT3A-mutated CHIP was associated with reduced relapse and increased CGVHD; no recipients of donors with CHIP with sole DNMT3A or TET2 developed donor cell leukemia.[225]
Donor cell leukemia was observed with a 10-year cumulative incidence of
0.7%; in 7/8 cases, donor cell leukemia evolved from donor CHIP with
rare TP53 or splicing factor mutations or from donors carrying germline DDX41 mutations.[225]
Other studies have reported cases of donor cell leukemia arising from
CHIP marked by somatic mutations in leukemia-related genes in donors
usually over age 60.[226,227]Gibson
et al. explored six patients in a group of 552 patients undergoing
allo-SCT, characterized by a condition of unexplained cytopenia
occurring after transplantation.[228] Five of these six patients displayed evidence of clonal hematopoiesis, all characterized by the presence of DNMT3A point mutations.[228]
In addition, four of these patients were followed through time: 3 did
not show any evidence of clonal expansion during the first three years
after transplantation; the fourth patient showed clonal expansion,
concomitantly with the acquisition of two additional mutations at the
level of TP53 and ASXL1 genes.[228]The
studies on allo-SCT with CHIP-positive donors showed a low risk of
leukemic transformation, limited to a minority of patients. However, a
debate is still open about the opportunity or not to screen stem cell
donors for clonal hematopoiesis.[229,230]
Heterogeneity of Clonal Hematopoiesis
The
heterogeneity of the mutational spectrum of clonal hematopoiesis is an
important driver of its potentiality for leukemic evolution. Thus, it
was proposed that according to the type and VAF of genes mutated and
the number and/or associations of multiple mutations, clonal
hematopoiesis can be divided into two different groups: CHIP and clonal
hematopoiesis with oncogenic potential (CHOP).[231,232]
Thus, the definition of CHIP implies the existence of clones of
hemopoietic cells with a minimal allele burden of 2%, the presence of
normal blood cell counts, the absence of persistent cytopenia, and the
exclusion of any pathology associated with the somatic alterations.[231,232]
CHOP is differentiated from CHIP for the presence of disease-related
mutations, cell differentiation, and/or cell proliferation.[231,232] A
notable example of the difference between CHIP and CHOP mutations is
given by a recent study by Cappelli et al., who studied 150 NPM1-mutated
AMLs treated with standard induction and consolidation therapy and
achieving a condition of complete molecular remission as assessed by
the absence of NPM1 transcripts at the end of treatment.[233]
These patients were explored by targeted NGS; at complete molecular
remission, 46% of patients displayed at least one mutation, with a VAF
cutoff of ≥ 1%, 27% had persistent DTA (DNMT3A, TET2, ASXL1)
mutations, and 15% persisting non-DTA mutations; patients with
persistence or acquisition of non-DTA mutations showed a worse
prognosis, while CHIP-like mutations did not affect the overall
survival.[233] Based on clonal evolution of longitudinal AML samples, it was concluded that in NPM1-mutated AMLs, CHIP-like mutations detectable from diagnosis to relapse were DNMT3A, TET2, ASXL1, IDH1, IDH2, SRSF2 and CHOP-like mutations, usually acquired at remission and/or persisting or acquired at relapse, include FLT3TKD, GATA2, NRAS, PTPN11, WT1, TP53, RUNX1.[233]
Clonal Hematopoiesis in Cytopenias
In
most individuals, clonal hematopoiesis is associated with a normal
hematologic phenotype, with normal blood cell counts and morphology.
However, in a minority of subjects, clonal hematopoiesis is associated
with cytopenia.[96] Cytopenia is defined as a
condition in which the patients must have, for at least six months,
hemoglobin, platelets and neutrophil counts less than 11g/dL, 100x109/L and 1.5x109/L,
respectively.[234] If a patient with cytopenia does not fulfil the
criteria for diagnosis of myelodysplastic syndrome, it is most probably
affected by the so-called idiopathic cytopenia of undetermined
significance (ICUS).[235] When a subject with ICUS
displays a somatic mutation in myelodysplasia-associated genes, in the
absence of diagnostic criteria for MDS, the condition is considered a
clonal cytopenia of undetermined significance (CCUS).[235]The
incidence of peripheral blood cytopenias was explored in the general
aging population. Thus, the analysis of the incidence of cytopenias in
the aging, making use of the prospective and population-based Lifelines
cohort comprising 167,729 community-dwelling individuals living in the
northern part of The Netherlands, showed anemia in 4,2% of cases,
thrombocytopenia in 1.6% and neutropenia in 4.8%.[236]
Anemia and thrombocytopenia increased with older age, while neutropenia
showed no increase in prevalence with older age; anemia and
thrombocytopenia and particularly the concomitant presence of anemia
and thrombocytopenia were associated with reduced overall survival; for
individuals aged ≥60 years the incidence of hematological malignancies
was higher among individuals with anemia or thrombocytopenia but not
neutropenia.[236] The highest incidence and mortality of hematological malignancies were observed in individuals with >1 cytopenia.[236] It
is important to point out that the diagnosis of ICUS does not imply
evidence of a clonal disorder. However, the exploration of ICUS
subjects in various studies showed that a part of these patients
displayed somatic mutations. Two initial investigations revealed that a
significant proportion of ICUS subjects have somatic mutations, partly
overlapping with those observed in myelodysplastic syndromes.[237-238]
Kwok et al. explored 369 patients with ICUS by targeted NGS and
observed that 28% of patients with ICUS displayed one or more somatic
mutations: these patients were subdivided into a dysplastic (patients
with rare dysplastic features), and non-dysplastic group and somatic
mutations were observed in 62% of patients with rare dysplastic
morphology and 20% of patients with no evidence of dysplasia.[237]
Patients with ICUS displayed fewer mutations than those with
myelodysplasia and less frequently two or more mutations; the spectrum
of mutated genes in ICUS patients was similar to that observed in
myelodysplastic syndromes, except for SF3B1 mutations and other splicing factors that are less frequent in ICUS patients without dysplasia.[237]
Cargo et al. showed that patients developing at later stages MDS or AML
display in pre-diagnostic samples somatic mutations with a spectrum
mirroring more than observed in MDS than in healthy individuals (CHIP).[238] Malcovati
et al. using a panel of 40 myeloid genes in a cohort of 683 patients
with myeloid neoplasms, including 154 ICUS patients, showed that about
36% of ICUS patients displayed one or more somatic mutations; the
number of somatic mutations per patient and their VAF was lower in ICUS
patients than in patients with myeloid neoplasms.[239] The most recurrent mutations observed in ICUS patients involved TET2 (14.9%), ASXL1 (8.4%), DNMT3A (8.4%), SRSF2 (7.1%), SF3B1, ZRSF2, IDH2, RUNX1 (all with a frequency of 2%).[239]
ICUS patients with clonal mutations (CCUS patients) displayed a
probability of developing a myeloid neoplasm that was 14 times higher
than that of patients with no evidence of clonal disease; ICUS patients
with spliceosome gene mutations or co-mutated gene patterns involving
epigenetic regulators had a risk of disease progression comparable to
patients with a myeloid neoplasm.[239] In addition
to somatic mutations, ICUSs, at least in some patients, display
structural aberrations. Thus, Mikkelsen et al. explored the possible
presence of structural aberrations (CNAs and CNLOHs) in a cohort of 153
patients with ICUS.[240] 23 of 153 ICUS patients
displayed structural aberrations, excluding LOY; mutations in
MDS-related genes were observed in 52% of these 23 patients; the
CAN/CNLOH identified in the ICUS patients were similar to those
observed in myeloid malignancies.[240] 10% of these ICUS patients
progressed to myeloid malignancy in a median follow-up time of 25
months; all patients but one who progressed displayed somatic
mutations. The presence of CAN/CNLOH was associated with reduced PFS
and OS, while the presence of somatic mutations was associated with
reduced PFS but not OS.[240] In the group of CCUS patients, after a
follow-up of 24 months, the median overall survival was 67 months in
the group of patients with CAN/CNLOH, compared to 104 months in the
group with or without CAN/CNLIH.[240]Cytotoxic
therapies may worsen a condition of cytopenia associated with clonal
hematopoiesis. Singh recently reported the study of 13 cases of
endocrine cancer patients undergoing treatment with peptide receptor
radionuclide therapy (PRRT). 62% of patients displayed CHIP at
baseline; persistent cytopenias were observed in 64% of the patients;
PRRT exposure resulted in clonal expansion of the mutant DNA damage
response genes TP53, PPM1D and CHEK2 and associated cytopenias in 75%
of the patients.[241]Some
CCUS patients may develop transfusion-dependent anemia; no large
clinical studies have explored these patients, but a recent
single-center study showed that transfusion-dependent CCUS patients
might benefit from MDS-type therapies, such as growth factors and
hypomethylating agents.[242]
Mouse Models of Clonal Hematopoiesis
Several mouse models involving the main genes implied in human CHIP have been developed. Many studies involved the analysis of DNMT3A, the gene most frequently mutated in CHIP. Knocking out DNMT3A in mice showed a consistent enhancement of LT-HSC self-renewal, with an expansion of the pool of these cells.[243] A sequential knocking experiment showed that: (i) DNMT3A mutation R878H (the equivalent of human R882H) induces an expansion of HSCs and MPPs; (ii) the induction of NPM1 mutation in these cells causes progression to a myeloproliferative disorder.[244] The cooperativity of DNMT3A and NPM1
mutations in inducing leukemia development is related to the capacity
of the mutant gene to alter chromatin structure at the level of HSCs
inducing increased accessibility of gene promoters of cell cycling,
stem cell, transcription factors, and PI3K/AKT/mTOR signaling members.[245] In addition to the effects on HSC expansion, DNMT3A knockout also induced a decreased bone mass via increased osteoclastogenesis.[246] In line with this observation, subjects with DNMT3A mutant CHIP showed increased incident osteoporosis and reduced bone mineral density.[246]A recent study systematically explored and characterized at a molecular level the different DNMT3A mutants observed in human hematopoietic cells and showed that 74% were loss-of-function mutations; half of DNMT3A variants exhibited reduced protein stability.[247] Several
recent studies based on the analysis of mouse knockout models or single
human hematopoietic cells bearing R882H mutations have supported the
conclusion that DNMT3A
mutants induce a condition of selective hypomethylation, largely
responsible for their pro-leukemic effect. Thus, Smith et al. developed
a somatic, inducible model of hematopoietic DNMT3A loss and showed that inactivation of DNMT3A in murine hematopoietic cells induces a relatively slow loss of methylation of some DNA sites throughout the genome.[248]
According to this observation, it was suggested that slow methylation
loss might explain the long latent period required for clonal expansion
and leukemic development in individuals with CHIP DNMT3A mutations.[248] Furthermore, Nam et al. reported a sing cell analysis of CD34+ cells purified from individuals bearing CHIP bearing DNMT3A
R882H mutation; multi-omics single-cell sequencing to detect the
mutational status of individuals cells was applied together with
downstream epigenetic and transcriptional information, thus enabling to
compare mutated cells with their wild-type counterparts from the same
individuals.[249] The results of this single-cell analysis showed that: (i) DNMT3A
mutations resulted in myeloid over lymphoid imbalance of HSCs, and in
an expansion of immature myeloid progenitors primed toward
megakaryocytic-erythroid fate; (ii) DNMT3A
R882H resulted in preferential hypomethylation of polycomb repressive
complex two targets and in the dysregulated expression of lineage and
leukemia stem cell markers and some key hematopoietic transcription
factors.[249]Other
studies have explored the effects of TET2 mutations on stem/progenitor
cells. Two initial studies showed that the conditional TET2 loss in the hematopoietic stem cell compartment leads to increased HSC self-renewal; in some animals, TET2 loss may also lead to a myeloproliferative condition.[250-251] While TET2 knockout mice exhibited expansion of HSCs and HPCs, TET2 mutant mice, engineered with a catalytic inactive TET2 variant, predominantly developed myeloid malignancies.[252]Studies in TET2-mutant
cells have characterized their methylation abnormalities. The
conversion of 5-methylcytosine (5mC) to hydroxymethylcytosine (5hmC) is
the key step of DNA demethylation catalyzed by TET2,
which requires ascorbate. 5hmC maps at the level of specific DNA sites
and is associated with active transcription and chromatin accessibility
of key hematopoietic regulators.TET2
mutations in primary human HSCs/HPCs determine an increased
self-renewal activity, increased colony-forming activity in vitro,
defective erythroid/megakaryocytic differentiation, and myeloid
skewing, associated with a decrease of 5hmC at the level of
erythroid-associated gene loci.[253] The study of the methylation profile of CHIP and CCUS subjects provided evidence that TET2
mutations are associated with DNA hypermethylation at enhancer DNA
sites: the hypermethylated sites are functionally related to genes
involved in leukocyte function and immune response and to ETS-related
and C/EBP-related transcription factor motifs. Most TET2 -associated
hypermethylation sites are shared between CHIP and AML; some
AML-specific hypermethylation sites are located at active enhancer DNA
elements in HSCs.[254]In conclusion, the studies on CHIP hematopoietic cells bearing mutated DNMT3A and/or TET2
display an altered phenotype resulting in specific changes in the DNA
methylome, which in turn lead to altered regulation of specific genes
or gene sets.Other studies have characterized the biological effects elicited by the induction of ASXL1 loss or of ASXL1 mutants. ASXL1 loss in vivo
induces progressive multilineage cytopenias and dysplasia with an
increased size of the pool of HSCs and impaired mature cell
differentiation, resulting in several changes resembling human MDS.[255-256] A similar phenotype was observed by retroviral overexpression of mutant ASXL1, thus suggesting that ASXL1 variants may inhibit the function of WT ASXL1.[257] Physiological expression of a C-terminal truncated ASXL1 mutant in vivo
using conditional knock-in resulted in HSC expansion, myeloid skewing,
age-dependent anemia, thrombocytosis, and morphological dysplasia.[258] Using this knockin mouse overexpressing a-terminally truncated form of ASXL1-mutant, the effect of mutant ASXL1 on physiological aging in HSCs was explored: HSCs expressing mutant ASXL1 acquire clonal advantage during aging; mutant ASXL1 cooperates with BAP1 to deubiquitinates and activate AKT.[259]An analysis carried out on a UK Biobank cohort provided evidence that ASXL1
loss of function mutations are strongly associated with current and
past smoking status; this finding strongly supports the hypothesis that
the inflammatory environment induced by smoking may promote the
outgrowth of ASXL1-mutant clones.[260]
Comments and Conclusions
The
studies carried out in the last two decades have elucidated in part the
mechanisms underlying the aging of HSCs and the development of clonal
hematopoiesis.
The most relevant findings can be summarized as follows:
- Even
though HSCs increase in number with age, they have a significantly
decreased self-renewal capacity and reconstitution potential upon
transplantation.
- By
aging, human HSCs become more myeloid-biased in their differentiation
potential, a phenomenon seemingly related to the increase of their
clonality with age, selecting for myeloid-biased HSC clones.
- It
was estimated that HSCs or MPPs accumulated a mean number of 17
mutations per year after birth and lost 30 base pairs per year of
telomere length.
- Hematopoiesis
in individuals under 65 years of age was largely polyclonal, implying a
population of 20,000-200,000 HSC/MPPs. By contrast, in individuals aged
75 years, 30-60% of hematopoiesis is sustained for 12-18 independent
clones. Thus, there is a progressive switch from polyclonal to
oligoclonal hematopoiesis during aging.
- CHIP is characterized by the presence of some recurrently mutated genes, such as DNMT3A, TET2 and ASXL1, all three acting as epigenetic modifiers. However, less frequently are mutated genes involved in DNA damage response (PPM1D, TP53, CHEK 2), spliceosome (SF3B1, SRSF2, U2AF1), or epigenetic control (IDH1, IDH2).
- DNMT3A-mutant clones preferentially expanded early in life, while splicing gene mutations expanded only in old individuals, and TET2-mutant clones expanded at all ages.
- CHIP
occurs with higher frequency in individuals with lymphoid or solid
tumors and is associated with expexposure genotoxic stress. PPM1D
mutations drive clonal hematopoiesis in response to cytotoxic
chemotherapy. Clonal hematopoiesis confers a greater risk of developing
therapy-related myeloid neoplasms.
- CHIP
is associated with a consistently increased risk (about 10-fold) of
hematopoietic malignancy; some CHIP features are associated with a
higher risk of transformation, such as the presence of TP53 or spliceosome gene mutations, a VAF> 10%, and the presence of multiple mutations.
- Some
individuals display clonal hematopoiesis in association with peripheral
blood cytopenia, a condition known as clonal cytopenia of undetermined
significance (CCUS)
- Individuals
with CHIP and CCUS have a markedly higher probability of leukemic
transformation than those with cytopenia alone.
References
- Ivanovs A, Rybtsov S, Ng ES, Stanley EG, Elefanty AG, Medvinsky A.
Human hematopoietic stem cell development: from the embryo to the dish.
Development 2017; 144: 2323-2337. https://doi.org/10.1242/dev.134866
PMid:28676567
- Tavian M, Robin C, Coulombel L,
Péault B. The human embryo, but not its yolk sac, generates
lympho-myeloid stem cells: mapping multipotent hematopoietic cell fate
in intraembryonic mesoderm. Immunity 2001; 15(3): 487-495.
https://doi.org/10.1016/S1074-7613(01)00193-5
- Perdiguero
EG, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H,
Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident
macrophages originate from yolk-sac-derived erythron-myeloid
progenitors. Nature 2015; 518(7540): 547-551.
https://doi.org/10.1038/nature13989 PMid:25470051
PMCid:PMC5997177
- Bian Z, Gong Y, Huang T, Lee C,
Bian L, Bai Z, Shi H, Zeng Y, Liu C, He J, Zhou J, Li X, Li Z, Ni Y, Ma
C, Cui L, Zhang R, Chan J, Ng LG, Lan Y, Ginhoux F, Liu B. Deciphering
human macrophage development at single-cell resolution. Nature 2020;
582(7813): 571-576. https://doi.org/10.1038/s41586-020-2316-7
PMid:32499656
- Dick SA, Wong A, Hamidzada H, Nejat
S, Nechanitzky R, Vohra S, Mueller B, Zaman R, et al. Three tissue
resident macrophage subsets coexist across organs with conserved
origins and life cycles. Sci Immunol 2022; 7(67): eabf7777.
https://doi.org/10.1126/sciimmunol.abf7777 PMid:34995099
- Atkins
MH, Scarfò R, McGrath KE, Yang D, Palis J, Ditadi A, Keller GM.
Modeling human yolk sac hematopoiesis with multipotent stem cells. J
Exp Med 2022; 219(3): e20211924. https://doi.org/10.1084/jem.20211924
PMid:34928315
- Zhao S, Feng S, Tian Y, Wen Z.
Hemogenic and aortic endothelium arise from a common hemogenic
angioblast precursor and are specified by the ETV2 dosage. Proc Natl
Acad Sci USA 2022; 119(13): e2119051119.
https://doi.org/10.1073/pnas.2119051119 PMid:35333649
PMCid:PMC9060440
- Baron CS, Kester L, Klaus A,
Boisset JC, Thambyrajah R, Yvernogeau L, Kouskoff V, Lacaud G, van
Oudenaarden A, Robin C. Single-cell transcriptomics reveal the dynamic
of haematopoietic stem cell production cell production in the aorta.
Nature Commun 2018; 9: 2517. https://doi.org/10.1038/s41467-018-04893-3
PMid:29955049 PMCid:PMC6023921
- Zhu Q, Gao P,
Tober J, Bennett L, Chen C, Uzun Y, Li Y, Howell ED, Mumau M, Yu W, He
B, Speck NA, Tan K. Developmental trajectory of prehematopoietic stem
cell formation from endothelium. Blood 2020; 136(7): 845-856.
https://doi.org/10.1182/blood.2020004801 PMid:32392346
PMCid:PMC7426642
- Hou S, Li Z, Zheng X, Gao Y,
Dong J, Ni Y, Wang X, LI Y, Ding X, Chang Z, Li S, Hu Y, Fan X, Hou Y,
Wen L, Liu B, Tang F, Lan Y. Embryonic endothelial evolution towards
first hematopoietic stem cells revealed by single-cell transcriptomic
and functional analyses. Cell Res 2020; 30(3): 376-392.
https://doi.org/10.1038/s41422-020-0300-2 PMid:32203131
PMCid:PMC7196075
- Zeng Y, He J, Bai Z, Li Z, Gong Y, Liu
C, Ni Y, Du J, Ma C, Bian L, Lan Y, Liu B. Tracing the firs
haematopoietic stem cell generation in human embryo by single-cell RNA
sequencing. Cell Res 2019; 29(11): 881-894.
https://doi.org/10.1038/s41422-019-0228-6 PMid:31501518
PMCid:PMC6888893
- Barcena A, Muench MO, Kapidzic
M, Fisher SJ. A new role for the human placenta as a hematopoietic site
throughout gestation. Reprod Sci 2009; 16(2): 178-187.
https://doi.org/10.1177/1933719108327621 PMid:19208786 PMCid:PMC2731631
- Van Handel B, Prashad SL, Hassanzadeh-Kiabi N, Huang A,
Magnusson M, Atanassova B, Chen A, Hamalainen EI, Mikkola H. The first
trimester human placenta is a site for terminal maturation of primitive
erythroid cells. Blood 2010; 116(17): 3321-3330.
https://doi.org/10.1182/blood-2010-04-279489 PMid:20628147
PMCid:PMC2995359
- Robin C, Bollerot K, Mendes S, Haak E,
Crisa M, Cerisoli F, Lauw I, Kaimakis P, Jorna R, Vermeulen M, Kayser
M, van der Linden R, Imarinad P, Verstegen M, Nawaz-Yousaf H, Papazian
N, Steegers E, Cupedo T, Dzierzak E. Human placenta is a potent
hematopoietic niche containing hematopoietic stem and progenitor cells
throughout development. Cell Stem Cell 5(19): 385-395.
https://doi.org/10.1016/j.stem.2009.08.020 PMid:19796619
PMCid:PMC2812802
- Muench MO, Kapidzic M, Gormley
M, Gutierrez AG, Ponder KL, Fomin ME, Beyer AI, Stolp H, Qi Z, Fisher
SJ, Barcena A. The human chorion contains definitive hematopoietic stem
cells from the fifteenth week of gestation. Development 2017; 144(8):
1399-1411. https://doi.org/10.1242/dev.138438 PMid:28255007
PMCid:PMC5399660
- Calvanese V, Capellera-Garcia S,
Ma F, Fares I, Liebscher S, Ng E, Ekstrand S, Aguadé-Grogorio J,
Vavilina A, Lefaudeux D, Nadel B, Li JY, Wang Y, Lee KL, Ardehali R,
Iruela-Arispe ML, Pellegrini M, Stanley EG, Elefanty AG,
Schenke-Layland K, Mikkola H. Mapping human haematopoietic stem cells
from haemogenic endothelium to birth. Nature 2022; 604(7106): 534-540.
https://doi.org/10.1038/s41586-022-04571-x PMid:35418685
- Holyoake
TL, Nicolini FE, Eaves CJ. Functional differences between
transplantable human hematopoietic stem cells from fetal liver, cord
blood, and adult marrow. Exp Hematol 1999; 29: 927-936.
https://doi.org/10.1016/S0301-472X(99)00078-8
- Walter
D, Lier A, Geiselhart A, Thalheimer FB, Huntscha S, Sobotta MC, Moehrle
B, Brocks D, Bayindir I, Kaschutnig P, et al.Exit from dormancy
provokes DNA-damage-induced attrition in haematopoietic stem cells.
Nature 2015; 520(7548): 549-552. https://doi.org/10.1038/nature14131
PMid:25707806
- Rodriguez-Fraticelli AE, Weinreb
CS, Wang SW, Migueles RP, Jankovic M, Usart M, Klein AM, Lowell S,
Camargo FD. Single-cell lineage tracing unveils a role for Tcf15 in
hematopoiesis. Nature 2020; 583(7817): 585-589.
https://doi.org/10.1038/s41586-020-2503-6 PMid:32669716
PMCid:PMC7579674
- Biswas A, Roy IM, Babu PC,
Manesia J, Schouteden S, Vijakakurup V, Anto RJ, Huelsken J,
Levy-Hulbert A, Varfaille CM, Khurana S. The periostin/integrin-αV axis
regulates the size of hematopoietic stem cell pool in the fetal liver.
Stem Cell Rep 2020; 15(2): 340-357.
https://doi.org/10.1016/j.stemcr.2020.06.022 PMid:32735820
PMCid:PMC7419718
- Vanuytsel K, Villacorta-Martin
C, Lindstrom-Vautrin J, Wang Z, Garcia-Beltran WF, Vrbanac V, Parsons
D, Lam EC, Matte TM, Dowrey TW, et al. Multi-modal profiling of human
fetal liver hematopoietic stem cells reveals the molecular signature of
engraftment. Nat Commun 2022; 13: 1103.
https://doi.org/10.1038/s41467-022-28616-x PMid:35232959
PMCid:PMC8888592
- Jardine L, Webb S, Goh I,
Quiraga Londono M, Reynolds G, Mather M, Olabi B, Stephenson E, Botting
RA, Horsfall D, et al. Blood and immune development in human fetal bone
marrow and Down syndrome. Nature 2021; 598(7880): 327-331.
https://doi.org/10.1038/s41586-021-03929-x PMid:34588693
PMCid:PMC7612688
- Li Y, Kong W, Yang W, Patel RM,
Casey EB, Okeyo-Owuor T, White JM, Porter SN, Morris SA, Magee JA.
Single-cell analysis of neonatal HSC ontogeny reveals gradual and
uncoordinated transcriptional reprogramming that begins before birth.
Cell Stem Cell 2020; 27(5): 732-747.
https://doi.org/10.1016/j.stem.2020.08.001 PMid:32822583
PMCid:PMC7655695
- Catlin SN, Busque L, Gale RE, Guttorp
P, Abkowitz JL. The replication of human hematopoietic stem cells in
vivo. Blood 2011; 117(17): 4460-4466.
https://doi.org/10.1182/blood-210-08-303537 PMid:21343613
PMCid:PMC3099568
- Laurenti E, Frelin C, Xie S,
Ferrari R, Dunant CF, Zandi S, Neumann A, Plumb I, Doulatov S, Chen J,
April C, Iscove N. CDK6 levels regulate quiescence exit in human
hematopoietic stem cells. Cell Stem Cell 2015; 16(3): 302-313.
https://doi.org/10.1016/j.stem.2015.01.017 PMid:25704240
PMCid:PMC4359055
- Takayama N, Murison A,
Takayanagi SI, Arlidge C, Zhou S, Garcia-Prat L, Chan-Sng-Yue M, Zandi
S, Gand OI, Boutzen H, Kaufmann KB, et al. The transition from
quiescent to activated states in human hematopoietic stem cells is
governed by dynamic 3D genome reorganization. Cell Stem Cell 2020;
28(3): 488-501. https://doi.org/10.1016/j.stem.2020.11.001
PMid:33242413
- Liang R, Arif T, Kalmykova S,
Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, Lin F, Benson
DL, Tzavaras N, Mahajan M, Papatsenko D, Ghaffari S. Restraining
lysosomal activity preserves hematopoietic stem cell quiescence and
potency. Cell Stem Cell 2020; 26(3): 359-376.
https://doi.org/10.1016/j.stem.2020.01.013 PMid:32109377
PMCid:PMC8075247
- Garcia-Prat L, Kaufmann KB,
Schneiter F, Voisin V, Murison A, Chen J, Chan-Sen-Yue M, Gan OI,
McLeod JL, Smith SA, Shoong MC, et al. TFEB-mediated endolysosomal
activity controls human hematopoietic stem cell fate. Cell Stem Cell
2021; 28(10): 1838-1850. https://doi.org/10.1016/j.stem.2021.07.003
PMid:34343492
- Waehstedt M, Ladopoulos V, Hidalgo I,
Sanchez Castillo M, Hannah R, Sawan P, Wan H, Dudenhoffer-Pfeifer M,
Magnusson M, Norddahl GL, Gottgens B, Bryder D. Critical modulation of
hematopoietic lineage fate by hepatic leukemia factor. Cell Rep 2017;
21(8): 2251-2263. https://doi.org/10.1016/j.celrep.2017.10.112
PMid:29166614 PMCid:PMC5714592
- Komorowska K,
Doyle A, Wahlestedt M, Bubramaniam A, Debnath S, Chen J, Soneji S, Van
Hendel B, Mikkola H, Miharada K, Bryder D, Larsson J, Magnusson M.
Hepatic leukemic factor maintains quiescence of hematopoietic stem
cells and protects the stem cell pool during regeneration. Cell Rep
2017; 21(12): 3514-3523. https://doi.org/10.1016/j.celrep.2017.11.084
PMid:29262330
- Yokomizo T, Watanabe N, Umemoto T,
Matsuo J, Harai R, Kihara Y, Nakamura E, Tada N, Sato T, Takaku T,
Shimono A, Takizawa H, Nakagata N, Mori S, Kurokawa M, Tenen DG, Osato
M, Suda T, Komatsu N. Hlf marks the developmental pathway for
hematopoietic stem cells but not for erythroid-myeloid progenitors. J
Exp Med 2019; 216(7): 1599-1614. https://doi.org/10.1084/jem.20181399
PMid:31076455 PMCid:PMC6605751
- Lehnertz B,
Chagraoui J, MacRae T, Tomellini E, Corneau S, Mayotte N, Boivin I,
Durand A, Gracias D, Sauvageau G. HLF expression defines the human
hematopoietic stem cell state. Blood 2021; 138(25): 2642-2654.
https://doi.org/10.1182/blood.2021010745 PMid:34499717
- Calvanese
V, Nguyen AT, Bolan TJ, Vavilina A, Su T, Lee LK, Wang Y, Lay FD,
Magnusson M, Crooks GM, Kudistan SK, Mikkola H. MLLT3 governs human
haematopoietic stem-cell self-renewal and engraftment. Nature 2019;
576(7786): 281-286. https://doi.org/10.1038/s41586-019-1790-2
PMid:31776511 PMCid:PMC7278275
- Rentas S,
Holzapfel N, Belew MS, Pratt G, Voisin V, Wilhelm BJ, Bader GD, Yeo GW,
Hope KJ. Musashi-2 attenuates AHR signalling to expand human
haematopoietic stem cells. Nature 532(7600): 508-511.
https://doi.org/10.1038/nature17665 PMid:27121842
PMCid:PMC4880456
- Belew MS, Bhatia S, Keyvani
Chani A, Rentas S, Draper JS, Hope KJ. PLAG1 and USF2 co-regulate
expression of Musashi-2 in human hematopoietic stem and progenitor
cells. Stem Cell Report 208; 10(4): 1384-1397.
https://doi.org/10.1016/j.stemcr.2018.03.006 PMid:29641991
PMCid:PMC5998603
- Wagner JE, Brunstein CG, Boitano
AE, DeFor TE, McKenna D, Sumstad D, Blazar BR, Tolar J, Le C, Jones J,
Cooke MP, Bleul CC. Phase I/II trial of StemRegenin-1 expanded
umbilical cord blood hematopoietic stem cells supports testing as a
stand-alone graft. Cell Stem Cell 2016; 18(1): 144-155.
https://doi.org/10.1016/j.stem.2015.10.004 PMid:26669897
PMCid:PMC4881386
- Cohen S, Roy J, Lachance S,
Delisle JS, Marinier A, Busque L, Roy DC, Barabé F, Ahmad I, Bambase N,
et al. Hematopoietic stem cells transplantation using single
UM171-expanded cord blood: a single-arm, phase 1-2 safety and
feasibility study. Lancer Hematol 2020; 7(2): e134-e145.
https://doi.org/10.1016/S2352-3026(19)30202-9
- Dumont-Lagace
M, Feghaly A, Meunier MC, Finney M, Van't Hof W, Masson Frenet E,
Sauvageau G, Cohen S. UM171 expansion of cord blood improves donor
availability and HLA matching for all patients, including minorities.
Transplant Cell Ther 2022; S2666-6367(22): in press.
https://doi.org/10.1016/j.jtct.2022.03.016 PMid:35311667
- Laurenti
E, Gottgrns B. From hematopoietic stem cells to complex differentiation
landscapes. Nature 2018; 553(7689): 418-426.
https://doi.org/10.1038/nature25022 PMid:29364285
PMCid:PMC6555401
- Kato H, Igarashi K. To be red or
white: lineage commitment and maintenance of the hematopoietic system
by the "inner myeloid". Haematologica 2019; 104(10): 1919-1927.
https://doi.org/10.3324/haematol.2019.216861 PMid:31515352
PMCid:PMC6886412
- Ligett LA, Sankaran VG.
Unraveling hematopoiesis through the lens of genomics. Cell 2020;
182(6): 1384-1400. https://doi.org/10.1016/j.cell.2020.08.030
PMid:32946781 PMCid:PMC7508400
- Yamamoto R, Morita
Y, Oehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H, Nakauchi H.
Clonal analysis unveils self-renewing lineage-restricted progenitors
generated directly from hematopoietic stem cells. Cell 2013; 154(5):
1112-1126. https://doi.org/10.1016/j.cell.2013.08.007
PMid:23993099
- Yamamoto R, Wilkinson AC, Oehara J,
Lan X, Lai CY, Nakauchi Y, Pritchard JK, Nakauchi H. Large-scale clonal
analysis resolves aging of the mouse hematopoietic stem cell
compartment. Cell Stem Cell 2018; 22(4): 600-607.
https://doi.org/10.1016/j.stem.2018.03.013 PMid:29625072
PMCid:PMC5896201
- Carrelha J, Meng Y, Kettyle LM,
Luis TC, Norfo R, Alcolea V, Boukarabila H, Grasso F, Gambardella A,
Grover A, Hogstrand K, Lord AM, Sanjuan-Pla A, Woll PS, Nerlov C,
Jacobsen SE. Hierarchically related lineage-restricyed fates of
multipotent haematopoietic stem cells. Nature 2018; 554(7690): 106-111.
https://doi.org/10.1038/nature25455 PMid:29298288
- Rodriguez-Fraticelli
AE, Wolock SL, Weinreb CS, Panero R, Patel SH, Jankovic M, Sun J,
Calogero RA, Klein AM, Camargo FD. Clonal analysis of lineage fate in
native haematopoiesis. Nature 2018; 553(7687): 212-216.
https://doi.org/10.1038/nature25168 PMid:29323290
PMCid:PMC5884107
- Upadhaya S, Sawai CM, Papalexi
E, Rashidfarrokhi A, Jang G, Chattopadhyay P, Satihja R, Raizis B.
Kinetics of adult hemopoietic stem cell differentiation in vivo. J Exp
Med 2018; 215(11): 2815-2832. https://doi.org/10.1084/jem.20180136
PMid:30291161 PMCid:PMC6219744
- Weinreh C,
Rodriguez-Fraticelli A, Camargo FD, Klein AM. Lineage tracing on
trasncriptional landscapes links state to fate during differentiation.
Science 2020; 367(6479): eaaw3381.
https://doi.org/10.1126/science.aaw3381 PMid:31974159
PMCid:PMC7608074
- Pei W, Shang F, Wang X, Fanti
AK, Greco A, Busch K, Klapproth K, Zhang Q, Quedenau C, Sauer S,
Feyerband TB, Hofer T, Rodewald HR. Resolving fates and single-cell
transciptomes of hematopoietic stem cell clones by PolyloxExpress
barcoding. Cell Stem Cell 2020; 27(20): 383-395.
https://doi.org/10.1016/j.stem.2020.07.018 PMid:32783885
- Velten
L, Haas SF, Raffel S, Blasziewicz S, Islam S, Hennig BP, Hirche C, Lutz
C, Buss EC, Nowak D, Boch T, Hofmann WK, Ho AD, Huber W, Trumpp A,
Essers M, Steinmetz LM. Human hematopoietic stem cell lineage
commitment is a continuous process. Nat Cell Biol 2017; 19(4): 271-281.
https://doi.org/10.1038/ncb3493 PMid:28319093 PMCid:PMC5496982
- Buenrostro
JD, Corces MR, Lareau CA, Wu B, Schep AN, Aryee MJ, Majeti R, Chang HY,
Greenleaf WJ. Integrated single-cell analysis maps the continuous
regulatory landscape of human hematopoietic differentiation. Cell 2018;
173(6): 1535-1548. https://doi.org/10.1016/j.cell.2018.03.074
PMid:29706549 PMCid:PMC5989727
- Psaila B, Mead AJ.
Single-cell approaches reveal novel cellular pathways for megakaryocyte
and erythroid differentiation. Blood 2019; 133(13): 1427-1435.
https://doi.org/10.1182/blood-2018-11-835371 PMid:30728145
PMCid:PMC6443046
- Karamitros D, Stoilova B,
Aboukhalil Z, Hamey F, Reinisch A, Samitsch M, Quek L, Otto G, Repapi
E, Doondeea J, et al. Single-cell analysis reveals the continuum of
human lympho-myeloid progenitor cells. Nat Immunol 2018; 19(1): 85-97.
https://doi.org/10.1038/s41590-017-0001-2 PMid:29167569
PMCid:PMC5884424
- Pellin D, Loperfido M, Baricordi
C, Wolock SL, Montepeloso A, Weinberg OK, Biffi A, Klein AM, Biasco L.
A A comprehensive single cell transcriptional landscape of human
hematopoietic progenitors. Nat Commun 2019; 10(1): 2395.
https://doi.org/10.1038/s41467-019-10291-0 PMid:31160568
PMCid:PMC6546699
- Notta F, Doulatov S, Laurenti E,
Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic
stem cells capable of long-term multilineage engraftment. Science 2011;
333(6039): 218-221. https://doi.org/10.1126/science.1201219
PMid:21737740
- Anjos-Afonso F, Buettner F, Mian
SA, Rhys H, Perez-Llorat J, Garcia-Albornoz M, Rastogi N,
Ariza-McNaughton L, Bonnet D. Single cell analyses identify a highly
regenerative and homogeneous human CD34+ hematopoietic stem cell
population. Nat Commun 2022; 13(5): 2048.
https://doi.org/10.1038/s41467-022-29675-w PMid:35440586
PMCid:PMC9018830
- Triana S, Vonfitch D, Jopp-Saile
L, Raffel S, Lutz R, Leonce D, Antes M, Hernandez-Malmierca P,
Ordonez-Rueda D, Ramasz B, et al. Single-cell proteo-genomic reference
maps of the hematopoietic system enable the purification and massive
profiling of precisely defined cell states. Nat Immunol 2021; 22(12):
1577-1589. https://doi.org/10.1038/s41590-021-01059-0 PMid:34811546
PMCid:PMC8642243
- Aksoz M,Gafencu GA, Stoilova B, Buono
M, Meng Y, Jakobsen NA, Metzner M, Clark SA, Beveridge R, Thongjuea S,
Vyas P, Narlov C. Identification and age-independent increase of
platelet biased human hematopoietic stem cells. BioRχIV 2022; in press.
https://doi.org/10.1101/2022.01.14.475546 PMid:35802706
- Loeffler
D, Wehling A, Schneiter F, Zhang Y, Muller-Botticher N, Hoppe PS,
Hilsenbeck O, Kakkaliaris KD, Endele M, Schroeder T. Asymmetric
lysosome inheritance predicts activation of haematopoietic stem cells.
Nature 2019; 573(7774): 426-429.
https://doi.org/10.1038/s41586-019-1531-6 PMid:31485073
- Loeffler
D, Schneiter F, Wang W, Wehling A, Kull T, Lengerke C, Manz MG,
Schroeder T. Asymmetric organelle inheritance predicts human blood stem
cell fate. Blood 2022; 139(13): 2011-2023.
https://doi.org/10.1182/blood.2020009778 PMid:34314497
- Sun
J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho YJ, Klein A, Hofmann O,
Camargo FD. Clonal dynamics of native haematopoiesis. Nature 2014;
14(7522): 322-327. https://doi.org/10.1038/nature13824 PMid:25296256
PMCid:PMC4408613
- Bush K, Klapproth K, Barile M,
Flossdorf M, Holland-Letz T, Schlenner SM, Reth M, Hofer T, Rodewald
HR. Fundamental properties of unperturbed haematopoiesis from stem
cells in vivo. Nature 2015; 518(7540): 542-546.
https://doi.org/10.1038/nature14242 PMid:25686605
- Sawai
C, Babovic S, Upadhaya S, Knapp D, Lavin Y, Lau C, Goloborodko A, Feng
J, Fujisaki J, Ding L, Mirny LA, Merad M, Eaves CJ, Reizis B.
Hematopoietic stem cells are the major source of multilineage
hematopoiesis in adult animals. Immunity 2016; 45(16): 597-609.
https://doi.org/10.1016/j.immuni.2016.08.007 PMid:27590115
PMCid:PMC5054720
- Chapple RH, Tseng YJ, Hu T, Kitano A,
Takeichi M, Hoegenauer KA, Nakada D. Lineage tracing of murine adult
hematopoietic stem cells reveals active contribution to steady-state
hematopoiesis. Blood Adv 2018; 2(11): 1220-1230.
https://doi.org/10.1182/bloodadvances.2018016295 PMid:29848758
PMCid:PMC5998934
- Sawen P, Eldeeb M, Erlandsson E,
Kristiansen TA, Laterza C, Kokaia Z, Karlssonb G, Yuan J, Soneji S,
Mandal PK, Rossi DJ, Bryder D, Murine HSCs contribute actively to
native hematopoiesis but with reduced differentiation capacity upon
aging. Elife 2018; 7: e41258. https://doi.org/10.7554/eLife.41258
PMid:30561324 PMCid:PMC6298771
- Biasco L, Pellin
D, Scala S, Dionisio F, Basso-Ricci L, Leonardelli L,Scaramuzza S,
Baricordi C, Ferrua F, Cicalese MP, et al. In vivo tracking of human
hematopoiesis reveals patterns of clonal dynamics during early and
steady-state reconstitution phases. Cell Stem Cell 2016; 19(1):
107-119. https://doi.org/10.1016/j.stem.2016.04.016 PMid:27237736
PMCid:PMC4942697
- Scala S, Basso-Ricci L, Dionisio
F, Pellin D, Giannelli S, Salerio FA, Leonardelli L, Cicalese MP,
Ferrua F, Aiuti A, Biasco L. Dynamics of genetically engineered
hematopoietic stem and progenitor cells after autologous
transplantation in humans. Nat Med 2018; 24(11): 1683-1690.
https://doi.org/10.1038/s41591-018-0195-3 PMid:30275570
- Lu
R, Neff NF, Quake SR, Weissman IL. Tracking single hematopoietic stem
cells in vivo using high-throughput sequencing in conjunction with
viral genetic barcoding. Nat Biotechnol 2011; 29(10): 928-933.
https://doi.org/10.1038/nbt.1977 PMid:21964413 PMCid:PMC3196379
- Lu
R, Czechowicz A, Seita J, Jiang D, Weissman IL. Clonal-level lineage
commitment pathways of hematopoietic stem cells in vivo. Proc Natl Acad
Sci USA 2019; 116(4): 1447-1456.
https://doi.org/10.1073/pnas.1801480116 PMid:30622181
PMCid:PMC6347684
- Koelle S, Espinoza DA, Wu C, Xu
J, Lu R, Li B, Donahue RE, Dunbar CE. Quantitative stability of
hematopoietic stem and progenitor cell clonal output in rhesus macaques
receiving transplants. Blood 2017; 129(11): 1448-1457.
https://doi.org/10.1182/blood-2016-07-728691 PMid:28087539
PMCid:PMC5356453
- Six E, Guilloux A, Denis A,
Lecoules A, Magnami A, Vilette R, Male F, Cagnard N, Delville M,
Caccavelli L, et al. Clonal tracking in gene therapy patients reveals a
diversity of human hematopoietic differentiation programs. Blood 2000;
135(15): 1219-1231. https://doi.org/10.1182/blood.2019002350
PMid:32040546 PMCid:PMC7146019
- Kollman C, Howe
CW, Anasetti C, Antin JH, Davies SM, Filipovich AH, Hegland J, Kamani
N, Kernan NA, King R, Ratanatharathorn V, Weidorf D, Confer DL. Donor
characteristics as risk factors in recipients after transplantation of
bone marrow from unrelated donors: the effect of donor age. Blood 2001;
98(7): 2043-2051. https://doi.org/10.1182/blood.V98.7.2043
PMid:11567988
- Kollman C, Spellman SR, Zhang MJ,
Hassebroek A, Anasetti C, Antin JH, Champlin RE, Confer DL, DiPersio
JF, Fernandez-Vina M, et al. The effect of donor characteristics on
survival after unrelated donor transplantation for hematologic
malignancy. Blood 2016; 127(2): 260-267.
https://doi.org/10.1182/blood-2015-08-663823 PMid:26527675
PMCid:PMC4713163
- DeZern AE, Franklin C, Tsai HL, Imus
PH, Cooke KR, Varadhan R, Jones RJ. Relationship of donor age and
relationship to outcomes of haploidentical transplantation with
posttransplant cyclophosphamide. Blood Adv 2021; 5(5): 1360-1368.
https://doi.org/10.1182/bloodadvances.2020003922 PMid:33661299
PMCid:PMC7948266
- Poletto E, Colella P, Pimentel
Vera LN, Khan S, Tomatsu S, Baldo G, Gomez-Ospina N. Improved
engraftment and therapeutic efficacy by human genome-edited
hematopoietic stem cells with Busulfan-based myeloablation. Mol Ther:
Methods & Clinical Dev 2022; in press.
https://doi.org/10.1016/j.omtm.2022.04.009 PMid:35573043
PMCid:PMC9065050
- Marktel S, Scaramuzza S,
Cicalese MP, Giglio F, Galimberti S, Lidonnici MR, Calbi V, Assanelli
A, Bernardo ME, Rossi C, et al. Intrabone hematopoietic stem cell gene
therapy for adult and pediatric patients affected by
transfusion-dependent β-thalassemia. Nat Med 2019; 25(2): 234-241.
https://doi.org/10.1038/s41591-018-0301-6 PMid:30664781
- Felker
S, Shrestha A, Bailey J, Pillis DM, Siniard D, Malik P. Differential
CXCR4 expression on hematopoietic progenitor cells versus stem cells
directs homing and engraftment. JCI Insight 2022; 7(9): e151847.
https://doi.org/10.1172/jci.insight.151847 PMid:35531956
PMCid:PMC9090236
- Rossi DJ, Bryder D, Zahn JM,
Ahlenius H, Sonu R, Wagers AJ. Cell intrinsuic alterations underlie
hematopoietic stem cell aging. Proc Natl Acad Sci USA 2005; 102(26):
9194-9199. https://doi.org/10.1073/pnas.0503280102 PMid:15967997
PMCid:PMC1153718
- Rossi DJ, Bryder D, Seita J,
Nussenzweig A, Hoieijmakers J, Weissman IL. Deficiencies in DNA damage
repair limit the function of haematopoietic stem cells with age. Nature
2007; 447(7145): 725-729. https://doi.org/10.1038/nature05862
PMid:17554309
- Chambers SM, Shaw CA, Gatza C, Fisk
CJ, Donehower LA. Aging hematopoietic stem cells decline in function
and exhibit epigenetic dysregulation. PLoS Biol 2007; 5(8): e201.
https://doi.org/10.1371/journal.pbio.0050201 PMid:17676974
PMCid:PMC1925137
- Ganuza M, Hall T, Finkelstein D, Wang
YD, Chabot A, Kang G, Wu B, Wu G, McKinney-Freeman S. The global clonal
complexity of the murine blood system declines throughout life and
after serial transplantation. Blood 2019; 133(18): 1927-1942.
https://doi.org/10.1182/blood-2018-09-873059 PMid:30782612
PMCid:PMC6497513
- Pang WW, Price EA, Sahoo D, Beerman I,
Maloney WJ, Rossi DJ, Schrier SL, Weissman IL. Human bone marrow
hematopoietic stem cells are increased in frequency and myeloid-biased
with age. Proc Natl Acad Sci USA 2011; 108(50): 20012-20017.
https://doi.org/10.1073/pnas.1116110108 PMid:22123971 PMCid:PMC3250139
- Nilsson
AR, Soneji S, Adolfsson S, Bryder D. Human and murine hematopoietic
stem cell aging is associataed with functional impairments and
intrinsic megakaryocytic/erythroid bias. PLoS ONE 2016; 11(7): e158369.
https://doi.org/10.1371/journal.pone.0158369 PMid:27368054
PMCid:PMC4930192
- Kuranda K, Vargaftig J, de la
Rochere P, Dosquet C, Charron D, Bardin F, Tonnelle C, Goodhardt M.
Age-related changes in human hematopoietic stem progenitor cells. Aging
Cell 2011; 10(3): 542-546.
https://doi.org/10.1111/j.1474-9726.2011.00675.x PMid:21418508
- Kowalczyk
M, Tirosh I, Hechi D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers
AJ, Ebert BL, Regev A. Single-cell RNA-seq reveals changes in cell
cycle and differentiation programs upon aging of hematopoietic stem
cells. Genome Res 2015; 25(12): 1860-1872.
https://doi.org/10.1101/gr.192237.115 PMid:26430063
PMCid:PMC4665007
- Salic A, Mitchison TJ. A
chemical method for fast and sensitive detection of DNA synthesis. Proc
Natl Acad Sci USA 2008; 105(7): 2415-2420.
https://doi.org/10.1073/pnas.0712168105 PMid:18272492
PMCid:PMC2268151
- Kovtonyuk LV, Ashcroft P,
Spaltro G, Tata NR, Skoda RC, Bonhoeffer S, Manz MG. Hematopoietic stem
cells increase quiescence during aging. Blood 2019; 134(suppl.1): 2484.
https://doi.org/10.1182/blood-2019-130668
- Florian MC,
Dorr K, Niebel A, Daira D, Schrezenmeier H, Rojewski M, Filippi MD,
Hasenberg A, Gunzer M, Scharffetter-Kochanek K, Zheng Y, Geiger H.
Cdc42 activity regulates hematopoietic stem cell aging and
rejuvenation. Cell Stam Cell 2012; 10(5): 520-530.
https://doi.org/10.1016/j.stem.2012.04.007 PMid:22560076
PMCid:PMC3348626
- Florian MC, Klose M, Sacma M,
Jablanovic J, Kundson L, Nattamai KJ, Marks G, Vollmer A, Soller K,
Sakk V, et al. Aging alters the epigenetic asymmetry of HSC division.
PLos Biology 2018; 16(9): e2003389.
https://doi.org/10.1371/journal.pbio.2003389 PMid:30235201
PMCid:PMC6168157
- Amoah A, Keller A, Emini R,
Hoenicka M, Libelold A, Vollmer A, Eiwen K, Soller K, Sakk V, Zheng Y,
Florian C, Geiger H. Aging of human hematopoietic stem cells is linked
to changes in Cdc42 activity. Haematologica 2022; 107(2): 393-402.
https://doi.org/10.3324/haematol.2020.269670 PMid:33440922
PMCid:PMC8804569
- Schmacher B, Pathof J, Vijg J,
Hoeimakers J. The central role of DNA damage in the ageing process.
Nature 2021; 592(7856): 695-703.
https://doi.org/10.1038/s41586-021-03307-7 PMid:33911272
- Mohrin
M, Bouorke E, Alexander D, Warr MR, Barry-Holson K, LeBeau MM, Morrison
CG, Passagué E. Hematopoietic stem cell quiescence promotes error-prone
DNA repair and mutagenesis. Cell Stem Cell 2010; 7(2): 174-185.
https://doi.org/10.1016/j.stem.2010.06.014 PMid:20619762
PMCid:PMC2924905
- Beerman I, Seitu J, Inlay MA,
Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate
DNA damage during aging that is repaired upon entry into cell cycle.
Cell Stem Cell 2014; 15(1): 37-50.
https://doi.org/10.1016/j.stem.014.04.016 PMid:24813857
PMCid:PMC4082747
- Flach J, Bakker ST, Mohrin M,
Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F,
Forsberg EC, et al. Replication stress is a potent driver of functional
decline in ageing haematopoietic stem cells. Nature 2014; 512(7513):
198-202. https://doi.org/10.1038/nature13619 PMid:25079315
PMCid:PMC4456040
- Walter D, Lier A, Geiselhart A,
Thalheimer FB, Huntscha S, Sobotta MC, Mehrle B, Brocks D, Bayindir I,
Kraschutnig P, et al. Exit from dormancy provokes DNA-damage-induced
attrition in haematopoietic stem cells. Nature 2015; 520(7548):
549-552. https://doi.org/10.1038/nature14131 PMid:25707806
- Pan
Y, Zuo H, Wen F, Huang F, Zhu Y, Cao L, Sha QQ, Li Y, Zhang H, ShiM, et
al. HMCES safeguards genome integrity and long-term self-renewal of
hematopoietic stem cells during stress responses. Leukemia 2022; 36(4):
1123-1131. https://doi.org/10.1038/s41375-021-01499-5 PMid:35039639
- Heylmann
D, Ponath V, Kindler T, Kaina B. Comparison of DAN repair and
radiosensitivity of different blood cell populations. Scient Rep 2021;
11(11): 2478. https://doi.org/10.1038/s41598-021-81058-1 PMid:33510180
PMCid:PMC7843614
- Biechonski S, Olender L, Zipin-Roitman
A, Yassin M, Aqaqe N, Marcu-Malina V, Rall-Scharpf M, Trottier M, Meyn
MS, Wiesmuller L, Beider K, Raz Y, Grisaru D, Nagler A, Milyavsky M.
Attenuated DNA damage responses and increased apoptosis characterize
human hematopoietic stem cells exposed to irradiation. Scient Rep 2018;
8(1): 6071. https://doi.org/10.1038/s41598-018-24440-w PMid:29666389
PMCid:PMC5904119
- Yizhak K, Aguet F, Kim J, Hess J,
Kubler K, Grimsby J, Frazer R, Zhang H, Haradhvaia NJ, Rosebrock D,
Livitz D, Li X, Arich-Landkof E, Shoresh N, Stewart C, Segrè AV,
Branton PA, Polak P, Ardle K, Getz G. RNA sequence analysis reveals
macroscopic somatic clonal expansion across normal tissues. Science
2019; 364(6444): eaaw0726. https://doi.org/10.1126/science.aaw0726
PMid:31171663 PMCid:PMC7350423
- Milholland B, Dong X,
Zhang L, Hao X, Vijg J. Differences between germline and somatic
mutation rates in human and mice. Nat Commun 2017; 8(5): 15183.
https://doi.org/10.1038/ncomms15183 PMid:28485371
PMCid:PMC5436103
- Lee-Six H, Oebro NF, Shepherd
MS, Grossmann S, Dawson K, Belmonte M, Osborne RJ, Huntlky B,
Martincorena I, Anderson E, O'Neill L, Streatton MR, Laurent E, Green
AR, Kent DG, Campbell PJ. Population dynamics of normal human blood
inferred from somatic mutations. Nature 2018; 561: 473-478.
https://doi.org/10.1038/s41586-018-0497-0 PMid:30185910
PMCid:PMC6163040
- Osorio FG, Rosendahl Huber A,
Oka R, VerheulnM, Patel SH, Hasaart K, de la Fonteijne L, Varela I,
Camargo FD, van Boxtel R. Somatic mutations reveal lineage
relationships and age-related mutagenesis in human hematopoiesis. Cell
Rep 2018; 25(11): 2308-2316.
https://doi.org/10.1016/j.celrep.2018.11.014 PMid:30485801
PMCid:PMC6289083
- Brandsma AM, Bertrums E, van
Rooismalen MJ, Hofman DA, Oka R, Verheulen M, Manders F, Ubels J,
Bbelderbos ME, van Boxtel R. Mutation signatures of pediatric acute
myeloid leukemia and normal blood progenitors associated with
differential patient outcomes. Blood Cancer Discov 2021; 2(9): 484-499.
https://doi.org/10.1158/2643-3230.BCD-21-0010 PMid:34642666
PMCid:PMC7611805
- Mura F, Degasperi A, Nadeu F,
Leongamornlert D, Davies H, Moore L, Royo R, Ziccheddu B, Puente XS,
Avet-Loiseau, H, Campbell, PJ, Nik-Zainal S, Campo E, Munshi N, Bolli
N. A practical guide for mutational signature analysis in
haematological malignancies. Nat Commun 2019; 10: 2969.
https://doi.org/10.1038/s41467-019-11037-8 PMid:31278357
PMCid:PMC6611883
- Alexandrov LB, Kim J, Haradhvala
NJ, Huang MN, Ng AWT, Wu Y, Boot A, Covington KR, Gordenin DA,
Bergstromn EN, Islam A, Lopez-Bigas N, Klimczak LJ, McPherson JR,
Morganella S, Sabarinathan R, Wheeler DA, Mustonen V, PCAWG Mutational
Signatures Woking Group, Getz G, Rozen SG, Startton MR, PCAWG
Consortium. The repertoire of mutational signatures in humanb cancer.
Nature 2020; 578: 94-101. https://doi.org/10.1038/s41586-020-1943-3
PMid:32025018 PMCid:PMC7054213
- Vijg J, Dong X.
Pathogenic mechanisms of somatic mutation and genome mosaicism in
aging. Cell 2020; 182: 12-23.
https://doi.org/10.1016/j.cell.2020.06.024 PMid:32649873
PMCid:PMC7354350
- Mustjoki S, Young NS. Somatic
mutations in "benign" disease. N Engl J Med 2021; 384(21): 2039-2052.
https://doi.org/10.1056/NEJMra2101920 PMid:34042390
- Steensma
DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, Ebert
BL. Clonal hematopoiesis of indeterminate potential and its distinction
from myelodysplastic syndromes. Blood 2015; 126(1): 9-16.
https://doi.org/10.1182/blood-2015-03-631747 PMid:25931582
PMCid:PMC4624443
- Busque L, Patel JP, Figueroa M,
Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy
A, Hassimi M, Socci N, Bhatt PK, Gonen M, Mason CE, Melnick A, Godley
LA, Brennan C, Abdel-Wahab O, Levine RL. Recurrent somatic TET2
mutations in normal elderly individuals with clonal hematopoiesis. Nat.
Genet. 2012; 44: 1179-1181. https://doi.org/10.1038/ng.2413
PMid:23001125 PMCid:PMC3483435
- Genovese G, Kahler
AK, Handsaker RE, Clonal hematopoiesis and blood-cancer risk inferred
from blood DNA sequence. New England J. Med. 2014; 371: 2477-2487.
https://doi.org/10.1056/NEJMoa1409405 PMid:25426838 PMCid:PMC4290021
- Xie
M, Lu C, Wang J, Age-related mutations associated with clonal
hematopoietic expansion and malignancies. Nat. Med. 2014; 20:
1472-1478. https://doi.org/10.1038/nm.3733 PMid:25326804
PMCid:PMC4313872
- Jaiswal S, Fontanillas P, Flannick J,
Age-related clonal hematopoiesis associated with adverse outcomes. N.
Engl. J. Med. 2014; 371: 2488-2498.
https://doi.org/10.1056/NEJMoa1408617 PMid:25426837 PMCid:PMC4306669
- McKerrell
T, Park N, Moreno T, Grove CS, Postingl H,Stephens J, Understanding
Society Scientific Group, Crawley C, Craig J, Scoot MA, Hodkinson C,
Baxter J. Rad R, Forsyth DR, Quail MA, Zeggini E, Ouwehand W, Varela I,
Vassiliou GS. Leukemia-associated somatic mutations drive distinct
patterns of age-related clonal hemopoiesis. Cell Rep. 2015; 10(3):
1239-1245. https://doi.org/10.1016/j.celrep.2015.02.005 PMid:25732814
PMCid:PMC4542313
- Buscarlet M, Provost S, Zada YF,
Barhdadi A, Bourgoin V, Lépine G, Mollica L, Szuber N, Dubé MP, Busque
L. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign
phenotypes and different genetic predispositions. Blood 2017; 130(6):
753-762. https://doi.org/10.1182/blood-2017-04-777029 PMid:28655780
- Young
AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring
AML-associated mutations is ubiquitous in healthy adults. Nat. Commun.
2016; 7: 12484. https://doi.org/10.1038/ncomms12484 PMid:27546487
PMCid:PMC4996934
- Arends CM, Galan-Sousa J, Hoyer
K, Chan W, Jager M, Yoshida K, Seeman R, Noerenberg D, Waldhueter N,
Fleisher-Notter H, Christen F, Schmitt CA, Dorken B, Pelzer U, Sinn M,
Zemojtel T, Ogawa S, Mardan S, Schreiber A, Kunitz A, Kruger U,
Bullinger L, Mylonas E, Frick M, Damm F. Hematopoietic lineage
distribution and evolutionary dynamics of clonal hematopoiesis.
Leukemia 2018; 32(9): 1908-1919.
https://doi.org/10.1038/s41375-018-0047-7 PMid:29491455
- Zink
F, Stacey SN, Nordahl GL, Frigge ML, agnusson OT, Jondottir I,
Thorgeirsson TE, Sigursson A, Gudjonsson SA, Gudmundsson J, Jonasson
JN, Tryggvadottir L, Jonsson T, Helagason A, Gylfason A, Sulem P,
Rafnar T, Thorsteinsdottir U, Gudbjartsson DF, Masson G, Kong A,
Stefansson K. Clonal hematopoiesis, with and without candidate driver
mutations, is common in the elderly. Blood 2017; 130(6): 742-752.
https://doi.org/10.1182/blood-2017-02-769869 PMid:28483762
PMCid:PMC5553576
- Van Zeventer IA, Salzbrunn JB,
de Graaf AO, van der Reijden BA, Boezen HM, Vonk JM, van der Harst P,
Schuringa JJ, Jansen JH, Huls G. Prevalence, predictors, and outcomes
of clonal hematopoiesis in individuals aged ≥80 years. Blood Adv. 2021;
5(8): 2115-2122. https://doi.org/10.1182/bloodadvances.2020004062
PMid:33877299 PMCid:PMC8095141
- Rossi M, Meggendorfer M,
Zampini M, Tettamanti M, Riva E, Travaglino E, Bersanelli M, Mandelli
S, Galbussera AA, osca E, et al. Clinical relevance of clonal
hemopoiesis in persons aged ≥80 years. Blood 2021; 138(21): 2093-2105.
https://doi.org/10.1182/blood.2021011320 PMid:34125889
- Kar
SP. Quiros PM, Gu M, Jiang T, Lagdon R, Iyer V, Barcena C, Vijayabaskar
MS, Fabre MA, Carter P, Burgess S, Vassiliou GS. Genome-wide analyses
of 200,453 individuals yeld new insights into the causes and
consequences of clonal hematopoiesis. BioRxIV 2022;
10.1101/2022.01.06.22268846.
https://doi.org/10.1101/2022.01.06.22268846
- Fabre
MA, De Almeida JG, Fiorillo E, Mitchell E, Damaskou A, Rak J, Orrù V,
Maronglu M, Vijayabaskar MS, Baxter J, et al. The longitudinal dynamics
and natural history of clonal hematopoiesis. BioRxIV
10.1101/2021.
- Pich O, Reyes-Salazar I,
Gonzalez-Perez A, Lopez-Bigas N. Discovering the drivers of clonal
hematopoiesis. BioRxIV 2020, 10.22.350140.
https://doi.org/10.1101/2020.10.22.350140
- Beauchamp
EM, Leventhal M, Bernard E, Hoppe ER, Todisco G, Creignou M, Galli A,
Castellano CA, McConkey M, Tarun A, et al. ZBTB33 is mutated in clonal
hematopoiesis and myelodysplastic syndromes and impacts RNA splicing.
Blood Cancer Discov 2021, 2 :500-517.
https://doi.org/10.1158/2643-3230.BCD-20-0224 PMid:34568833
PMCid:PMC8462124
- Poon G, Watson CJ, Fisher DS,
Blundell JR. Synonymous mutations reveal genome-wide levels of positive
selection in healthy tissues. Nat Genet 2021; 53(11): 1597-1605.
https://doi.org/10.1038/s41588-021-00957-1 PMid:34737428
- Mitchell
E, Chapmen MS, Williams N, Dawson K, Mende N, Caldferbank EF, Jung H,
Mitchell T, Coorens T, Spencer D, et al. Clonal dynamics of
haematopoiesis across the human lifespan. Nature 2022; 606(7913):
343-350. https://doi.org/10.1038/s41586-022-04786-y PMid:35650442
PMCid:PMC9177428
- Abascal F, Harvey L, Mitchell E,
Lawson A, Lensing S, Ellis P, Russell A, Alcantara R, Baez-Ortega A,
Wang Y, et al. Somatic mutation landscapes at single-molecule
resolution. Nature 2021; 593(7859): 405-410.
https://doi.org/10.1038/s41586-021-03477-4 PMid:33911282
- Wong
WH, Tong S, Druley TE. Error-corre3cted sequencing of cord bloods
identifies pediatric AMNL associated clonal hematopoiesis. Blood 2017;
130, suppl.1, 2687.
- Hasaart K, Manders F, van der
Hoorn ML, Verheul M, Popolonski T, Kruijk E, Chuva de Lopes S, van
Boxtel R. Mutation accumulation and developmental lineages in normal
and Down syndrome human fetal hematopoiesis. Scient Rep 2020; 10:
12991. https://doi.org/10.1038/s41598-020-69822-1 PMid:32737409
PMCid:PMC7395765
- Chapman MS, Ranzoni AM, Myers B,
Williams N, Coorens T, Mitchell E, Butler T, Dawson K, Hooks Y, Moore
L, Nangalia J, Robinson PS, Yoshida K, Hook E, Campbell PJ, Cvejic A.
Lineage tracing of human development through somatic mutations. Nature
2021; 595: 85-90. https://doi.org/10.1038/s41586-021-03548-6
PMid:33981037
- Hansen JW, Pedersen DA, Larsen LA,
Husby S, Clemmensen SB, Hjelmborg J, Favero F, Weischenfeldt J,
Christensen K, Gronbaek K. Clonal hematopoiesis in elderly twins:
concordance, discordance, and mortality. Blood 2020; 135: 261-268.
https://doi.org/10.1182/blood.2019001793 PMid:31697811
PMCid:PMC6978157
- Niroula A, Sekar A, Murakami MA,
Trinder M, Agrawal M, Wong WJ, Bick AG, Uddin M, Gibson CJ, Griffin GK,
Honigberg MC, Sekavat SM, Parachuri K, Natarajan P, Ebert BL.
Distinction of lymphoid and myeloid clonal hematopoiesis. Nature
Medicine 2021; in press. https://doi.org/10.1038/s41591-021-01521-4
PMid:34663986 PMCid:PMC8621497
- Challen GA, Goodell MA.
Clonal hematopoiesis: mechanisms driving dominance of stem cell clones.
Blood 2020; 136: 1590-1598. https://doi.org/10.1182/blood.2020006510
PMid:32746453 PMCid:PMC7530644
- Chin DWL,
Yoshizato T, Culleton SV, Grasso F, Barbachowska M, Ogawa S, Jacobsen
SEW, Woll PS. Aged healthy mice acquire clonal hematopoietic mutations.
Blood 2022; 139(4): 629-634. https://doi.org/10.1182/blood.2021014235
PMid:34665864 PMCid:PMC8832470
- Christen F,
Hablesreiter R, Hoyer K, Hennch C, Maluck-Bottcher A, Segler A, Madadi
A, Frick M, Bullinger L, Briest F, Damm F. Modeling clonal
hematopoiesis in umbilical cord blood cells by CRISPR/Cas9. Leukemia
2022; 36(4): 1102-1110. https://doi.org/10.1038/s41375-021-01469-x
PMid:34782715 PMCid:PMC8979818
- Boettcher S, Wilk
CM, Singer J, Beier F, Burcklen E, Biesel C, Ventura Ferreira MS,
Gourri E, Gassner C, Frey BM, Schanz U, Skoda RC, Ebert BL, Brummendorf
TH, Beerenwinkel, N, Manz MG. Clonal hematopoiesis in donors and
long-term survivors of related allogeneic hematopoietic stem cell
transplantation. Blood 2020; 135(18): 1548-1559.
https://doi.org/10.1182/blood.2019003079 PMid:32181816
- Wong
WH, Bhatt S, Trinkaus K, Pusic I, Elliott K, Mahajan N, Wan F, Switzer
GE, Confer DL, DiPersio J, Pulsipher MA, Shah NN, Seses J, Bystry A,
Blundell JR, Shaw BE, Druley TE. Enfraftment of rare, pathogenic donor
hematopoietic mutations in unrelated hematopoietic stem cells
transplantation. Sci. Transl Med 2020; 12(526): doi:10.1126.
https://doi.org/10.1126/scitranslmed.aax6249 PMid:31941826
PMCid:PMC7521140
- Heini AD, Porret N, Zenhaeusen R,
Winkler A, Bacher U, Pabst T. Clonal hematopoiesis after autologous
stem cell transplantation does not confer adverse prognosis in patients
with AML. Cancers 2021; 13: 3190.
https://doi.org/10.3390/cancers13133190 PMid:34202404 PMCid:PMC8267699
- Oran
B, Champlin RE, Wang F, Tanaka T, Saliba RM, Al-Atrash G, Garcia-Manero
G, Kantarjian H, Cao K, Shpall EJ, Alousi AM, Melita RS, Popat U,
Futreal U, Takahashi K. Donor clonal hematopoiesis increases graft
versus host disease after matched sibling transplantation. Leukemia
2021; in press. https://doi.org/10.1038/s41375-021-01430-y
PMid:34876697
- Nevejan L, Nollet F, Devos H, Vynck M,
Van Vlieberghe P, Tajdar M, Lodewyck T, Selleslag D. Malignant
progression of donor-engrafted clonal hematopoiesis in sibling
recipients after stem cell transplantation. Blood Adv 2020; 4(22):
5631-5634. https://doi.org/10.1182/bloodadvances.2020003168
PMid:33186460 PMCid:PMC7686891
- Laurie CC, Laurie
CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh
EW, Amos C, et al. Detectable clonal mosaicism from birth to old age
and its relationship to cancer. Nat. Genet. 2012; 44(6): 642-650.
- Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z,
Rodriguez-Santiago B, Hutchinson A, Deng X, Liu C, Horner MJ, et al.
Detectable clonal mosaicism and its relationship to aging and cancer.
Nat. Genet. 2012; 44(6): 651-658.
- Loh PR,
Genovese G, Handsaker RE, Finucane HK, Reshf JA, Palamara PF, Birmann
BM, Talkowski ME, Bakhoum SF, McCarroll SA, Price AL. Insights into
clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature
2018; 559: 350-355. https://doi.org/10.1038/s41586-018-0321-x
PMid:29995854 PMCid:PMC6054542
- Terao C, Suzuki A,
Mmomozawa Y, Akiytama M, Ishigaki K, Yamamoto K, Matsuda K, Murakami Y,
McCarroll SA, Kubo M, Loh PR, Kamatami Y. Chromosomal alterations among
age-related haematopoietic clones in Japan. Nature 2020; 584: 130-134.
https://doi.org/10.1038/s41586-020-2426-2 PMid:32581364
PMCid:PMC7489641
- Zekavat SM, Lin SH, Bick AG, Liu
A, Paruchuri K, Wang C, Uddin M, Ye Y, Yu Z, Liu X, Kamatani Y, et al.
Hematopoietic mosaic chromosomal alterations increase the risk for
diverse types of infection. Nat Med 2021; 27(6): 1012-1024.
https://doi.org/10.1038/s41591-021-01371-0 PMid:34099924
PMCid:PMC8245201
- Thompson DJ, Genovese G,
Halvardson J, Ulirsch JC, Wright DJ, Terao C. Genetic predisposition to
mosaic Y chromosome loss in blood. Nature 2019; 575: 652-657.
https://doi.org/10.1038/s41586-019-1765-3 PMid:31748747
PMCid:PMC6887549
- Ljungstrom V, Mattisson J, Halvardson
J, Pandzic T, Davies H Rychlicka-Buniowska E, Danielsson M, Lacaze P,
Cavelier L, Dumanski JP, Baliakis P, Forsberg LA. Loss of Y and clonal
hematopoiesis in blood-two sides of the same coin? Leukemia 2021; in
press. https://doi.org/10.1038/s41375-021-01456-2 PMid:34725452
PMCid:PMC8885420
- Ouseph MM, Hasserjian RPDal Cin P,
Lovitch SB, Steensma DP, Nardi V, Weinberg OK. Genomic alterations in
patients with somatic loss of the Y chromosome as the sole cytogenetic
finding in bone marrow cells. Haematologica 2021; 106(2): 555-564.
https://doi.org/10.3324/haematol.2019.240689 PMid:32193254
PMCid:PMC7849577
- Lin SH, Loftfield E, Sampson JN,
Zhou W, Yeager M, Freedman ND, Chanock SJ, Machiela MJ. Mosaic
chromosome Y loss is associated with alterations in blood cell counts
in the UK Biobank men. Scient Rep. 2020; 10: 3655.
https://doi.org/10.1038/s41598-020-59963-8 PMid:32108144
PMCid:PMC7046668
- Saiki R, Momozawa Y, Nannya Y,
Nakagawa MM, Ochi Y, Yoshizato T, Terao C, Kuroda Y, Shiraishi Y, Chiba
K, Tanaka H, Niida A, Imoto S, Matsuda K, Morisaki T, Murakami Y,
Kamatani Y, Matsuda S, Kubo M, Miyano S, Makishima H, Ogawa S. Combined
landscape of single-nucleotide variants and copy number alterations in
clonal hematopoiesis. Nature Med. 2021; 27(7): 1239-1249.
https://doi.org/10.1038/s41591-021-01411-9 PMid:34239136
- Brown
DW, Cato LD, Zhao Y, Nandakumar SK, Bao EL, Rehling T, Song L, Yu K,
Chanock SJ, Perry J, Sankaran VG, Machiela MJ. Shared and distinct
genetic etiologies for different types of clonal hematopoiesis. BioRxiv
2022; in press. https://doi.org/10.1101/2022.03.14.483644
- Van
Zeventer IA, de Graaf A, Koorenhof-Scheele TN, van der Reijden BA, van
der Klauw MM, Dinmohamed AG, Diepstra A, Schuringa JJ, Malcovati L,
Huls G, Jansen JH. Monocytosis and its association with clonal
hematopoiesis in community-dwelling individuals. Blood Adv 2022; in
press. https://doi.org/10.1182/bloodadvances.2021006755 PMid:35561316
PMCid:PMC9327556
- Silver AJ, Bick AG, Savona MR.
Germline risk of clonal haematopoiesis. Nat. Rev Genet. 2021; 22(9):
603-617. https://doi.org/10.1038/s41576-021-00356-6 PMid:33986496
PMCid:PMC8117131
- Bick AG, Weinstock JS,
Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, Szeto MD, Liao X,
Leventhal MJ, Nasser J, et al. Inherited causes of clonal
haematopoiesis in 97,691 whole genomes. Nature 2020; 586: 763-768.
https://doi.org/10.1038/s41586-020-2819-2 PMid:33057201
PMCid:PMC7944936
- Cohen Aubart F, Roos-Weil D, Armand M,
Marceau-Renault A, Emile JF, Duployez N, Charlotte F, Poulain S, Lhote
R, Helias-Rodzewicz Z, Della-Valle V, Bernard O, Maloum K, Ngueyen-Khac
F, Donadieu J, Amoura Z, Abdel-Wahab O, Heroche J. High frequency of
clonal hematopoiesis in Erdheim-Chester disease. Blood 2021; 137(4):
485-492. https://doi.org/10.1182/blood.2020005101 PMid:33067622
PMCid:PMC8555377
- Ferris MA, Smith AM, Heath SE,
Duncavage EJ, Oberley MJ, Freyer D, Wynn R, Douzgou S, Maris JM, Reilly
AF, Wu M, Choo F, Fiets RB, Koene S, Spencer DH, Miller CA, Shinawi M,
Ley TJ. DNMT3A overgrowth syndrome is associated with the development
of hematopoietic malignancies in children and young adults. Blood 2022;
139(3): 461-464. https://doi.org/10.1182/blood.2021014052 PMid:34788385
- Smith AM, LaValle TA, Shinawi M, Ramakrishan SM, Abel
HJ, Hill CA, Kirkland NM, Rettig MP, Helton NM, Heath SE, Ferraro F,
Chen DY, Adak S, Semenkovich CF, Christian DL, Martin JR, Gabel HW,
Miller CA, Ley TJ. Functional and epigenetic phenotypes of humans and
mice with DNMT3A overgrowth syndrome. Nat Commun 2021; 12: 4549.
https://doi.org/10.1038/s41467-021-24800-7 PMid:34315901
PMCid:PMC8316576
- Tovy A, Rosas C, Gaikwad AS, Medrano
G, Zhang L, Reyes JR, Huang YH, Arakawa T, Kurtz K, Conneely SE, Guzman
AG, Aguilar R, Gao A, Chen CW, Kim JJ, Carter MT, Lasa-Aranzasti A,
Valenzuela I, Maldergem LV, Brunetti L, Hicks MJ, Marcogliese AN,
Goodell MA, Rau RE. Perturbed hematopoiesis in individuals with germile
DNMT3A overgrowth Tatton-Brown-Rahman syndrome. Haematologica 2022,
107(4): 887-898. https://doi.org/10.3324/haematol.2021.278990
PMid:34092059 PMCid:PMC8968878
- Xia J, Miller CA, Baty
J, Ramesh A, Jotte M, Fulton RS, Vogel TP, Cooper MA, Walcovich KJ,
Maaaaakarayan V, Bolyard AA, Dinauer MC, Wilson DB, Vlachos A, myers
KC, Rothbaum RJ, Bertuch AA, Dale DC, Shimamura A, Boxer LA, Link DC.
Somatic mutations and clonal hematopoiesis in congenital neutropenia.
Blood 2018; 131(4): 408-416.
https://doi.org/10.1182/blood-2017-08-801985 PMid:29092827
PMCid:PMC5790127
- Kennedy AL, Myers KC, Bowman J, Gibson
CJ, Camarda ND, Furutani E, Muscato GM, Klein RH, Ballotti K, Liu S, et
al. Distinct genetic pathways define pre-malignant versus compensatory
clonal hematopoiesis in Shwachman-Diamond syndrome. Nat Commun 2021;
12: 1334. https://doi.org/10.1038/s41467-021-21588-4 PMid:33637765
PMCid:PMC7910481
- Furutani E, Liu S, Galvin A, Steltz S,
Malsch MM, Loveless SK, Mount L, Larson JH, Queenan K, Bertuch AA, et
al. Hematologic complications with age in Schwachman-Diamond syndrome.
Blood Adv 2022; 6(1): 297-306.
https://doi.org/10.1182/bloodadvances.2021005539 PMid:34758064
PMCid:PMC8753194
- Narumi S, Amano N, Ishii T, Katumata
N, Muroya K, Adachi M, Toyoshima K, Tanaka Y, Fukuzawa R, Miyako K, et
al. SAMD9 mutations cause a novel mulisystem dosorder, MIRAGE syndrome,
and are associated with loss of chromosome 7. Nat. Genet. 2016; 48(7):
792-797. https://doi.org/10.1038/ng.3569 PMid:27182967
- Tesi
B, Daudsson J, Voss M, Rahikkala E, Holmes TD, Chang S,
Komulainen-Ebrahim J, Gorcenco S, Rundberg Nilsson A, Ripperberg T, et
al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia,
immunodeficiency, MDS, and neurological symptoms. Blood 2017; 129(16):
2266-2279. https://doi.org/10.1182/blood-2016-10-743302 PMid:28202457
PMCid:PMC5399482
- Wong JC, Bryant V, Lamprecht T, Ma J,
Walsh M, Schwartz J, del pilar Alzamora M, Mullighan CC, Loh ML,
Ribeiro R, et al. Germline SAMD9 and SAMD9L mutations are associated
with extensive genetic evolution and diverse hematologic outcomes. JCI
Insight 2018; 3(14): e121086.
https://doi.org/10.1172/jci.insight.121086 PMid:30046003
PMCid:PMC6124395
- Schwartz JR, Ma J, Lamprecht T, Walsh
M, Wang S, Bryany V, Song G, Wu G, Easton J, Kesserwan C, Nichols KE,
Mullighan CG, Ribeiro RC, Klco JM. The genomic lanscape of pediatric
myelodysplastic syndromes. Nat Commun 2017; 8: 1557.
https://doi.org/10.1038/s41467-017-01590-5 PMid:29146900
PMCid:PMC5691144
- Sahoo SS, Pastor VB, Goodings C,
Voss RK, Kozyra RJ, Szvetnik A, Noellke P, Dworzak M, Stary J,
Locatelli F, et al. Clinical evolution, genetic landscape and
trajectories of clonal hematopoiesis in SMD9/SAMD9L syndromes. Nat Med
2021; 27(10): 1806-1817. https://doi.org/10.1038/s41591-021-01511-6
PMid:34621053 PMCid:PMC9330547
- Thomas ME, Abdelhamed S,
Hiltenbrand R, Schwartz JR, Sakurada SM, Walsh M, Song G, Ma J,
Pruett-Miller SM, Klco JM. Pediatric MDS and bone marrow
failure-associated germline mutations in SAMD9 and SMAD9L impair
multiple pathways in primary hematopoietic cells. Leukemia 2021; 35(1):
3232-3244. https://doi.org/10.1038/s41375-021-01212-6 PMid:33731850
PMCid:PMC8446103
- Jasra S, Giricz O, Zeig-Owens R,
Pradhan K, Goldfarb DG, Barreto-Galvez A, Silver AJ, Chen J, Sahu S,
Gordon-Mitchell S, et al. High burden of clonal hematopoiesis in first
responders exposed to the World Trade Center disaster. Nat Med 2022;
28(3): 468-471. https://doi.org/10.1038/s41591-022-01708-3
PMid:35256801
- Coombs CC, Zehir A, Devlin SM, Kishtagari
A, Syed A, Jonsson P, Hyman DM, Solit DB, Robson ME, Baselga J, Arcila
ME, Ladanyi M, Tallman MS, Levine RL, Berger MF. Therapy-related clonal
hematopoiesis in patients with non-hematologic cancers is common and
associated with adverse clinical outcomes. Cell Stem Cell 2017; 21(9):
374-382. https://doi.org/10.1016/j.stem.2017.07.010 PMid:28803919
PMCid:PMC5591073
- Ptashkin RN, Mandelker DL, Coombs CC,
Bolton K, Yelskaya Z, Hyman DM, Solit DB, Baselga J, Arcila ME, Ladanyi
M, et al. Prevalence of clonal hematopoiesis mutations in tumor-only
clinical genomic profiling of solid tumors. JAMA Oncol. 2028; 4(11):
1589-1593. https://doi.org/10.1001/jamaoncol.2018.2297 PMid:29872864
PMCid:PMC6224316
- Coombs CC, Gillis NK, Tan X, Berg JS,
Ball M, Balasis ME, Montgomery ND, Bolton KL, Parker JS, Mesa TE, Yoder
SJ, Hayward M, Patel NM, Richards KL, Walko CM, Knepper TC, Earp III
HS, Levine RL, Papaemmanuil E, Zehir A, Hayes DN, Padron E.
Identification of clonal hematopoiesis mutations in solid tumor
patients undergoing unpaired next-generation sequencing assays. Clin
Cancer Res 2018; 24(23): 5918-5924.
https://doi.org/10.1158/1078-0432.CCR-18-1201 PMid:29866652
PMCid:PMC6812550
- Severson EA, Riedinger GM, Connelly CF,
Vergilio CA, Golfinger M, Ramkisson S, Frampton GM, Ross JS,
Fratella-Calabrese A, Gay L, Ali S, Miller V, Elvin J, Hadigol M,
Hirshfield KM, Rodriguez-Rodriguez L, Ganesan S, Khiabanian H.
Detection of clonal hematopoiesis of indeterminate potential in
clinical sequencing of solid tumor specimens. Blood 2018; 131(22):
2501-2505. https://doi.org/10.1182/blood-2018-03-840629 PMid:29678827
PMCid:PMC5981171
- Gao T, Ptashkin R, Bolton KL,Sirenko
M, Fong C, Spitzer B, Menghrajani K, Arango Ossa JE, Zhou Y, Bernard E,
Levine M, Medina Martinez JS, Zhang Y, Franch-Esposito S, Patel M,
Braunstein LZ, Kelly D, Yabe M, Benayed R, Caltabellotta NM, Philip J,
Paraiso E, Mantha S, Solit DB, Diaz LA, Berger MF, Klimek V, Levine RL,
Zehir A, Devlin SM, Papaemmanuil E. Interplay between chromosomal
alterations and gene mutations shapes the evolutionary trajectory of
clonal hematopoiesis. Nat Commun 12, 338 (2021).
https://doi.org/10.1038/s41467-020-20565-7
- McNerney ME,
Godley LA, Le Beau MM. Therapy-related myeloid neoplasm: when genetics
and environment collide. Nat. Rev. Cancer 2017; 17(9): 513-527.
https://doi.org/10.1038/nrc.2017.60 PMid:28835720 PMCid:PMC5946699
- Tiruneh
T, Enawgaw B, Shiferaw E. Genetic pathway in the pathogenesis of
therapy-related myeloid neoplasms: a literature review. Oncol Ther
2020; 8: 45-57. https://doi.org/10.1007/s40487-020-00111-7
PMid:32700075 PMCid:PMC7360004
- Pich T, Muinos F,
Lolkema MP, Steeghs N, Gonzalez-Perez A, Lopez-Bigas N. The mutational
fottprints of cancer therapies. Nature Genet 2019; 51: 1732-1740.
https://doi.org/10.1038/s41588-019-0525-5 PMid:31740835
PMCid:PMC6887544
- Pich O, Bullich-Cortes A, Muinos
F, Pratcorona M, Gonzalez-Perez A, Lopez-Bigas N. The evolution of
hematopoietic cells under cancer therapy. Nat Commun 2021; 12: 4803.
https://doi.org/10.1038/s41467-021-24858-3 PMid:34376657
PMCid:PMC8355079
- Diamond B, Zicheddu B,
Maclachlan K, Taylor J, Boyle E, Ossa JA, Jahn J, Affer M, Totiger TM,
Coffey D, et al. Chemotheray signatures map evolution of
therapy-related myeloid neoplasms. BioRxiv 2022; in press.
https://doi.org/10.1101/2022.04.26.489507
- Tariq H,
Stonim LB, Coty Fattal Z, Alikan MB, Segal J, Gurbuxani S, Helenowski
IB, Zhang H, Sukhanova M, Lu X, Altman JK, Chen QC, Bahdad A.
Therapy-related myeloid neoplasms with normal karyotype show distinct
genomic and clinical characteristics compared to their counterparts
with abnormal karyotype. Br J Haematol 2022; in press.
https://doi.org/10.1111/bjh.18154 PMid:35304738
- Wong
TN, Miller CA, Jotte M, Bagegni N, Baty JD, Schmidt AP, Cashen AF,
Duncavage DJ, Helton NM, Fiala M, Fulton RS, Heath SE, Janke M, Luber
K, Westervelt P, Vij R, DiPersio JF, Welch JS, Graubert TA, Walter MJ,
Ley TJ, Link DC. Cellular stressors contribute to the expansion of
hematopoietic clones of varying leukemic potential. Nat. Commun. 2018;
9(1): 455. https://doi.org/10.1038/s41467-018-02858-0 PMid:29386642
PMCid:PMC5792556
- Husby S, Favero F, Nielsen C, Sorensen
BS, Baech J, Grell K, Hansen JW, Rodriguez-Gonzalez FG, Haastrup EK,
Fischer-Nielsen A, et al. Clinical impact of clonal hematopoiesis in
patients with lymphoma undergoing ASCT: a national population-based
cohort study. Leukemia 2020; 34: 3256-3268.
https://doi.org/10.1038/s41375-020-0795-z PMid:32203146
- Gibson
CJ, Lindslay RC, Tchekmedyan V, Mar BG, Shi J, Jaiswal S, Bosworth A,
Francosco L, He J, Bansal A, Morgan EA, Lacasce AS, Preedman AS, Fisher
DC, Jacobsen E, Armand P, Alyea EP, Koreth J, Ho V, Soiffer RJ, Antin
JH, Ritz J, Nikiforow S, Forman SJ, Michor F, Neuberg D, Bhatia R,
Bhatia S, Ebert BL. Clonal hematopoiesis associated with adverse
outcomes after autologous stem-cell trasnplantation for lymphoma. J.
Clin. Oncol. 2017; 35( 14): 1598-1605.
https://doi.org/10.1200/JCO.2016.71.6712 PMid:28068180 PMCid:PMC5455707
- Eskelund CW, Husby S, Favero F, Wirenfeldt Klausen T,
Rodriguez-Gonzalez FG, Kolstadt A, Pedersen LB, Raty RK, Geisler CH,
Jerkeman M, Weischenfeldt J, Grobaek K. Clonal hematopoiesis evolves
from pretreatment clones and stabilizes after end of chemotherapy in
patients with MCL. Blood 2020; 135: 2000-2003.
https://doi.org/10.1182/blood.2019003539 PMid:32181815
- Venanzi
A, Marra A, Schiavoni G, Millner SG, Limongello R, Santi A, Pettirossi
V, Ultimo S, Tasselli L, Puccairini A, et al. Dissecting clonal
hematopoiesis in tissues of patients with classic Hodgkin lymphoma.
Blood Cancer Discov 2021; 2: 216-225.
https://doi.org/10.1158/2643-3230.BCD-20-0203 PMid:34164626
PMCid:PMC7611041
- Saini NY, Swoboda DM, Greenbaum
U, Ma J, Patel RD, Devashish K, Das K, Tanner MR, Strati P, Nair R, et
al. Clonal hematopoiesis is associated with increased risk of severe
neurotoxicity in axicabtagene ciloleucel therapy of large B-cell
lymphoma. Blood Cancer Discov 2022; in press.
https://doi.org/10.1158/2643-3230.BCD-21-0177 PMid:35533245
- Hatakeyama
K, Hieda M, Semba Y, Moriyama S, Wang Y, Maeda T, Kato K, Miyamoto T,
Akashi K, Kilkushige Y. TET2 clonal hematopoiesis is associated with
anthracycline-induced cardiotoxicity in patients with lymphoma. JACC:
Cardioncol 2022; 4: 141-143.
https://doi.org/10.1016/j.jaccao.2022.01.098 PMid:35492814
PMCid:PMC9040099
- Lewis NE, Petrova-Drus K, Huet
S, Epstein-Petersen ZD, Gao Q, Sigler AE, Baik J, Ozkaya N, Moskowitz
AJ, Kuamr A, et al. Clonal hematopoiesis in angioimmunoblastic T-cell
lymphoma with divergent evolution to myeloid neoplasms. Blood Adv 2020;
4: 2291-2300. https://doi.org/10.1182/bloodadvances.2020001636
PMid:32442302 PMCid:PMC7252546
- Khanlari M, Yin CC,
Takahashi K, Lachowiecz C, Tang G, Loghavi S, Bah I, Wang W, Konoplev
S, Medeiros LJ, Pammaraju N, Khoury JD, Wang SA. Bone marrow clonal
hematopoiesis is highly prevalent in blastic plasmocytoid dendritic
cell neoplasms and frequently sharing a clonal origin in elderly
patients. Leukemia 2022; 36: 1343-1350.
https://doi.org/10.1038/s41375-022-01538-9 PMid:35279700
- Mouhieddine,
TH, Sperling AS, Redd R, Park J, Leventhal M, Gibson CJ, Manier S,
Nassar AH, Capelletti M, Huynh D, et al. Clonal hematopoiesis is
associated with adverse outcomes in multiple myeloma patients
undergoing transplant. Nature Commun 2020; 11: 2996.
https://doi.org/10.1038/s41467-020-16805-5 PMid:32533060
PMCid:PMC7293239
- Maia C, Puig N, Cedena MT,
Goicoechea I, Valdes-Mas R, Vazquez I, Chillon MC, Aguirre P, Sarvide
S, Grazia-Aznarez FJ, et al. Biological and clinical significance of
dysplatic hematopoiesis in patients with newly diagnosed multiple
myeloma. Blood 2020; 25: 2375-2387.
https://doi.org/10.1182/blood.2019003382 PMid:32299093
- Tahri
S, Mouhieddine TH, Redd R, Lampe L, Nilsson KI, El-Khoury H, Su Nk,
Nassar AH, Adib E, Bindra G, et al. Clonal hematopoiesis is associated
with increased risk of progression or asymptomatic Waldenstrom
macroglobulinemia. Blood Adv 2022; 6: 2230-2238.
https://doi.org/10.1182/bloodadvances.2021004926 PMid:34847227
PMCid:PMC9006277
- Zajkowicz A, Butkiewicz D, Drosik A,
Giglok M, Suwinski R, Rusin M. Truncating mutations of PPM1D are found
in blood DNA samples of lung cancer patients. Brit J Cancer 2015; 112:
1114-1120. https://doi.org/10.1038/bjc.2015.79 PMid:25742468
PMCid:PMC4366904
- Pharoah P, Song H, Dirks E,
Intermaggio MP, Harrington P, Baynes C, Alsop K, Australian Ovarian
Cancer Group, Bogdanova D, Cicek MS, et al. PPM1S mosaic truncating
variants in ovarian cancer cases may be treatment-related somatic
mutations. J. Natl. Cancer Inst. 2016; 108(3): djv347.
https://doi.org/10.1093/jnci/djv347 PMid:26823519
PMCid:PMC5072371
- Swisher EM, Harrell M, Norquist
BM, Walsh T, Brady M, Lee M, Hershberg R, Kalli KR, Lankes L, Konnick
EQ, Pritchard CC, Monk BJ, Chan JK, Burger R, Kaufmann SH, Birerr MJ.
Somatic mosaic mutations in PPM1D and TP 53 in the blood of women with
ovarian carcinoma. JAMA Oncol. 2(3): 370-372.
https://doi.org/10.1001/jamaoncol.2015.6053 PMid:26847329
PMCid:PMC4865293
- Kahn JD, Miller PG, Silver AJ,
Sellar RB, Bhatt S, Gibson C, McConkey M, Adams D, Mar B, Mertins P,
Fereshetian S, Krug K, Zhu H, Letai A, Carr SA, Doench J, Jaiswal S,
Ebert BL. PPM1D-truncating mutations confer resistance to chemotherapy
and sensitivity to PPM1D inhibition in hematopoietic cells- Blood 2018;
132(11): 1095-1105. https://doi.org/10.1182/blood-2018-05-850339
PMid:29954749 PMCid:PMC6137556
- Bolton KL,
Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D, Patel M, Berthon
A, Syed A, Yabe M, et al. Cancer therapy shapes the fitness landscape
of clonal hematopoiesis. Nature Genetics 2020; 52(11): 1219-1226.
https://doi.org/10.1038/s41588-020-00710-0 PMid:33106634
PMCid:PMC7891089
- Leone G, Pagano L, Ben-yehuda D, Voso
MT. Therapy-related leukemia and myelodysplasia: susceptibility and
incidence. Haematologica 2007; 92(10): 1389-1398.
https://doi.org/10.3324/haematol.11034 PMid:17768113
- Leone
G, Fianchi K, Pagano L, Voso MT. Incidence and susceptibility to
therapy-related myeloid neoplasms. Chem. Biol. Interact. 2010;
184(1-2): 39-45. https://doi.org/10.1016/j.cbi.2009.12.013
PMid:20026017
- Leone G, Fianchi L, Voso MT.
Therapy-related myeloid neoplasms. Curr. Opin. Oncology 2011; 23(6):
672-680. https://doi.org/10.1097/CCO.0b013e32834bcc2a PMid:21918440
- Wong
TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Lamprecht TL,
Shen D, Hundai J, Fulton RS, Heath S, Baty JD, Klco JM, Ding L, Mardis
ER, Westervelt P, DiPersio JF, Walter MJ, Graubert TA, Ley TJ, Druley
T, Link DC, Wilson RK. The role of TP53 mutations in the origin and
evolution of therapy-related AML. Nature 2015; 518(7540): 552-555.
https://doi.org/10.1038/nature13968 PMid:25487151 PMCid:PMC4403236
- Takahashi
K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E, Zhao L, Patel K,
Neelapu S, Gumbs C, Bueso-Ramos C, DiNardo CD, Colla S, Ravandi F,
Zhang J, Huang X, Wu X, Samaniego F, Garcia-Menero G, Futreal PA.
Pre-leukemic clonal hematopoiesis and the risk of therapy-related
myeloid neoplasms: a case-control study. Lancet Oncol. 2017; 18(1):
100-111. https://doi.org/10.1016/S1470-2045(16)30626-X
- Gillis
NK, Ball M, Zhang Q, Ma Z, Zhao YL, Yoder SJ, Balasis ME, Mesa TE,
Saliman DA, Lancet JE, Kromokji RS, List AF, McLeod HL, Alsina DA, Baz
R, Shaln KH, Rollison DE, Padron E. Clonal haemopoiesis and
therapy-related myeloid malignancies in elderly patients: a
proof-of-concept, case-control study. Lancet Oncol. 2017; 18(1):
112-121. https://doi.org/10.1016/S1470-2045(16)30627-1
- Kwan
TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, Aghajanian C, Lorusso
D, Colombo N, Dean A, Weberpals J, Severson E, Vo LT, Goble S, Maloney
S, Harding T, Kaufmann SH, Ledermann JA, Coleman RL, McNeish IA, Lin
KK, Swisher EM. Preexisting TP53-variant clonal hematopoiesis and risk
of secondary myeloid neoplasms in patients with high-grade ovarian
cancer treated with rucapirib. JAMA Oncol. 2021; e214664.
https://doi.org/10.1001/jamaoncol.2021.4664 PMid:34647981
- Khalife-Hachem
S, Saleh K, Pasquier F, Willekens C, Taraby A, Antoun L, Grinda T,
Castilla-Llorente C, Duchmann M, Quivoron C, Auger N, Saada V, Delaloge
S, Leary A, Renneville A, Antony-Debre J, Rosselli F, De Botton S,
Salviat F, Marzac C, Micol JB. Molecular landscape of therapy-related
myeloid neoplasms in patients previously treated for gynecologic and
breast cancer. HemaSphere 2021; 5(9): e632.
https://doi.org/10.1097/HS9.0000000000000632 PMid:34423258
PMCid:PMC8373540
- Lindsley RC, Mar BG, Mazzola E,
Grauman PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba
HP, Damon LE, Powell BL, Lindeman N, Steensma DP, Wadleigh M, DeAngelo
DJ, Neuberg D, Stone RM, Ebert BL. Acute myeloid ontogeny is defined by
distinct somatic mutations. Blood 2015; 125(9): 1367-1376.
https://doi.org/10.1182/blood-2014-11-610543 PMid:25550361
PMCid:PMC4342352
- Coorens T, Collord G, Lu W, Mitchell
E, Ijaz J, Roberts T, Oliver T, Burke A, Gattens M, Dickens E, Nangalia
J, Tischkowitz M, Anderson J, Shlien A, Godfrey AL, Murray MJ, Behjati
S. Clonal hematopoiesis and therapy-related myeloid neoplasms following
neuroblastoma treatment. Blood 2021; 137(21): 2992-2997.
https://doi.org/10.1182/blood.2020010150 PMid:33598691
PMCid:PMC8160503
- Chen S, Wang Q, Yu H, Capitano
ML, Vermula S, Nabinger SC, Gao R, Yao C, Kobayashi M, Geng Z, Fahey A,
Henley D, Liu SZ, Barajas S, Cai W, Wolf ER, Ramdas B, Cai Z, Gao H,
Luo N, Sun Y, Wong TN, Link DC, Liu Y, Boswell HS, Mayo LD, Huang G,
Kapur R, Yoder MC, Broxmeyer HE, Gao Z, Liu Y. Mutant p53 drives clonal
hematopoiesis through modulating epigenetic pathway. Nature Commun
2019; 10: 5649. https://doi.org/10.1038/s41467-019-13542-2
PMid:31827082 PMCid:PMC6906427
- Abelson S, Collord G, Ng
SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, Barda N, Zuzarte PC,
Heister L, Sundaravadanam Y, et al. Prediction of acute myeloid
leukemia risk in healthy individuals. Nature 2018; 559(7714): 400-404.
https://doi.org/10.1038/s41586-018-0317-6 PMid:29988082
PMCid:PMC6485381
- Desai P, Mencia-Trinchant N, Savenkov
O, Simon MS, Cheang G, Lee S, Samuel M, Ritchie EK, Guzman ML, Ballman
KV, Roboz GJ, Hassane DC. Somatic mutations precede acute myeloid
leukemia years before diagnosis. Nature Med. 24(7): 1015-1023.
https://doi.org/10.1038/s41591-018-0081-z PMid:29988143
PMCid:PMC6849383
- Young AL, Tong RS, Birmann BM, Druley
TE. Clonal hematopoiesis and risk of acute myeloid leukemia.
Haematologica 2019; 104(12): 2410-2417.
https://doi.org/10.3324/haematol.2018.215269 PMid:31004019
PMCid:PMC6959179
- Watson CJ, Papula A, Poon Y, Wong WH,
Young AL, Druley TE, Fisher DS, Blundell JR. The evolutionary dynamics
and fitness landscape of clonal haematopoiesis. Science 2020;
367(6485): 1449-1454. https://doi.org/10.1126/science.aay9333
PMid:32217721
- Sill H, Zebisch A, Haase D. Acute myeloid
leukemia and myelodysplastic syndromes with TP53 aberrations - a
distinct stem cell disorder. Clin Cancer Res 2020; 26(20): 5304-5309.
https://doi.org/10.1158/1078-0432.CCR-20-2272 PMid:32816950
PMCid:PMC7116522
- Boettcher S, Miller PG, Sharma
R, McConkey M, Leventhal M, Krivstov AV, Giacomelli AO, Wong W, Kim J,
Chao S, Kurppa KJ, Yang X, Milenkowic K, Piccioni F, Root DE, Rucker
FG, Flamand Y, Neuberg D, Lindsley RC, Janne PA, Hahn WC, Jacks T,
Dohner H, Armstrong SA, Ebert BL. A dominant-negative effect drives
selection of TP53 missense mutations in myeloid malignancies. Science
2019; 365(6453): 599-604. https://doi.org/10.1126/science.aax3649
PMid:31395785 PMCid:PMC7327437
- Zebisch A, Lai R,
Muller M, Lind K, Kashofer K, Girschikofsky M, Fuchs D, Wolfler A,
Geigl JB, Sill H. Acute myeloid leukemia with TP53 germ line mutations
Blood 2016; 128(18): 2270-2272.
https://doi.org/10.1182/blood-2016-08-732610 PMid:27621308
PMCid:PMC5095760
- Stengel A, Kern W, Haferlach T,
Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and
TP53 deletions on survival varies between AML, ALL, MDS and CLL: an
analysis of 3307 cases. Leukemia 2017; 31(3): 705-711.
https://doi.org/10.1038/leu.2016.263 PMid:27680515
- Prochazka
K, Pregartner G, Rucker FG, Heltzer E, Pabst G, Wolfler A, Zerbish A,
Berghold A, Dohner K, Sill H. Clinical implications of subclonal TP53
mutations in acute myeloid leukemia. Haematologica 2018; 104(3):
516-523. https://doi.org/10.3324/haematol.2018.205013 PMid:30309854
PMCid:PMC6395341
- Goel S, Hall J, Pradhan K, Hirsch C,
Przychodzen B, Shastri A, Mantzaris I, Janakiram M, Battini R, Kornblum
N, Derman O, Gristman K, Al-Hadifh J, Wang Y, Halmos B, Steidl U,
Macijewski JP, Braunschweig I, Verma A. High prevalence and allele
burden-independent prognostic importance of p53 mutations in an
inner-city MDS/AML cohort. Leukemia 2016; 30: 1793-1795.
https://doi.org/10.1038/leu.2016.74 PMid:27125205
- Lal
R, Lind K, Heitzer H, Ulz P, Aubell K, Kashofer K, Middeke JM, Thiede
C, Schutz E, Rosenberger A, Hofer S, Felhauer B, Rinner B, Svendova V,
Schimek MG, Rucker FG, Hoefler G, Dohner K, Zebisch A, Wolfler A, Sill
H. Somatic TP53 mutations characterize preleukemic stem cells in acute
myeloid leukemia. Blood 2017; 129(18): 2587-2591.
https://doi.org/10.1182/blood-2016-11-751008 PMid:28258055
- Pabst
G, Lind K, Graf R, Zebisch A, Stolzel F, Dohner K, Heltzer E, Reinisch
A, Sill H. TP53 mutated AML subclones exhibit engraftment in a
humanized bone marrow ossicle mouse model. Ann Hematol 2020; 99:
653-655. https://doi.org/10.1007/s00277-020-03920-y PMid:32002654
PMCid:PMC7060155
- Quintas-Cardama A, Hu C, Qutub A, Qiu
YH, Zhang X, Post SM, Zhang N, Coombes K, Kornblau SM. P53 pathway
dysfunction is highly prelavent in acute myeloid leukemia independent
of TP53 mutational status. Leukemia 2017; 31(6): 1296-1305. https://doi.org/10.1038/leu.2016.350 PMid:27885271
- Carvajal
LA, Ben Neriah D, Senecal A, Bernard L, Tiruthuvanathan V, Yatsenko T,
Narayanagari SR, Wheat JC, Todorova TI, Mitchell K, Kenworthy C,
Guerlavis V, Annis DA, Bartholdy B, Will B, D'Anampa J, Mantzaris I,
Alvado M, Singer RH, Coleman RA, Verma A, Steidl U. Dual inhibition of
MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Trans Med
2018; 10(436): eaao3003. https://doi.org/10.1126/scitranslmed.aao3003 PMid:29643228 PMCid:PMC6130841
- Yang
M, Pan Z, Huang K, Busche G, Liu H, Gohring G, Rumpel R,
Dittrich-Breiholz O, Talbot S, Scherr M, Chaturvedi A, Eder M, Skokowa
J, Zhou J, Welte K, von Neurhoff N, Liu L, Ganser A, Li Z. A unique
role of p53 haploinsufficiency or loss in the development of acute
myeloid leukemia with FLT3-ITD mutation. Leukemia 2021; in press. https://doi.org/10.1038/s41375-021-01452-6 PMid:34732858 PMCid:PMC8885416
- Ortmann
CA, Dorsheimer L, Abou-El.Ardat K, Hoffrichter J, Assmus B, Bonig H,
Scholz A, Pfeifer H, Martin H, Schmid T, Brune B, Scheich S, Steffen B,
Riemann J, Hermann S, Dukat A, Bug G, Brandts CH, Wagner S, Serve H,
Rieger MA. Functional dominance of CHIP-mutated hematopoietic stem
cells in patients undergoing autologous transplantation. Cell Rep.
2019; 27(9): 2022-2028. https://doi.org/10.1016/j.celrep.2019.04.064 PMid:31091442
- Soerensen
JF, Aggerholm A, Kendrup GB, Hansen MC, Ewald IK, Bill M, Ebbesen LH,
Rosenberg CA, Hokland P, Ludvigsen M, Roug AS. Clonal hematopoiesis
predicts development of therapy-related myeloid neoplasms
post-autologous stem cell transplantation. Blood Adv 2020; 10(4):
885-892.https://doi.org/10.1182/bloodadvances.2019001157 PMid:32150606 PMCid:PMC7065480
- Berger
G, Kroeze LI, Koorenhof-Scheele TN, de Graaf AO, Yoshida K, Ueno H,
Shiraishi Y, Miyano S, van den Berg E, Schepers H, van der Reijden BA,
Ogawa S, Vellenga E, Jansen JH. Early detection and evolution of
preleukemic clones in therapy-related myeloid neoplasms following
autologous SCT. Blood 2018; 131(16): 1846-1857. https://doi.org/10.1182/blood-2017-09-805879 PMid:29311096
- Frick
M, Chan W, Arends CM, Hablesreiter R, Halik A, Heuser M, Michonneau D,
Blau O, Hoyer K, Christen F, et al. Role of donor clonal hematopoiesis
in allogeneic hematopoietic stem-cell transplantation. J Clin Oncol
2018; 36: 1-10.
- Gibson CJ, Kim HT, Zhao L, Murdock M,
Hambley B, Ogata A, Madero-Marroquin R, Wang S, Green L, Fleharty M,
Dougan T, et al. Donor clonal hematopoiesis and recipient outcomes
after transplantation. J Clin Oncol 2021; in press. https://doi.org/10.1101/2021.09.25.21263697
- Gondek
LP, Zheng G, Ghiaur G, DeZern AE, Matsui W, Yegnasubramanian S, Lin MT,
Levis M, Eshleman JR, Varadhan R, Ticker N, Jones R, Gocke CD. Donor
cell leukemia arising from clonal hematopoiesis after bone marrow
transplantation. Leukemia 2016; 30(4): 1916-1920. https://doi.org/10.1038/leu.2016.63 PMid:26975880 PMCid:PMC5014666
- Herold
S, Kuhn M, Bonin MV, Stange T, Platzbecker U, Radke J, Lange T, Sockel
K, Gutsche K, Schetelig J, Rollig C, Schuster C, Roeder I, Dahl A, Mohr
B, Serve H, Brandts C, Ahninger G, Bornhauser M, Thiede C. Donor cell
leukemia: evidence for multiple preleukemic clones and parallel long
term clonal evolution in donor and recipient. Leukemia 2017; 31(7):
1637-1640. https://doi.org/10.1038/leu.2017.104 PMid:28348390
- Gibson
CJ, Kennedy JA, Nikiforow S, Kuo FC, Alyea EP, Ho, Ritz J, Soiffer R,
Antin JH, Lindsley RC. Donor-engrafted CHIP is common among stem cell
transplant recipients with unexplained cytopenias. Blood 2016; 130(1):
91-94. https://doi.org/10.1182/blood-217-01-764951 PMid:28446434 PMCid:PMC5501150
- Gibson CJ, Lindsley RC. Stem cell donors should not be screened for clonal hematopoiesis. Blood Adv 2020; 4(4): 789-792. https://doi.org/10.1182/bloodadvances.2019000395 PMid:32097457 PMCid:PMC7042999
- DeZern AE, Gondek LP. Stem cell donors should be screened for CHIP. Blood Adv 2020; 4(4): 784-788. https://doi.org/10.1182/bloodadvances.2019000394 PMid:32097458 PMCid:PMC7042978
- Valent
P, Akin C, Arock M, Bock C, George TI, Galli SJ, Gotlib J, Haferlach T,
Hoermann G, Hermine O, et al. Preposed terminology and classification
of pre-malignant neoplastic conditions: a consensus proposal.
EBioMedicine 2017; 26: 17-24. https://doi.org/10.1016/j.ebiom.2017.11.024 PMid:29203377 PMCid:PMC5832623
- Valent
P, Kern W, Hoermann G, Milosevic Feenstra JD, Sotlar K, Pfeilstocker M,
Germing U, Sperr WR, Reietr A, Wolf D, Arock M, Haferlach T, Horny HP.
Clonal hematopoiesis with oncogenic potential (CHOP): separation from
CHIP and roads to AML. Int Mol Sci 2019; 20: 789. https://doi.org/10.3390/ijms20030789 PMid:30759825 PMCid:PMC6387423
- Cappelli
LV, Meggendorfer M, Baer C, Nadarajah N, Hutter S, Jeromin S, Dicker F,
Kem W, Haferlach T, Haferlach C, Hollein A. Indeterminate and oncogenic
potential: CHIP and CHOP mutations in AML with NPM1 alteration.
Leukemia 2021; in press. https://doi.org/10.1038/s41375-021-01368-1 PMid:34376804 PMCid:PMC8807394
- Gurnari
C, Fabiani E, Falconi G, Travaglini S, Ottone T, Cristiano A, Voso MT.
From clonal hematopoiesis to therapy-related myeloid neoplasms: the
silent way of cancer progression. Biology 2021; 10: 128. https://doi.org/10.3390/biology10020128 PMid:33562056 PMCid:PMC7914896
- DeZern
AE, Malcovati L, Ebert BL. CHIP, CCUS, and other acronyms: definition,
implications, and impact on practice. ASCO Educ Book 2019; 400-410. https://doi.org/10.1200/EDBK_239083 PMid:31099654
- Van
Zeventer IA, de Graaf AO, van der Klauw MM, Vellenga E, van der Reijden
BA, Schuringa JJ, Diepstra A, Malcovati L, Jansen JH, Huls G.
Peripheral blood cytopenias in the aging general population and risk of
incident hematological disease and mortality. Blood Adv 2021; 5(17):
3266-3278. https://doi.org/10.1182/bloodadvances.2021004355 PMid:34459888 PMCid:PMC8525229
- Kwok
B, Hall JM, Witte JS, Xu Y, Reddy P, Lin K, Flamholz R, Dabbas B, Yung
A, Al-Hadif J, Balmert E, Vaupei C, El Hader C, McGinnis MJ, Nahas SA,
Kines J, Bejar R. MDS-associated somatic mutations and clonal
hematopoiesis are common in idiopathic cytopenias of undetermined
significance. Blood 2015; 126(21): 2355-2361. https://doi.org/10.1182/blood-2015-08-667063 PMid:26429975 PMCid:PMC4653764
- Cargo
CA, Rowbotham N, Evans PA, Barrans SL, Bowen DT, Crouch S, Jack AS.
Targeted sequencing identifies patients with preclinical MDS at high
risk of disease progression. Blood 2015; 126(21): 2362-2365. https://doi.org/10.1182/blood-2015-08-663237 PMid:26392596
- Malcovati
L, Galli A, Travaglino E, Ambaglio I, Rizzo E, Molteni E, Elena C,
Ferretti VV, Catricalà S, Bono E, Todisco G, Bianchessi A, Rumi E,
Zibellini S, Pietra D, Boveri E, Camaschella C, Toniolo D, Papaemmanuil
E, Ogawa S, Cazzola M. Clinical significance of somatic mutation in
unexplained blood cytopenia. Blood 2017; 129(25): 3371-3378. https://doi.org/10.1182/blood-2017-01-763425 PMid:28424163 PMCid:PMC5542849
- Mikkelsen
SU, Safavi S, Dimopoulos K, O'Rourke CJ, Andersen MK, Holm MS, Marcher
CW, Andersen JB, Hansen JW, Grinboeck K. Structural aberrations are
associated with poor survival in patients with clonal cytopenia of
undetermined significance. Haematologica 2021; 106(6): 1762-1766. https://doi.org/10.3324/haematol.2020.263319 PMid:33179473 PMCid:PMC8168501
- Singh
A, Mencia-Trinchant N, Griffiths EA, Altahan A, Swaminathan M, Gupta M,
Gravina M, Tajammal R, Faber MG, Yan LB, et al. Mutant PPM1D- and
TP53-driven hematopoiesis populates the hematopoietic compartment in
response to peptide receptor radionuclide therapy. JCO Precis Oncol
2022; 6: e2100309. https://doi.org/10.1200/PO.21.00309 PMid:35025619
- Xie
Z, Nanaa A, Saliba AN. Treatment outcome of clonal cytopenias of clonal
cytopenias of undetermined significance: a single-institution
retrospective study. Blood Cancer J 2021; 11(3): 3. https://doi.org/10.1038/s41408-021-00439-x PMid:33649321 PMCid:PMC7921651
- Jeong
M, Park HJ, Celik H, Ostrander EL, Reyes JM, Guzman A, Rodriguez B, Lei
Y, Lee Y, Ding L, et al. Loss of Dnmt3A immortalizes hematopoietic stem
cells in vivo. Cell Rep 2018; 23(1): 1-10. https://doi.org/10.1016/j.celrep.2018.03.025 PMid:29617651 PMCid:PMC5908249
- Loberg
MA, Bell RK, Goodwin LO, Eudy E, Miles LA, SanMiguel JM, Young K,
Bergstrom DE, Levine R, Schneider RK, Trowbridge JJ. Sequential
inducible mouse models reveal that Nmp1 mutation causes malignant
transformation of Dnmt3A-mutant clonal hematopoiesis. Leukemia 2019;
33(7): 1635-1649. https://doi.org/10.1038/s41375-018-0368-6 PMid:30692594 PMCid:PMC6609470
- SanMiguel
JM, Eudy E, Loberg MA, Miles LA, Steams T, Mistry JJ, Rauh MJ, Levine
RL, Trowbridge JJ. Cell origin-dependent cooperativity of mutant Dnmt3A
and Npm1 in clonal hematopoiesis and myeloid malignancy. Blood Adv
2022; 6(12): 3666-3677. https://doi.org/10.1182/bloodadvances.2022006968 PMid:35413095
- Kim
PG, Niroula A, Shkolnik V, McConkey M, Lin AE, Stabicki M, Kemp JP,
Bick A, Gibson CJ, Griffin G, et al. Dnmt3a-mutated clonal
hematopoiesis promotes osteoporosis. 2021; 218(12): e20211872. https://doi.org/10.1084/jem.20211872 PMid:34698806 PMCid:PMC8552148
- Huang
YH, Chen CW, Sundaramurthy V, Stabicki M, Hao D, Watson CJ, Tovy A,
Reyes JM, Dakhova O, Grovetti BR, et al. Systematic profiling of DNMT3A
variants reveals protein instability mediated by the DCAF8 E3 ubiquitin
ligase adaptor. Cancer Discov 2022; 12: 220-235. https://doi.org/10.1158/2159-8290.CD-21-0560 PMid:34429321 PMCid:PMC8758508
- Smith
AM, Verdoni AM, Abel HJ, Chen DY, Ketkar S, Leight ER, Miller CA, Ley
TJ. Somatic Dnmt3a inactivation leads to slow, canonical DNA
methylation loss in murine hematopoietic cells. iScience 2022; 25:
104004. https://doi.org/10.1016/j.isci.2022.104004 PMid:35313694 PMCid:PMC8933692
- Nam
AS, Dusaj N, Izzo F, Murali R, Myers RM, Mouhieddine T, Sotelo J,
Benbarche S, Waarts M, Gaiti F, et al. Single-cell multi-omics of human
clonal hematopoiesis reveals that DNMT3A R882 mutations perturb early
progenitor states through selective hypomethylation. BioRxiv 2022;
10.1101/2022.01.14.476225. https://doi.org/10.1101/2022.01.14.476225
- Moran-Crusio
K, Reavie L, Shih A, Abdel-Wahab O, Ndiaye-Lobry D, Lobry C, Figueroa
ME, Vasanthakumar A, Patel J, Zhao X, et al. Tet2 loss leads to
increased hematopoietic stem cell self-renewal and myeloid
differentiation. Cancer Cell 2011; 20(1): 11-24. https://doi.org/10.1016/j.ccr.2011.06.001 PMid:21723200 PMCid:PMC3194039
- Li
Z, Cai X, Cai CL, Wang J, Zhang W, Petersen BE, Yang FC, Xu M.
Detection of Tet2 in mice leads to dysregulated hematopoietic stem
cells and subsequent development of myeloid malignancies. Blood 2011;
118(17): 4509-4518. https://doi.org/10.1182/blood-2010-12-325241 PMid:21803851 PMCid:PMC3952630
- Ito
K, Lee J, Chrysanthou S, Zhao Y, Josephs K, Sato H, Teruya-Feldstein J,
Zheng D, Dawlaty MM, Ito K. Non-catalytic role of Tet2 are essential to
regulate hematopoietic stem and progenitor cell homeostasis. Cell Rep
2019; 28(10): 1480-2490. https://doi.org/10.1016/j.celrep.2019.07.094 PMid:31484061 PMCid:PMC6750732
- Nakauchi
Y, Azizi A, Thomas D, Corces MR, Reisch A, Sharma R, Hernandez DC,
Kohnke T, Karigane D, Fan A, et al. The cell type specific 5hmC
landscape and dynamics of healthy hematopoiesis and TET2-mutant
pre-leukemia. Blood Cancer Discov 2022; in press. https://doi.org/10.1158/2643-3230.BCD-21-0143 PMid:35532363
- Tulstrup
M, Soerensen M, Werner Hansen J, Gillberg L, Needhamsen M, Kaastrup K,
Helin K, Christensen K, Weischenfeldt J, Gronbaeck K. TET2 mutations
are associated with hypermethylation at key regulatory enhancers in
normal and malignant hematopoiesis. Nature Commun 2021; 2:6061. https://doi.org/10.1038/s41467-021-26093-2 PMid:34663818 PMCid:PMC8523747
- Abdel-Wahab
O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, Kuscu T, Hricik T,
Ndiaye-Lobry D, Lafave LM, et al. Deletion of Asxl1 results in
myelodysplasia and severe developmental defects in vivo. J Exp Med
2013; 210: 2641-2659. https://doi.org/10.1084/jem.20131141 PMid:24218140 PMCid:PMC3832937
- Wang
J, Li Z, He Y, Pan F, Chen S, Rhodes S, Nguyen L, Yuan J, Jiang L, Yang
X, et al. Loss of Asxl1 leads to myelodysplastic syndrome-like
syndrome. Blood 2014; 123: 541-553. https://doi.org/10.1182/blood-2013-05-500272 PMid:24255920 PMCid:PMC3901067
- Inoue
DJ, Kitaura J, Togami K, Nishimura K, Enomoto Y, Uchida T, Kagiyama Y,
Kawabata KC, Nakahara F, Izawa K, et al. Myelodysplastic syndromes are
induced by histone methylation-altering ASXL1 mutations. J Clin Invest
2013; 123: 4627-4640. https://doi.org/10.1172/JCI70739 PMid:24216483 PMCid:PMC3809801
- Nagase
R, Inoue D, Pastore A, Fujino T, Hou HN, Yamasaki N, Goyama S, Saika M,
Kanai A, Sera Y, et al. Expression of mutant Asxl1 perturbs
hematopoiesis and promotes susceptibility to leukemic transformation. J
Exp Med 2018; 215(6): 1729-1747. https://doi.org/10.1084/jem.20171151 PMid:29643185 PMCid:PMC5987913
- Fujino
T, Goyama S, Sugiura Y, Inoue D, Asada S, Yamasaki S, Matsumoto A,
Yamaguchi K, Isobe Y, Tsuchya A, Shikata S, et al. Mutant ASXL1 induces
age-related expansion of phenotypic hematopoietic stem cells through
activation of Akt/mTOR pathway. Nature Commun 2021; 12: 1826. https://doi.org/10.1038/s41467-021-22053-y PMid:33758188 PMCid:PMC7988019
- Dawoud
A, Tapper WJ, Cross N. Clonal myelopoiesis in the UK Biobank cohort:
ASXL1 mutations are strongly associated with smoking. Leukemia 2020;
34(10): 2660-2672. https://doi.org/10.1038/s41375-020-0896-8 PMid:32518416
[TOP]