Francesco Fabozzi1 and Angela Mastronuzzi1*
1 Department of Pediatric Hematology/Oncology and Cellular and Gene Therapy, Bambino Gesù Children's Hospital IRCCS, Rome, Italy
Correspondence to: Dr.
Angela Mastronuzzi. Department of Pediatric Hematology/Oncology and
Cellular and Gene Therapy, Bambino Gesù Children's Hospital IRCCS,
Rome, Italy. E-mail:
angela.mastronuzzi@opbg.net
Published: May 1, 2023
Received: March 14, 2023
Accepted: April 19, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023032 DOI
10.4084/MJHID.2023.032
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Advances
in molecular biology and genetic testing have greatly improved our
understanding of the genetic basis of hematologic malignancies and have
enabled the identification of new cancer predisposition syndromes.
Recognizing a germline mutation in a patient affected by a hematologic
malignancy allows for a tailored treatment approach to minimize
toxicities. It informs the donor selection, the timing, and the
conditioning strategy for hematopoietic stem cell transplantation, as
well as the comorbidities evaluation and surveillance strategies.
This
review provides an overview of germline mutations that predispose to
hematologic malignancies, focusing on those most common during
childhood and adolescence, based on the new International Consensus
Classification of Myeloid and Lymphoid Neoplasms.
|
Introduction
Advances
in molecular biology and genetic technologies have significantly
improved our knowledge about the genetic landscape of major cancer
types in children and adults.[1] Aside from offering
valuable diagnostic and prognostic insights from somatic alterations,
assessing non-tumor or germline material using comprehensive sequencing
techniques has revolutionized our understanding of how germline
mutation affects cancer development. According to several large-scale
studies involving pediatric cancer patients, the frequency of
potentially harmful germline mutations was estimated to be around 8.5%.[2,3]
Hematologic
malignancies represent the most frequent neoplasm affecting children
and adolescents, with acute lymphoblastic leukemia (ALL) being the most
common type of childhood cancer.[4] In hematologic
malignancies, most efforts have focused on identifying acquired genetic
alterations to guide prognostic stratification and tailored treatment
strategies.[5,6] Although the role of germline genetic
alterations in the development of hematologic malignancies has for a
long time been underestimated, the inclusion of the category "Myeloid
neoplasms with germline predisposition" in the fourth edition of the
World Health Organization (WHO) Classification of Tumors of
Hematopoietic and Lymphoid Tissues has underscored the utmost
importance of germline assessment in patients with myeloid tumors.[7]
Furthermore, it is becoming increasingly clear that these observations
can now be extended to lymphoid malignancies, as demonstrated by the
recent International Consensus Classification (ICC) of Myeloid and
Lymphoid Neoplasms.[8] Thus, the title is changed from "myeloid neoplasms" to "hematologic neoplasms" with germline predisposition.
Even though many patients lack a family history consistent with a cancer predisposition syndrome,[3] some clues can help us suspect a germline mutation in patients with a hematologic malignancy[6] (Figure 1). In particular, several associated clinical features may point toward specific syndromes (Table 1).[6]
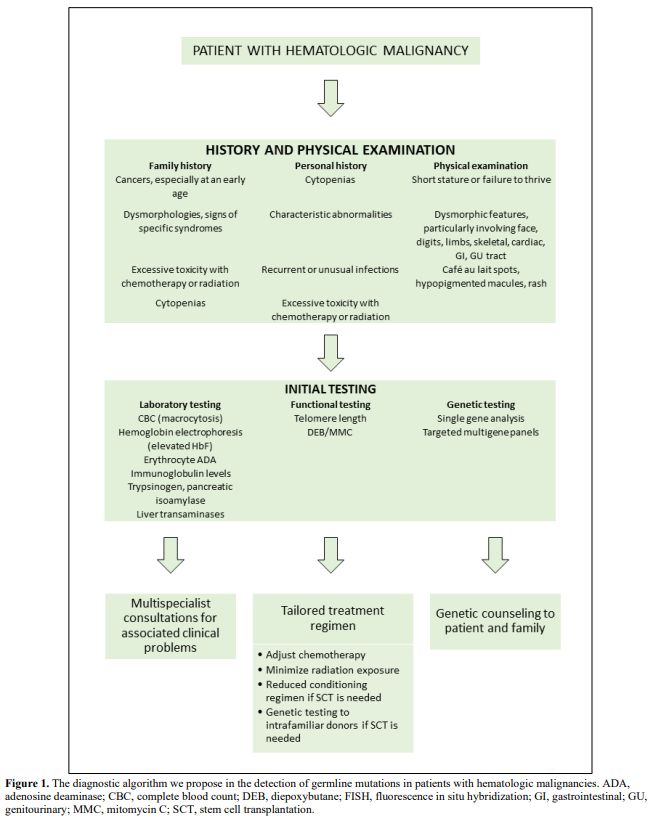 |
Figure
1. The diagnostic algorithm we propose in the detection of
germline mutations in patients with hematologic malignancies. ADA,
adenosine deaminase; CBC, complete blood count; DEB, diepoxybutane;
FISH, fluorescence in situ hybridization; GI, gastrointestinal;
GU,genitourinary; MMC, mitomycin C; SCT, stem cell transplantation. |
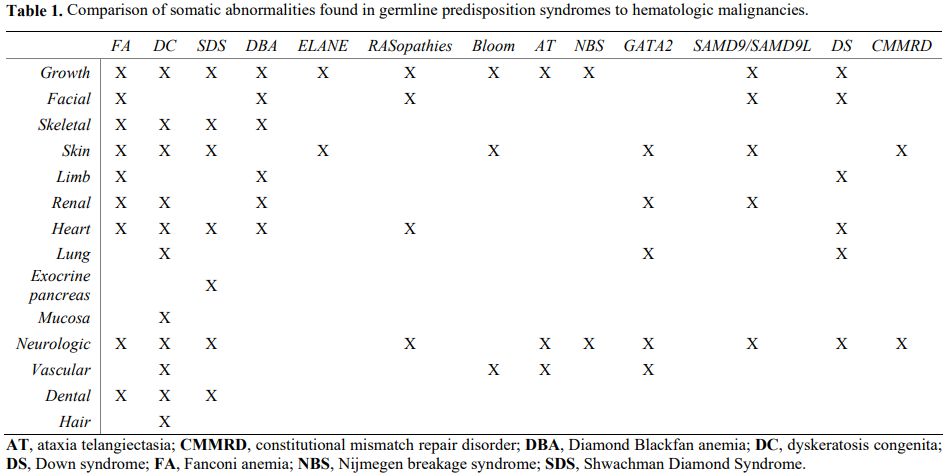 |
Table 1. Comparison of somatic abnormalities found in germline predisposition syndromes to hematologic malignancies.
|
The discovery of a germline mutation in a patient affected by a hematologic malignancy has significant
implications, impacting the patient's psychosocial well-being and
family relationships, and may lead to important decisions regarding
reproductive planning and genetic counseling. Furthermore, it may
influence the type and intensity of treatment and the risk of
recurrence and secondary cancers. In fact, several of these conditions
carry an increased risk of severe toxicity with standard chemotherapy
or radiation dosages. Such toxicity can result in prolonged or
permanent cytopenias, organ damage, or significant mucositis; thus,
early detection of such patients enables tailored treatment using less
intense regimens.[6,9] Finally,
discovering an inherited mutation in a patient with a hematologic
malignancy inevitably impacts the selection of a donor when
hematopoietic stem cell transplantation (SCT) is indicated. Even though
HLA-matched sibling donors are usually the preferred donors, they may
share the same mutation with the affected individual.
Consequently,
screening must be performed even if the sibling appears asymptomatic.
Several questions also arise regarding the ideal timing for performing
SCT as well as the intensity of the conditioning regimen to be
preferred, which must be evaluated on a case-by-case basis considering
the specific disease.[10] For example, in patients
with germline mutations carrying a high penetrance of leukemia, a
preemptive SCT may represent a wise option; on the other hand, in cases
with a lower probability of developing leukemia, a watch-and-wait
strategy may be preferred. Similarly, a reduced-intensity conditioning
regimen may benefit patients at high risk of transplant-related
toxicities, such as syndromic conditions characterized by numerous
comorbidities.
This review provides an overview of genetic
mutations predisposing to hematologic malignancies, focusing on those
most common among children and young adults. For convenience, we have
grouped genes according to the new ICC (Table 2),
which includes 4 major subgroups with new entities added in comparison
with the 2016 WHO classification: hematologic neoplasms with germline
predisposition without a constitutional disorder, including CEBPA,
DDX41, and TP53 alterations; those associated with thrombocytopenia or
platelet dysfunction including RUNX1, ANKRD26, and ETV6 alterations;
those associated with constitutional disorders affecting multiple organ
systems including GATA2, SAMD9, and SAMD9L mutations, inherited genetic
mutations associated with classic bone marrow failure (BMF) syndromes
and juvenile myelomonocytic leukemia (JMML), and Down syndrome; ALL
with germline predisposition. These classifications should not be
considered rigid as they can sometimes overlap; for example, Down
syndrome and germline mutations in ETV6 or TP53 predispose to ALL.
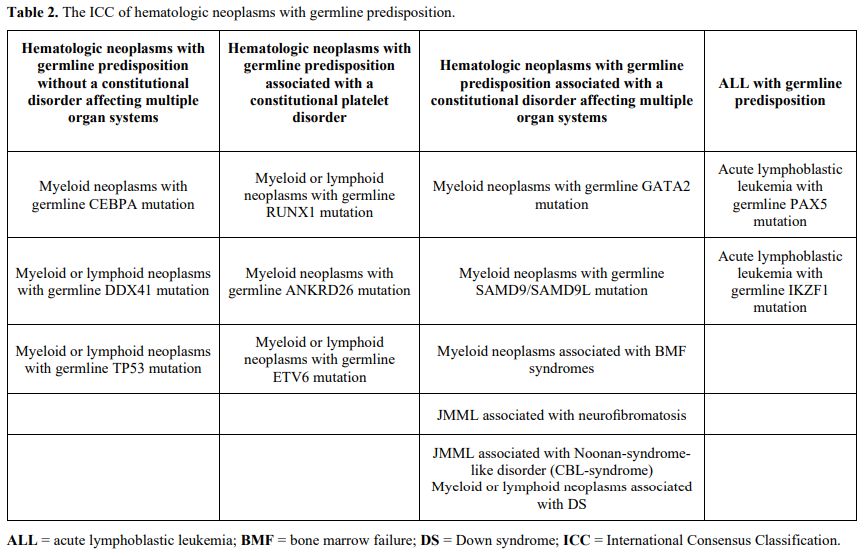 |
- Table 2. The ICC of hematologic neoplasms with germline predisposition.
|
Hematologic neoplasms with germline predisposition without a constitutional disorder affecting multiple organ systems
Myeloid neoplasm with germline CEBPA mutation. CEBPA is a single exon gene in the chromosomal region of 19q13.1 encoding for a granulocyte differentiation factor.[11] Biallelic mutations are often recognized in acute myeloid leukemias (AMLs), defining a unique subtype with good outcome.[12,13]
It has been shown that nearly 10% of these cases also carry a germline
CEBPA mutation, typically a frameshift or nonsense mutation near the
amino terminus of the encoded protein.[14]
Progression to AML occurs with a near complete penetrance, often in the
second or third decade of life, and may develop without a previous
myelodysplastic syndrome (MDS). It is commonly associated with an
acquired mutation in the remaining wild-type CEBPA allele.[14,15]
One of the peculiar features of this entity is that when these patients
have disease recurrence after chemotherapy, they present new clones
with a different spectrum of acquired mutations, including new somatic
CEBPA mutations, demonstrating that these second leukemias are not true
relapses.[15]
Myeloid or lymphoid neoplasms with germline TP53 mutation.
TP53 is commonly considered the guardian of the genome, as it plays a
pivotal role in the cell cycle, DNA repair, and apoptosis.[16]
Germline mutations are the defining feature of Li-Fraumeni syndrome
(LFS) and predispose to a diverse range of tumors in adults and
children, particularly breast cancer, sarcomas, and brain tumors. In
contrast, hematological malignancies are relatively uncommon.[17-19]
Leukemias occur with an estimated incidence of 4% and are predominantly
hypodiploid ALL and therapy-related myeloid disorders, including AML
and MDS.[20-22] In particular, germline TP53
alterations are a hallmark of low hypodiploid ALL, as found in more
than half of the children affected.[23] Leukemic transformation is associated with somatic alterations of IKZF2, CDKN2A, and CDKN2B.[23]
Due
to the very increased susceptibility to second cancers, patients with
LFS and a hematological malignancy should avoid exposure to radiation
therapy when possible.
Myeloid or lymphoid neoplasms with germline DDX41 mutation.
Unlike the other genes cited in this review, germline DDX41 mutations
predispose to neoplasm arising during adulthood, typically in the 6th
decade.[24-26] These alterations probably underlie more than 5% of AMLs, making them the most common predisposing events reported in AML.[27]
Patients carrying DDX41 germline mutations represent a unique AML
subset with male sex skewing, older age, low leukocyte count, few
somatic genetic events, and high response rates to intensive
chemotherapy leading to prolonged survival.[28] A second somatic DDX41 mutation represents the main driver for AML progression.[28] Lymphoid neoplasms have also been described but are less common.[25]
Hematologic neoplasms with germline predisposition associated with a constitutional platelet disorder
Myeloid or lymphoid neoplasms with germline RUNX1 mutation. RUNX1
is a transcription factor that plays a critical role in regulating
blood cell development and differentiation, especially involved in
megakaryocyte maturation, differentiation, ploidization, and
proplatelet formation.[29] Whereas somatic
alterations in RUNX1 are among the most common mutations in both adults
and children with ALL, AML and MDS, germline mutations define familial
platelet disorder with predisposition to myeloid malignancy (FDP-MM),
initially described in 1999.[30] Several mutations
have been identified to date, including larger gene deletions, nonsense
or frameshift mutations, and point mutations acting by
haploinsufficiency with dominant negative effects.[31]
All these alterations result in an autosomal dominant disorder with a
variable penetrance, characterized by quantitative and/or qualitative
platelet defects with a predisposition to developing hematological
malignancies. The symptomatic patients typically present with
mild-to-moderate thrombocytopenia. Platelet morphology is normal but is
associated with a severe decrease in platelet aggregation due to
decreased dense granules.[31] The risk of malignant transformation into MDS and AML usually occurs in adulthood and is estimated to be 30%-40%;[32]
patients carrying RUNX1 mutations with a dominant-negative effect
appear to have a higher risk than patients carrying loss-of-function
alleles.[31,33] The progression is
associated with the acquisition of somatic mutations in the remaining
wild-type RUNX1 allele, as well as GATA2 mutations, and less commonly,
other genes recurrently mutated in AML and MDS. More rarely, a
malignant transformation in other hematological malignancies may occur,
T-ALL being the most frequent.[31,34-37]
Myeloid neoplasms with germline ANKRD26 mutation.
Gain-of-function single nucleotide substitutions in the ANKRD26 gene,
typically in the promoter region, lead to increased gene transcription
and signaling through the MPL pathway and impaired proplatelet
formation by megakaryocytes.[38] Carriers present with moderate thrombocytopenia, a normal mean platelet volume, and an absent or mild bleeding tendency.[39]
The risk of progression to malignancies is estimated at 5% for AML,
2.2% for MDS, and 1.3% for chronic myeloid leukemia (CML).[40]
Myeloid or lymphoid neoplasms with germline ETV6 mutation.
ETV6 is a tumor suppressor gene frequently mutated by somatic
alterations, such as the ETV6-RUNX1 fusion commonly seen in childhood
ALL.[41] Germline mutations are associated with mild
to moderate thrombocytopenia with normal-sized platelets and mild to
moderate bleeding tendency.[42,43] They can be found in approximately 1% of pediatric ALL cases[44]
and are predominantly missense variants. Other than ALLs, ETV6 germline
mutations are also associated with MDS/AML, mixed-phenotype acute
leukemia, chronic myelomonocytic leukemia (CMML), plasma cell myeloma
and polycythemia vera, as well as with solid tumors including
colorectal, breast, kidney, and skin cancers, and meningioma.[42,43]
Hematologic neoplasms with germline predisposition associated with a constitutional disorder affecting multiple organ systems
Myeloid neoplasms with germline GATA2 mutation.
GATA2 is a transcription factor that plays a leading role in
hematopoiesis but can also be expressed in endothelial cells, central
nervous system, placenta, fetal liver, and fetal heart.[45,46]
This ubiquitous expression is reflected in the wide range of clinical
features that patients carrying germline mutations may present, like
pulmonary alveolar proteinosis, lymphedema and sensorineural deafness,
and miscarriages. However, bone marrow dysfunction represents the
hallmark of the disease, leading to recurrent infections (mainly
atypical mycobacterial infections and recurrent HPV-related warts) and
hematological malignancies.[47-49] Patients carry
loss-of-functions mutations, involving mostly the second zinc finger
domain and resulting in GATA2 haploinsufficiency.[50] GATA2 deficiency underlies 15% of advanced forms and 7% of all primary MDS in childhood.[51,52]
Clinical onset can occur over a highly variable time frame, at a median
age of 18 years, whereas some carriers may remain asymptomatic for life
though the penetrance at age 60 is 90%.[53]
Therefore, intrafamily donor genetic testing, even if asymptomatic,
must be warranted before proceeding to SCT. At birth, carriers
typically have normal cell counts; however, a progressive reduction of
CD34+ cells in bone marrow occurs over time, resulting in
monocytopenia, dendritic cell deficiency, NK cell deficiency, B cell
deficiency, and, less commonly, neutropenia.[54,55] The progression into MDS is associated with monosomy 7 or trisomy 8,[46,56] whereas progression to AML is frequently driven by ASXL1 alterations.[51]
Currently, clear guidelines for managing patients with GATA2 mutations
are lacking. A possible algorithm for patient monitoring is proposed
in.[57]
Myeloid neoplasms with germline SAMD9 or SAMD9L mutation.
Together with GATA2, SAMD9/SAMD9L mutations, two interferoninducible
genes located on chromosome 7, are the most frequent germline mutations
in pediatric MDS.[52] They were initially recognized
to underlie MIRAGE (Myelodysplasia, Infection, Restriction of growth,
Adrenal hypoplasia, Genital phenotypes, and Enteropathy) syndrome and
ataxia-pancytopenia syndrome, respectively.[58,59] The penetrance is incomplete, and MDS can also arise in patients without syndromic features.[60]
SAMD9/SAMD9L mutations are typically gain-of-function mutations and
enhance the effects of the wild-type genes leading to growth arrest
when exogenously expressed in cells.[58] The strong selective pressure
to not express the mutant allele is responsible for losing the copy of
chromosome 7 carrying the altered gene. Together with the SAMD9/SAMD9L
gene, several genes on chromosome 7 (e.g., EZH2, SAMD9, SAMD9L, CUX1,
and KMT2C) resulted lost, perturbing hematopoiesis and ultimately
leading to progression into MDS and AML.[52,58] Importantly, somatic
revertant mosaicism that can restore correct hematopoiesis represents
another unique feature of SAMD9/9L syndromes. Two main mechanisms have
been observed so far: the acquisition of loss-of-function SAMD9/9L
mutations neutralizing the gain-of-function germline mutation or an
independent uniparental disomy of 7q (UPD7q).[52,56]
The timing for performing SCT must be decided on a case-by-case basis,
taking into account that children with high expression of the MIRAGE
phenotype experience a high rate of transplant-related comorbidities.[61]
Myeloid neoplasms associated with bone marrow failure syndromes.
Inherited bone marrow failure syndromes (IBMFS) are a group of various
disorders characterized by failure in the production of one or more
blood lineages, usually associated with extra hematopoietic
abnormalities, that present during childhood in most cases.[62]
Different genes involved in diverse cellular functions, including DNA
repair, telomere maintenance, and ribosome biogenesis, underlie these
disorders (Table 3).
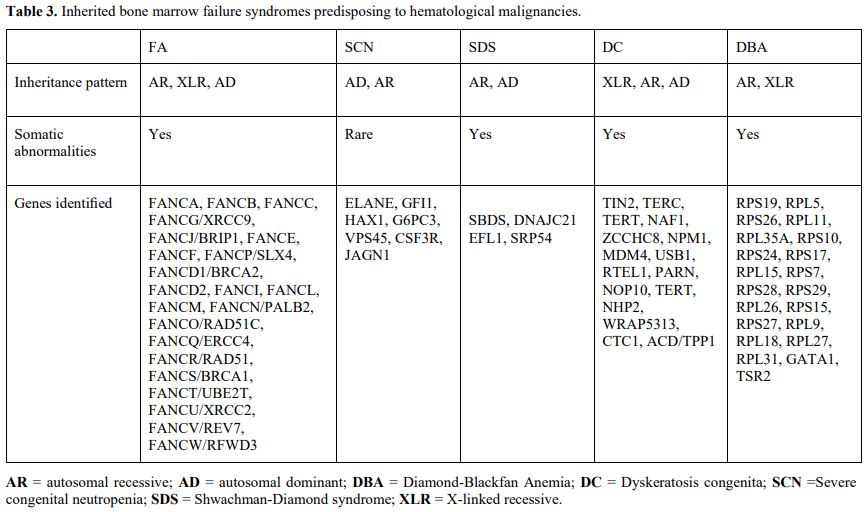 |
- Table 3. Inherited bone marrow failure syndromes predisposing to hematological malignancies.
|
Fanconi Anemia
Fanconi
anemia (FA) is a heterogeneous disorder characterized by BMF with a
predisposition to AML, increased risk of other solid tumors, growth
retardation, and congenital abnormalities, including kidney and urinary
tract malformations, thumb and radial ray abnormalities and café
au-lait spots.[63-65] It is mostly inherited as an AR trait but can
rarely be an X-linked or an AD disorder.[62] Overall, germline
mutations affecting 23 genes, all encoding proteins involved in DNA
repair, underlie the disease.[62] The cumulative incidence of AML at
40 years is estimated at 15-20%, and the cumulative incidence of MDS at
50 years is 40%.[66] The FANCD1/BRCA2 mutation carriers have a higher
risk of developing AML, with a cumulative incidence of 80% at age 10
years.[66] Due to the high toxicity, FA patients suffer when exposed to
irradiation and alkylating agents, fludarabine-based conditioning
regimens are currently preferred.[62]
Severe congenital neutropenia
Severe
peripheral neutropenia (< 0.2 x 10^9/L) is the hallmark of severe
congenital neutropenia (SCN), causing an increased risk for recurrent
and often life-threatening infections.[67] Several germline mutations
can underlie SCN, but it is most commonly caused by AD mutations in
ELANE, which encodes neutrophil elastase, and AR mutations in HAX1,
involved in the granulocyte-colony stimulating factor signaling pathway.[68,69] SCN patients have a high risk of developing MDS or AML, with a
median incidence of 21%.[70,71] Malignant transformation is often
driven by acquired mutations in CSF3R (encoding G-CSF receptor) and
subsequently in other leukemia-associated genes (such as RUNX1).[72]
Shwachman‑Diamond syndrome
Shwachman-Diamond
syndrome (SDS) is usually an AR disorder caused mostly by biallelic
mutations in the SBDS gene, encoding a protein involved in ribosome
biogenesis.[73] The disease is characterized by exocrine pancreatic
insufficiency, BMF, and extra hematopoietic abnormalities such as
metaphyseal dysostosis.[62] Patients have a cumulative risk of
developing MDS/AML reaching 36% by 30 years of age.[74]
Dyskeratosis congenita and telomere biology disorders
Dyskeratosis
congenita (DC) belongs to a spectrum of disorders caused by pathogenic
germline variants in telomere biology genes that share a high risk of
hematologic and solid malignancies. Only a minority of patients present
with the classical triad of mucosal leukoplakia, abnormal skin
pigmentation, and nail dystrophy.[75] Most patients carry X-linked
pathogenic variants in dyskerin, encoded by DKC1.[76] Other genes were
found to underlie these disorders, both AD and AR, while in a
significant percentage of cases, the gene responsible is not identified.[77] A cumulative incidence of 2% by age 50 years for leukemia has
been reported.[78] When they underwent HSCT, patients with DC suffer
from increased transplant-related mortality due to predisposition to
both pulmonary and endothelial disease as well as increased
susceptibility to alkylating agents and irradiation; therefore,
low-intensity fludarabine-based conditioning regimens are currently
preferred.[79,80]
Diamond‑Blackfan anemia
Diamond-Blackfan
anemia (DBA) is characterized by pure red blood cell aplasia, often
associated with congenital anomalies, including thumb abnormalities and
short stature.[81,82] Pathogenic AD variants in ribosomal proteins
underlie the disease, while X-linked pathogenic mutations in GATA1 can
be found in a minority of patients.[62,82] Patients with DBA have an
estimated 5-fold increased risk of cancer, including osteogenic
sarcoma, colon cancer, and AML.[83]
JMML and related disorders
The
ICC separates JMML from adult MDS/MPN. JMML is now considered a genetic
entity defined by the presence of molecular alteration of RAS pathway
genes,[8] including NRAS, KRAS, PTPN11, NF1, CBL, or rarely RRAS. As
might be expected, genetic syndromes associated with germline mutations
in these genes, known collectively as Rasopathies, have a significantly
increased risk of developing this disease.[84-90] In particular, two
JMML subtypes are now defined by germline events in either NF1 or CBL,
with malignant progression driven by acquired biallelic inactivation of
the respective genes in hematopoietic cells. Importantly, patients
harboring germline CBL mutations often experience spontaneous disease
resolution, unlike patients with germline NF1 mutations.[87,91-95]
In
addition, the ICC distinguishes another entity defined as Noonan
syndrome–associated myeloproliferative disorder, associated with
germline mutations in PTPN11, KRAS, NRAS, or RIT1. This disorder is
characterized by a myeloproliferative disorder occurring in the first
year of life and lacking acquired somatic mutations. Although it
resembles the typical clinical and hematological parameters of JMML,
the disorder generally has a self-limiting course.[90,96-98]
Myeloid or lymphoid neoplasms associated with Down syndrome.
Children with Down Syndrome (DS) have an increased risk of developing
hematological neoplasms, particularly AML, with nearly a 150-fold
increased risk in the first 5 years of life.[99] Morphologically it is
commonly a megakaryoblastic AML, with a favorable outcome compared to
the counterpart arising in non-DS patients.[100-102] Furthermore, a
transient myeloproliferative disorder (TMD) occurs in the neonatal
period in 10% of infants with DS, characterized by an accumulation of
immature megakaryoblasts in the fetal liver and peripheral blood.[103,104]. Despite TMD regressing, 20-30% of children that experienced
TMD will develop DS-AML within the first 4 years of life.[103] A
somatic GATA1 mutation is usually found in both TMD and DS-AML.[104-106]
Patients with DS also have an increased incidence of
B-ALL, often characterized by alterations in cytokine receptors or
kinase signaling pathways (e.g., Philadelphia chromosome-like ALL),
notably with CRLF2 dysregulation.[107,108] DS
patients are particularly susceptible to treatment-related toxicity,
especially with high-dose methotrexate.[109] Consequently, they
require tailored therapy with reduced doses of chemotherapy and reduced
intensity conditioning regimens when SCT is needed.[110,111]
Acute lymphoblastic leukemia with germline predisposition
Acute lymphoblastic leukemia with a germline PAX5 mutation.
PAX5 encodes a transcription factor involved in B-lymphoid lineage
maturation, commonly found as a target of somatic alterations in B-ALL.[41,112–114]
Germline mutations were recognized in families with increased incidence
of B-ALL, inherited as an autosomal dominant trait with variable
penetrance.[115,116] B-ALL develops as a result of the loss of 9p containing the wild-type copy.[114]
Acute lymphoblastic leukemia with germline IKZF1 mutation.
IKZF1 encodes for IKAROS, a zinc-finger transcription factor that acts
as a master transcription regulator in lymphoid development.[117,118]
Somatic IKZF1 alterations often occur as secondary events in
kinase-driven B-ALL (Ph+ or Ph-like ALL) and DUX4-rearranged ALL.[119,120] Importantly, in kinase-driven ALL, IKZF1 alterations are associated with poor outcome, unlike in DUX4-rearranged.[121-124]
Germline mutations have been found in several families affected by
immunodeficiency with B-cell lymphopenia and increased incidence of
B-ALL.[125-127] Similarly, germline mutations in
other members of IKAROS transcription factor, namely IKZF2 and IKZF3,
have been recognized as related to immunodeficiency syndromes with
immune dysregulation.[128]
Additional germline mutations associated with hematologic neoplasm predispositions
In
the context of hereditary syndromes, several germline mutations
predispose to the development of hematologic malignancies: Bloom's
syndrome (BLM), constitutional mismatch repair deficiency (MLH1, MSH2,
MSH6, EPCAM, PMS2), DNMT3A, ERCC6L2, MBD4, Ataxia-Telangiectasia,
Nijmegen breakage syndrome, and xeroderma pigmentosum (XPC).[129-138]
In addition, hematological malignancies can frequently arise in
patients affected by immunodeficiency or immune dysregulation.[139]
Conclusions
The
increasingly widespread availability of next-generation sequencing
techniques expands the knowledge of the genetic mechanisms underlying
cancer development. It enables the identification of a growing number
of germline variants associated with hematologic neoplasms. Early
identification of these variants at the time of diagnosis allows for
personalized treatment and optimized donor selection if SCT is needed.
On the other hand, this relatively easy access to genetic information
raises some ethical considerations. For example, related donors could
not want to know if they carry a pathogenetic germline mutation;
however, they may feel forced to do so unwillingly because of pressure
from other family members, although they might not be ready to handle
the results should they test positive. This situation is even more
challenging in the pediatric setting, where consent is expressed by
proxy from parents or guardians, and the child, once he or she becomes
an adult, may suffer the consequences of decisions not made by himself
or herself.[10]
References
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes
Consortium; Aaltonen, LA; Abascal, F.; Abeshouse, A.; Aburatani, H.;
Adams, D.J.; Agrawal, N.; Ahn, KS; Ahn, S.-M.; Aikata, H.; et al.
Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82-93,
doi:10.1038/s41586-020-1969-6. https://doi.org/10.1038/s41586-020-1969-6 PMid:32025007 PMCid:PMC7025898
- Gröbner,
S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.;
Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.;
Brabetz, S.; et al. The Landscape of Genomic Alterations across
Childhood Cancers. Nature 2018, 555, 321-327, doi:10.1038/nature25480. https://doi.org/10.1038/nature25480 PMid:29489754
- Zhang,
J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.;
Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations
in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015,
373, 2336-2346, doi:10.1056/NEJMoa1508054. https://doi.org/10.1056/NEJMoa1508054 PMid:26580448 PMCid:PMC4734119
- Pizzo
and Poplack's Pediatric Oncology; Blaney, S.M., Adamson, P.C., Helman,
L., Eds.; Eighth edition.; Wolters Kluwer Health: Philadelphia, 2021;
ISBN 978-1-975124-79-3.
- Klco,
J.M.; Mullighan, C.G. Advances in Gerline Predisposition to Acute
Leukaemias and Myeloid Neoplasms. Nat. Rev. Cancer 2021, 21, 122-137,
doi:10.1038/s41568-020-00315-z. https://doi.org/10.1038/s41568-020-00315-z PMid:33328584 PMCid:PMC8404376
- Furutani,
E.; Shimamura, A. Genetic Predisposition to MDS: Diagnosis and
Management. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019,
110-119, doi:10.1182/hematology.2019000021. https://doi.org/10.1182/hematology.2019000021 PMid:31808839 PMCid:PMC6913485
- Arber,
D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau,
MM; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to
the World Health Organization Classification of Myeloid Neoplasms and
Acute Leukemia. Blood 2016, 127, 2391-2405,
doi:10.1182/blood-2016-03-643544. https://doi.org/10.1182/blood-2016-03-643544 PMid:27069254
- Arber,
D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.;
Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et
al. International Consensus Classification of Myeloid Neoplasms and
Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data.
Blood 2022, 140, 1200-1228, doi:10.1182/blood.2022015850. https://doi.org/10.1182/blood.2022015850 PMid:35767897
- Rudelius,
M.; Weinberg, O.K.; Niemeyer, C.M.; Shimamura, A.; Calvo, K.R. The
International Consensus Classification (ICC) of Hematologic Neoplasms
with Germline Predisposition, Pediatric Myelodysplastic Syndrome, and
Juvenile Myelomonocytic Leukemia. Virchows Arch. 2023, 482, 113-130,
doi:10.1007/s00428-022-03447-9. https://doi.org/10.1007/s00428-022-03447-9 PMid:36445482
- Hamilton,
K.V.; Maese, L.; Marron, J.M.; Pulsipher, M.A.; Porter, C.C.; Nichols,
K.E. Stopping Leukemia in Its Tracks: Should Preemptive Hematopoietic
Stem-Cell Transplantation Be Offered to Patients at Increased Genetic
Risk for Acute Myeloid Leukemia? J. Clin. Oncol. Off. J. Am. Soc. Clin.
Oncol. 2019, 37, 2098-2104, doi:10.1200/JCO.19.00181. https://doi.org/10.1200/JCO.19.00181 PMid:31170028
- Keeshan,
K.; Santilli, G.; Corradini, F.; Perrotti, D.; Calabretta, B.
Transcription Activation Function of C/EBPα Is Required for Induction
of Granulocytic Differentiation. Blood 2003, 102, 1267-1275,
doi:10.1182/blood-2003-02-0477. https://doi.org/10.1182/blood-2003-02-0477 PMid:12702500
- Fröhling,
S.; Schlenk, R.F.; Stolze, I.; Bihlmayr, J.; Benner, A.; Kreitmeier,
S.; Tobis, K.; Döhner, H.; Döhner, K. CEBPA Mutations in Younger Adults
with Acute Myeloid Leukemia and Normal Cytogenetics: Prognostic
Relevance and Analysis of Cooperating Mutations. J. Clin. Oncol. Off.
J. Am. Soc. Clin. Oncol. 2004, 22, 624-633,
doi:10.1200/JCO.2004.06.060. https://doi.org/10.1200/JCO.2004.06.060 PMid:14726504
- Pabst,
T.; Mueller, B.U.; Zhang, P.; Radomska, H.S.; Narravula, S.;
Schnittger, S.; Behre, G.; Hiddemann, W.; Tenen, D.G. Dominant-Negative
Mutations of CEBPA, Encoding CCAAT/Enhancer Binding Protein-α (C/EBPα),
in Acute Myeloid Leukemia. Nat. Genet. 2001, 27, 263-270,
doi:10.1038/85820. https://doi.org/10.1038/85820 PMid:11242107
- Pabst,
T.; Eyholzer, M.; Haefliger, S.; Schardt, J.; Mueller, B.U. Somatic
CEBPA Mutations Are a Frequent Second Event in Families with Germline
CEBPA Mutations and Familial Acute Myeloid Leukemia. J. Clin. Oncol.
Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5088-5093,
doi:10.1200/JCO.2008.16.5563. https://doi.org/10.1200/JCO.2008.16.5563 PMid:18768433
- Tawana,
K.; Wang, J.; Renneville, A.; Bödör, C.; Hills, R.; Loveday, C.; Savic,
A.; Van Delft, F.W.; Treleaven, J.; Georgiades, P.; et al. Disease
Evolution and Outcomes in Familial AML with Germline CEBPA Mutations.
Blood 2015, 126, 1214-1223, doi:10.1182/blood-2015-05-647172. https://doi.org/10.1182/blood-2015-05-647172 PMid:26162409
- Lane, D.P. P53, Guardian of the Genome. Nature 1992, 358, 15-16, doi:10.1038/358015a0. https://doi.org/10.1038/358015a0 PMid:1614522
- Nigro,
J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hosteller, R.;
Cleary, K.; Signer, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al.
Mutations in the P53 Gene Occur in Diverse Human Tumour Types. Nature
1989, 342, 705-708, doi:10.1038/342705a0. https://doi.org/10.1038/342705a0 PMid:2531845
- Li,
FP; Fraumeni, J.F. Soft-Tissue Sarcomas, Breast Cancer, and Other
Neoplasms. A Familial Syndrome? Ann. Intern. Med. 1969, 71, 747-752,
doi:10.7326/0003-4819-71-4-747. https://doi.org/10.7326/0003-4819-71-4-747 PMid:5360287
- Gonzalez,
K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.;
Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li
Fraumeni Syndrome: Clinical Characteristics of Families with P53
Germline Mutations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009,
27, 1250-1256, doi:10.1200/JCO.2008.16.6959. https://doi.org/10.1200/JCO.2008.16.6959 PMid:19204208
- Bougeard,
G.; Renaux-Petel, M.; Flaman, J.-M.; Charbonnier, C.; Fermey, P.;
Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.;
Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53
Mutation Carriers. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015,
33, 2345-2352, doi:10.1200/JCO.2014.59.5728. https://doi.org/10.1200/JCO.2014.59.5728 PMid:26014290
- Link,
D.C. Identification of a Novel TP53 Cancer Susceptibility Mutation
Through Whole-Genome Sequencing of a Patient With Therapy-Related AML.
JAMA 2011, 305, 1568, doi:10.1001/jama.2011.473. https://doi.org/10.1001/jama.2011.473 PMid:21505135 PMCid:PMC3170052
- Zebisch,
A.; Lal, R.; Müller, M.; Lind, K.; Kashofer, K.; Girschikofsky, M.;
Fuchs, D.; Wölfler, A.; Geigl, J.B.; Sill, H. Acute Myeloid Leukemia
with TP53 Germ Line Mutations. Blood 2016, 128, 2270-2272,
doi:10.1182/blood-2016-08-732610. https://doi.org/10.1182/blood-2016-08-732610 PMid:27621308 PMCid:PMC5095760
- Holmfeldt,
L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.;
Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.-C.; et al. The
Genomic Landscape of Hypodiploid Acute Lymphoblastic Leukemia. Nat.
Genet. 2013, 45, 242-252, doi:10.1038/ng.2532.5 https://doi.org/10.1038/ng.232 PMid:23334668 PMCid:PMC3919793
- Polprasert,
C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono,
N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited
and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015,
27, 658-670, doi:10.1016/j.ccell.2015.03.017. https://doi.org/10.1016/j.ccell.2015.03.017 PMid:25920683 PMCid:PMC8713504
- Lewinsohn,
M.; Brown, A.L.; Weinel, L.M.; Phung, C.; Rafidi, G.; Lee, M.K.;
Schreiber, A.W.; Feng, J.; Babic, M.; Chong, C.-E.; et al. Novel Germ
Line DDX41 Mutations Define Families with a Lower Age of MDS/AML Onset
and Lymphoid Malignancies. Blood 2016, 127, 1017-1023,
doi:10.1182/blood-2015-10-676098. https://doi.org/10.1182/blood-2015-10-676098 PMid:26712909 PMCid:PMC4968341
- Li,
P.; Brown, S.; Williams, M.; White, T.; Xie, W.; Cui, W.; Peker, D.;
Lei, L.; Kunder, C.A.; Wang, H.-Y.; et al. The Genetic Landscape of
Germline DDX41 Variants Predisposing to Myeloid Neoplasms. Blood 2022,
140, 716-755, doi:10.1182/blood.2021015135. https://doi.org/10.1182/blood.2021015135 PMid:35671390
- Rio-Machin,
A.; Fitzgibbon, J. DDX41 : The Poster Child for Familial AML. Blood
2022, 140, 667-669, doi:10.1182/blood.2022016598. https://doi.org/10.1182/blood.2022016598 PMid:35980681
- Duployez,
N.; Largeaud, L.; Duchmann, M.; Kim, R.; Rieunier, J.; Lambert, J.;
Bidet, A.; Larcher, L.; Lemoine, J.; Delhommeau, F.; et al. Prognostic
Impact of DDX41 Germline Mutations in Intensively Treated Acute Myeloid
Leukemia Patients: An ALFA-FILO Study. Blood 2022, 140, 756-768,
doi:10.1182/blood.2021015328. https://doi.org/10.1182/blood.2021015328 PMid:35443031
- Bluteau,
D.; Glembotsky, A.C.; Raimbault, A.; Balayn, N.; Gilles, L.; Rameau,
P.; Nurden, P.; Alessi, M.C.; Debili, N.; Vainchenker, W.; et al.
Dysmegakaryopoiesis of FPD/AML Pedigrees with Constitutional RUNX1
Mutations Is Linked to Myosin II Deregulated Expression. Blood 2012,
120, 2708-2718, doi:10.1182/blood-2012-04-422337. https://doi.org/10.1182/blood-2012-04-422337 PMid:22898599
- Song,
W.-J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin,
D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al.
Haploinsufficiency of CBFA2 Causes Familial Thrombocytopenia with
Propensity to Develop Acute Myelogenous Leukaemia. Nat. Genet. 1999,
23, 166-175, doi:10.1038/13793. https://doi.org/10.1038/13793 PMid:10508512
- Latger-Cannard,
V.; Philippe, C.; Bouquet, A.; Baccini, V.; Alessi, M.-C.; Ankri, A.;
Bauters, A.; Bayart, S.; Cornillet-Lefebvre, P.; Daliphard, S.; et al.
Haematological Spectrum and Genotype-Phenotype Correlations in Nine
Unrelated Families with RUNX1 Mutations from the French Network on
Inherited Platelet Disorders. Orphanet J. Rare Dis. 2016, 11, 49,
doi:10.1186/s13023-016-0432-0. https://doi.org/10.1186/s13023-016-0432-0 PMid:27112265 PMCid:PMC4845427
- Owen,
C.J.; Toze, C.L.; Koochin, A.; Forrest, D.L.; Smith, C.A.; Stevens,
J.M.; Jackson, S.C.; Poon, M.-C.; Sinclair, G.D.; Leber, B.; et al.
Five New Pedigrees with Inherited RUNX1 Mutations Causing Familial
Platelet Disorder with Propensity to Myeloid Malignancy. Blood 2008,
112, 4639-4645, doi:10.1182/blood-2008-05-156745. https://doi.org/10.1182/blood-2008-05-156745 PMid:18723428
- Michaud,
J.; Wu, F.; Osato, M.; Cottles, G.M.; Yanagida, M.; Asou, N.;
Shigesada, K.; Ito, Y.; Benson, K.F.; Raskind, W.H.; et al. In Vitro
Analyses of Known and Novel RUNX1/AML1 Mutations in Dominant Familial
Platelet Disorder with Predisposition to Acute Myelogenous Leukemia:
Implications for Mechanisms of Pathogenesis. Blood 2002, 99, 1364-1372,
doi:10.1182/blood.v99.4.1364. https://doi.org/10.1182/blood.V99.4.1364 PMid:11830488
- Antony-Debré,
I.; Duployez, N.; Bucci, M.; Geffroy, S.; Micol, J.-B.; Renneville, A.;
Boissel, N.; Dhédin, N.; Réa, D.; Nelken, B.; et al. Somatic Mutations
Associated with Leukemic Progression of Familial Platelet Disorder with
Predisposition to Acute Myeloid Leukemia. Leukemia 2016, 30, 999-1002,
doi:10.1038/leu.2015.236. https://doi.org/10.1038/leu.2015.236 PMid:26316320
- Churpek,
J.E.; Pyrtel, K.; Kanchi, K.-L.; Shao, J.; Koboldt, D.; Miller, C.A.;
Shen, D.; Fulton, R.; O'Laughlin, M.; Fronick, C.; et al. Genomic
Analysis of Germ Line and Somatic Variants in Familial
Myelodysplasia/Acute Myeloid Leukemia. Blood 2015, 126, 2484-2490,
doi:10.1182/blood-2015-04-641100. https://doi.org/10.1182/blood-2015-04-641100 PMid:26492932 PMCid:PMC4661171
- Preudhomme,
C.; Renneville, A.; Bourdon, V.; Philippe, N.; Roche-Lestienne, C.;
Boissel, N.; Dhedin, N.; André, J.-M.; Cornillet-Lefebvre, P.;
Baruchel, A.; et al. High Frequency of RUNX1 Biallelic Alteration in
Acute Myeloid Leukemia Secondary to Familial Platelet Disorder. Blood
2009, 113, 5583-5587, doi:10.1182/blood-2008-07-168260. https://doi.org/10.1182/blood-2008-07-168260 PMid:19357396
- Shiba,
N.; Hasegawa, D.; Park, M.; Murata, C.; Sato-Otsubo, A.; Ogawa, C.;
Manabe, A.; Arakawa, H.; Ogawa, S.; Hayashi, Y. CBL Mutation in Chronic
Myelomonocytic Leukemia Secondary to Familial Platelet Disorder with
Propensity to Develop Acute Myeloid Leukemia (FPD/AML). Blood 2012,
119, 2612-2614, doi:10.1182/blood-2011-02-333435. https://doi.org/10.1182/blood-2011-02-333435 PMid:22138511
- Bluteau,
D.; Balduini, A.; Balayn, N.; Currao, M.; Nurden, P.; Deswarte, C.;
Leverger, G.; Noris, P.; Perrotta, S.; Solary, E.; et al.
Thrombocytopenia-Associated Mutations in the ANKRD26 Regulatory Region
Induce MAPK Hyperactivation. J. Clin. Invest. 2014, 124, 580-591,
doi:10.1172/JCI71861. https://doi.org/10.1172/JCI71861 PMid:24430186 PMCid:PMC3904625
- Noris,
P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.;
Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al.
Mutations in ANKRD26 Are Responsible for a Frequent Form of Inherited
Thrombocytopenia: Analysis of 78 Patients from 21 Families. Blood 2011,
117, 6673-6680, doi:10.1182/blood-2011-02-336537. https://doi.org/10.1182/blood-2011-02-336537 PMid:21467542
- Noris,
P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller,
P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et
al. ANKRD26-Related Thrombocytopenia and Myeloid Malignancies. Blood
2013, 122, 1987-1989, doi:10.1182/blood-2013-04-499319. https://doi.org/10.1182/blood-2013-04-499319 PMid:24030261
- Mullighan,
C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton,
J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-Wide
Analysis of Genetic Alterations in Acute Lymphoblastic Leukaemia.
Nature 2007, 446, 758-764, doi:10.1038/nature05690. https://doi.org/10.1038/nature05690 PMid:17344859
- Zhang,
M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.;
Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et
al. Germline ETV6 Mutations in Familial Thrombocytopenia and
Hematologic Malignancy. Nat. Genet. 2015, 47, 180-185,
doi:10.1038/ng.3177. https://doi.org/10.1038/ng.3177 PMid:25581430 PMCid:PMC4540357
- Noetzli,
L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.;
Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.L.; et al. Germline
Mutations in ETV6 Are Associated with Thrombocytopenia, Red Cell
Macrocytosis and Predisposition to Lymphoblastic Leukemia. Nat. Genet.
2015, 47, 535-538, doi:10.1038/ng.3253. https://doi.org/10.1038/ng.3253 PMid:25807284 PMCid:PMC4631613
- Moriyama,
T.; Metzger, M.L.; Wu, G.; Nishii, R.; Qian, M.; Devidas, M.; Yang, W.;
Cheng, C.; Cao, X.; Quinn, E.; et al. Germline Genetic Variation in
ETV6 and Risk of Childhood Acute Lymphoblastic Leukaemia: A Systematic
Genetic Study. Lancet Oncol. 2015, 16, 1659-1666,
doi:10.1016/S1470-2045(15)00369-1. https://doi.org/10.1016/S1470-2045(15)00369-1 PMid:26522332
- Collin,
M.; Dickinson, R.; Bigley, V. Haematopoietic and Immune Defects
Associated with GATA2 Mutation. Br. J. Haematol. 2015, 169, 173-187,
doi:10.1111/bjh.13317. https://doi.org/10.1111/bjh.13317 PMid:25707267 PMCid:PMC4409096
- Wlodarski,
M.W.; Collin, M.; Horwitz, M.S. GATA2 Deficiency and Related Myeloid
Neoplasms. Semin. Hematol. 2017, 54, 81-86,
doi:10.1053/j.seminhematol.2017.05.002. https://doi.org/10.1053/j.seminhematol.2017.05.002 PMid:28637621 PMCid:PMC5650112
- Spinner,
M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.;
Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2
Deficiency: A Protean Disorder of Hematopoiesis, Lymphatics, and
Immunity. Blood 2014, 123, 809-821, doi:10.1182/blood-2013-07-515528. https://doi.org/10.1182/blood-2013-07-515528 PMid:24227816 PMCid:PMC3916876
- Fabozzi,
F.; Strocchio, L.; Mastronuzzi, A.; Merli, P. GATA2 and Marrow Failure.
Best Pract. Res. Clin. Haematol. 2021, 34, 101278,
doi:10.1016/j.beha.2021.101278. https://doi.org/10.1016/j.beha.2021.101278 PMid:34404529
- Fabozzi,
F.; Mastronuzzi, A.; Ceglie, G.; Masetti, R.; Leardini, D. GATA 2
Deficiency: Focus on Immune System Impairment. Front. Immunol. 2022,
13. https://doi.org/10.3389/fimmu.2022.865773 PMid:35769478 PMCid:PMC9234111
- Crispino,
J.D.; Horwitz, M.S. GATA Factor Mutations in Hematologic Disease. Blood
2017, 129, 2103-2110, doi:10.1182/blood-2016-09-687889. https://doi.org/10.1182/blood-2016-09-687889 PMid:28179280 PMCid:PMC5391620
- Wlodarski,
M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.;
Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et
al. Prevalence, Clinical Characteristics, and Prognosis of
GATA2-Related Myelodysplastic Syndromes in Children and Adolescents.
Blood 2016, 127, 1387-1397, doi:10.1182/blood-2015-09-669937. https://doi.org/10.1182/blood-2015-09-669937 PMid:26702063
- Sahoo,
S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik,
A.; Noellke, P.; Dworzak, M.; Starý, J.; Locatelli, F.; et al. Clinical
Evolution, Genetic Landscape and Trajectories of Clonal Hematopoiesis
in SAMD9/SAMD9L Syndromes. Nat. Med. 2021, 27, 1806-1817,
doi:10.1038/s41591-021-01511-6. https://doi.org/10.1038/s41591-021-01511-6 PMid:34621053 PMCid:PMC9330547
- Donadieu,
J.; Lamant, M.; Fieschi, C.; de Fontbrune, F.S.; Caye, A.; Ouachee, M.;
Beaupain, B.; Bustamante, J.; Poirel, H.A.; Isidor, B.; et al. Natural
History of GATA2 Deficiency in a Survey of 79 French and Belgian
Patients. Haematologica 2018, 103, 1278-1287,
doi:10.3324/haematol.2017.181909. https://doi.org/10.3324/haematol.2017.181909 PMid:29724903 PMCid:PMC6068047
- Novakova,
M.; aliova, M.; Sukova, M.; Wlodarski, M.; Janda, A.; Fro kova, E.;
Campr, V.; Lejhancova, K.; Zapletal, O.; Pospi ilova, D.; et al. Loss
of B Cells and Their Precursors Is the Most Constant Feature of GATA-2
Deficiency in Childhood Myelodysplastic Syndrome. Haematologica 2016,
101, 707-716, doi:10.3324/haematol.2015.137711. https://doi.org/10.3324/haematol.2015.137711 PMid:27013649 PMCid:PMC5013954
- Ganapathi,
K.A.; Townsley, D.M.; Hsu, A.P.; Arthur, D.C.; Zerbe, C.S.;
Cuellar-Rodriguez, J.; Hickstein, D.D.; Rosenzweig, S.D.; Braylan,
R.C.; Young, N.S.; et al. GATA2 Deficiency-Associated Bone Marrow
Disorder Differs from Idiopathic Aplastic Anemia. Blood 2015, 125,
56-70, doi:10.1182/blood-2014-06-580340. https://doi.org/10.1182/blood-2014-06-580340 PMid:25359990 PMCid:PMC4281830
- Sahoo,
SS; Kozyra, E.J.; Wlodarski, M.W. Germline Predisposition in Myeloid
Neoplasms: Unique Genetic and Clinical Features of GATA2 Deficiency and
SAMD9/SAMD9L Syndromes. Best Pract. Res. Clin. Haematol. 2020, 33,
101197, doi:10.1016/j.beha.2020.101197. https://doi.org/10.1016/j.beha.2020.101197 PMid:33038986 PMCid:PMC7388796
- Bruzzese,
A.; Leardini, D.; Masetti, R.; Strocchio, L.; Girardi, K.; Algeri, M.;
Del Baldo, G.; Locatelli, F.; Mastronuzzi, A. GATA2 Related Conditions
and Predisposition to Pediatric Myelodysplastic Syndromes. Cancers
2020, 12, 2962, doi:10.3390/cancers12102962. https://doi.org/10.3390/cancers12102962 PMid:33066218 PMCid:PMC7602110
- Narumi,
S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.;
Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9
Mutations Cause a Novel Multisystem Disorder, MIRAGE Syndrome, and Are
Associated with Loss of Chromosome 7. Nat. Genet. 2016, 48, 792-797,
doi:10.1038/ng.3569. https://doi.org/10.1038/ng.3569 PMid:27182967
- Chen,
D.-H.; Below, J.E.; Shimamura, A.; Keel, S.B.; Matsushita, M.; Wolff,
J.; Sul, Y.; Bonkowski, E.; Castella, M.; Taniguchi, T.; et al.
Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L.
Am. J. Hum. Genet. 2016, 98, 1146-1158, doi:10.1016/j.ajhg.2016.04.009.
https://doi.org/10.1016/j.ajhg.2016.04.009 PMid:27259050 PMCid:PMC4908176
- Schwartz,
J.R.; Ma, J.; Lamprecht, T.; Walsh, M.; Wang, S.; Bryant, V.; Song, G.;
Wu, G.; Easton, J.; Kesserwan, C.; et al. The Genomic Landscape of
Pediatric Myelodysplastic Syndromes. Nat. Commun. 2017, 8, 1557,
doi:10.1038/s41467-017-01590-5. https://doi.org/10.1038/s41467-017-01590-5 PMid:29146900 PMCid:PMC5691144
- Ahmed,
I.A.; Farooqi, M.S.; Vander Lugt, M.T.; Boklan, J.; Rose, M.;
Friehling, E.D.; Triplett, B.; Lieuw, K.; Saldana, B.D.; Smith, C.M.;
et al. Outcomes of Hematopoietic Cell Transplantation in Patients with
Germline SAMD9/SAMD9L Mutations. Biol. Blood Marrow Transplant. 2019,
25, 2186-2196, doi:10.1016/j.bbmt.2019.07.007. https://doi.org/10.1016/j.bbmt.2019.07.007 PMid:31306780 PMCid:PMC7110513
- Dokal,
I.; Tummala, H.; Vulliamy, T. Inherited Bone Marrow Failure in the
Pediatric Patient. Blood 2022, 140, 556-570,
doi:10.1182/blood.2020006481. https://doi.org/10.1182/blood.2020006481 PMid:35605178 PMCid:PMC9373017
- Strocchio,
L.; Pagliara, D.; Algeri, M.; Li Pira, G.; Rossi, F.; Bertaina, V.;
Leone, G.; Pinto, R.M.; Andreani, M.; Agolini, E.; et al.
HLA-Haploidentical TCRαβ+/CD19+-Depleted Stem Cell Transplantation in
Children and Young Adults with Fanconi Anemia. Blood Adv. 2021, 5,
1333-1339, doi:10.1182/bloodadvances.2020003707. https://doi.org/10.1182/bloodadvances.2020003707 PMid:33656536 PMCid:PMC7948273
- Giardino,
S.; de Latour, R.P.; Aljurf, M.; Eikema, D.-J.; Bosman, P.; Bertrand,
Y.; Tbakhi, A.; Holter, W.; Bornhäuser, M.; Rössig, C.; et al. Outcome
of Patients with Fanconi Anemia Developing Myelodysplasia and Acute
Leukemia Who Received Allogeneic Hematopoietic Stem Cell
Transplantation: A Retrospective Analysis on Behalf of EBMT Group. Am.
J. Hematol. 2020, 95, 809-816, doi:10.1002/ajh.25810. https://doi.org/10.1002/ajh.25810 PMid:32267023
- Bogliolo,
M.; Surrallés, J. Fanconi Anemia: A Model Disease for Studies on Human
Genetics and Advanced Therapeutics. Curr. Opin. Genet. Dev. 2015, 33,
32-40, doi:10.1016/j.gde.2015.07.002. https://doi.org/10.1016/j.gde.2015.07.002 PMid:26254775
- Alter,
B.P.; Giri, N.; Savage, S.A.; Peters, J.A.; Loud, J.T.; Leathwood, L.;
Carr, A.G.; Greene, M.H.; Rosenberg, P.S. Malignancies and Survival
Patterns in the National Cancer Institute Inherited Bone Marrow Failure
Syndromes Cohort Study: Malignancies and Survival in IBMFS. Br. J.
Haematol. 2010, no-no, doi:10.1111/j.1365-2141.2010.08212.x. https://doi.org/10.1111/j.1365-2141.2010.08212.x PMid:20507306 PMCid:PMC3125983
- Skokowa,
J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe Congenital
Neutropenias. Nat. Rev. Dis. Primer 2017, 3, 17032,
doi:10.1038/nrdp.2017.32. https://doi.org/10.1038/nrdp.2017.32 PMid:28593997 PMCid:PMC5821468
- Dale,
D.C.; Person, R.E.; Bolyard, A.A.; Aprikyan, A.G.; Bos, C.; Bonilla,
M.A.; Boxer, L.A.; Kannourakis, G.; Zeidler, C.; Welte, K.; et al.
Mutations in the Gene Encoding Neutrophil Elastase in Congenital and
Cyclic Neutropenia. Blood 2000, 96, 2317-2322. https://doi.org/10.1182/blood.V96.7.2317.h8002317_2317_2322 PMid:11001877
- Klein,
C.; Grudzien, M.; Appaswamy, G.; Germeshausen, M.; Sandrock, I.;
Schäffer, A.A.; Rathinam, C.; Boztug, K.; Schwinzer, B.; Rezaei, N.; et
al. HAX1 Deficiency Causes Autosomal Recessive Severe Congenital
Neutropenia (Kostmann Disease). Nat. Genet. 2007, 39, 86-92,
doi:10.1038/ng1940. https://doi.org/10.1038/ng1940 PMid:17187068
- Rosenberg,
P.S. The Incidence of Leukemia and Mortality from Sepsis in Patients
with Severe Congenital Neutropenia Receiving Long-Term G-CSF Therapy.
Blood 2006, 107, 4628-4635, doi:10.1182/blood-2005-11-4370. https://doi.org/10.1182/blood-2005-11-4370 PMid:16497969 PMCid:PMC1895804
- Rosenberg,
P.S.; Zeidler, C.; Bolyard, A.A.; Alter, B.P.; Bonilla, M.A.; Boxer,
L.A.; Dror, Y.; Kinsey, S.; Link, D.C.; Newburger, P.E.; et al. Stable
Long-Term Risk of Leukaemia in Patients with Severe Congenital
Neutropenia Maintained on G-CSF Therapy: Short Report. Br. J. Haematol.
2010, no-no, doi:10.1111/j.1365-2141.2010.08216.x. https://doi.org/10.1111/j.1365-2141.2010.08216.x PMid:20456363 PMCid:PMC2906693
- Touw,
I.P. Game of Clones: The Genomic Evolution of Severe Congenital
Neutropenia. Hematology 2015, 2015, 1-7,
doi:10.1182/asheducation-2015.1.1. https://doi.org/10.1182/asheducation-2015.1.1 PMid:26637693
- Warren,
A.J. Molecular Basis of the Human Ribosomopathy Shwachman-Diamond
Syndrome. Adv. Biol. Regul. 2018, 67, 109-127,
doi:10.1016/j.jbior.2017.09.002. https://doi.org/10.1016/j.jbior.2017.09.002 PMid:28942353 PMCid:PMC6710477
- Donadieu,
J.; Fenneteau, O.; Beaupain, B.; Beaufils, S.; Bellanger, F.; Mahlaoui,
N.; Lambilliotte, A.; Aladjidi, N.; Bertrand, Y.; Mialou, V.; et al.
Classification of and Risk Factors for Hematologic Complications in a
French National Cohort of 102 Patients with Shwachman-Diamond Syndrome.
Haematologica 2012, 97, 1312-1319, doi:10.3324/haematol.2011.057489. https://doi.org/10.3324/haematol.2011.057489 PMid:22491737 PMCid:PMC3436231
- Calado, R.T.; Young, N.S. Telomere Diseases. N. Engl. J. Med. 2009, 361, 2353-2365, doi:10.1056/NEJMra0903373. https://doi.org/10.1056/NEJMra0903373 PMid:20007561 PMCid:PMC3401586
- Heiss,
N.S.; Knight, S.W.; Vulliamy, T.J.; Klauck, S.M.; Wiemann, S.; Mason,
P.J.; Poustka, A.; Dokal, I. X-Linked Dyskeratosis Congenita Is Caused
by Mutations in a Highly Conserved Gene with Putative Nucleolar
Functions. Nat. Genet. 1998, 19, 32-38, doi:10.1038/ng0598-32. https://doi.org/10.1038/ng0598-32 PMid:9590285
- Dokal,
I.; Vulliamy, T.; Mason, P.; Bessler, M. Clinical Utility Gene Card
for: Dyskeratosis Congenita - Update 2015. Eur. J. Hum. Genet. EJHG
2015, 23, doi:10.1038/ejhg.2014.170. https://doi.org/10.1038/ejhg.2014.170 PMid:25182133 PMCid:PMC4667501
- Alter,
B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the National
Cancer Institute Inherited Bone Marrow Failure Syndrome Cohort after
Fifteen Years of Follow-Up. Haematologica 2018, 103, 30-39,
doi:10.3324/haematol.2017.178111. https://doi.org/10.3324/haematol.2017.178111 PMid:29051281 PMCid:PMC5777188
- Tummala,
H.; Walne, A.; Dokal, I. The Biology and Management of Dyskeratosis
Congenita and Related Disorders of Telomeres. Expert Rev. Hematol.
2022, 15, 685-696, doi:10.1080/17474086.2022.2108784. https://doi.org/10.1080/17474086.2022.2108784 PMid:35929966
- Agarwal,
S. Evaluation and Management of Hematopoietic Failure in Dyskeratosis
Congenita. Hematol. Oncol. Clin. North Am. 2018, 32, 669-685,
doi:10.1016/j.hoc.2018.04.003. https://doi.org/10.1016/j.hoc.2018.04.003 PMid:30047419 PMCid:PMC7307713
- Vlachos, A.; Muir, E. How I Treat Diamond-Blackfan Anemia. Blood 2010, 116, 3715-3723, doi:10.1182/blood-2010-02-251090. https://doi.org/10.1182/blood-2010-02-251090 PMid:20651069 PMCid:PMC2981532
- Vlachos,
A.; Ball, S.; Dahl, N.; Alter, B.P.; Sheth, S.; Ramenghi, U.; Meerpohl,
J.; Karlsson, S.; Liu, J.M.; Leblanc, T.; et al. Diagnosing and
Treating Diamond Blackfan Anaemia: Results of an International Clinical
Consensus Conference. Br. J. Haematol. 2008, 142, 859-876,
doi:10.1111/j.1365-2141.2008.07269.x. https://doi.org/10.1111/j.1365-2141.2008.07269.x PMid:18671700 PMCid:PMC2654478
- Vlachos,
A.; Rosenberg, P.S.; Atsidaftos, E.; Alter, B.P.; Lipton, J.M.
Incidence of Neoplasia in Diamond Blackfan Anemia: A Report from the
Diamond Blackfan Anemia Registry. Blood 2012, 119, 3815-3819,
doi:10.1182/blood-2011-08-375972. https://doi.org/10.1182/blood-2011-08-375972 PMid:22362038 PMCid:PMC3335385
- Schubbert,
S.; Shannon, K.; Bollag, G. Hyperactive Ras in Developmental Disorders
and Cancer. Nat. Rev. Cancer 2007, 7, 295-308, doi:10.1038/nrc2109. https://doi.org/10.1038/nrc2109 PMid:17384584
- Stiller,
C.; Chessells, J.; Fitchett, M. Neurofibromatosis and Childhood
Leukaemia/Lymphoma: A Population-Based UKCCSG Study. Br. J. Cancer
1994, 70, 969-972, doi:10.1038/bjc.1994.431. https://doi.org/10.1038/bjc.1994.431 PMid:7947106 PMCid:PMC2033537
- Bollag,
G.; Clapp, D.W.; Shih, S.; Adler, F.; Zhang, Y.Y.; Thompson, P.; Lange,
B.J.; Freedman, M.H.; McCormick, F.; Jacks, T.; et al. Loss of NF1
Results in Activation of the Ras Signaling Pathway and Leads to
Aberrant Growth in Haematopoietic Cells. Nat. Genet. 1996, 12, 144-148,
doi:10.1038/ng0296-144. https://doi.org/10.1038/ng0296-144 PMid:8563751
- Locatelli,
F.; Niemeyer, C.M. How I Treat Juvenile Myelomonocytic Leukemia. Blood
2015, 125, 1083-1090, doi:10.1182/blood-2014-08-550483. https://doi.org/10.1182/blood-2014-08-550483 PMid:25564399
- Lipka,
D.B.; Witte, T.; Toth, R.; Yang, J.; Wiesenfarth, M.; Nöllke, P.;
Fischer, A.; Brocks, D.; Gu, Z.; Park, J.; et al. RAS-Pathway Mutation
Patterns Define Epigenetic Subclasses in Juvenile Myelomonocytic
Leukemia. Nat. Commun. 2017, 8, 2126, doi:10.1038/s41467-017-02177-w. https://doi.org/10.1038/s41467-017-02177-w PMid:29259247 PMCid:PMC5736667
- Niemeyer,
C.M.; Kang, M.W.; Shin, D.H.; Furlan, I.; Erlacher, M.; Bunin, N.J.;
Bunda, S.; Finklestein, J.Z.; Gorr, T.A.; Mehta, P.; et al. Germline
CBL Mutations Cause Developmental Abnormalities and Predispose to
Juvenile Myelomonocytic Leukemia. Nat. Genet. 2010, 42, 794-800,
doi:10.1038/ng.641. https://doi.org/10.1038/ng.641 PMid:20694012 PMCid:PMC4297285
- Niemeyer,
C.M.; Flotho, C. Juvenile Myelomonocytic Leukemia: Who's the Driver at
the Wheel? Blood 2019, 133, 1060-1070,
doi:10.1182/blood-2018-11-844688. https://doi.org/10.1182/blood-2018-11-844688 PMid:30670449
- Yoshida,
N.; Yagasaki, H.; Xu, Y.; Matsuda, K.; Yoshimi, A.; Takahashi, Y.;
Hama, A.; Nishio, N.; Muramatsu, H.; Watanabe, N.; et al. Correlation
of Clinical Features With the Mutational Status of GM-CSF Signaling
Pathway-Related Genes in Juvenile Myelomonocytic Leukemia. Pediatr.
Res. 2009, 65, 334-340, doi:10.1203/PDR.0b013e3181961d2a. https://doi.org/10.1203/PDR.0b013e3181961d2a PMid:19047918
- Wintering,
A.; Dvorak, C.C.; Stieglitz, E.; Loh, M.L. Juvenile Myelomonocytic
Leukemia in the Molecular Era: A Clinician's Guide to Diagnosis, Risk
Stratification, and Treatment. Blood Adv. 2021, 5, 4783-4793,
doi:10.1182/bloodadvances.2021005117. https://doi.org/10.1182/bloodadvances.2021005117 PMid:34525182 PMCid:PMC8759142
- Hecht,
A.; Meyer, J.A.; Behnert, A.; Wong, E.; Chehab, F.; Olshen, A.;
Hechmer, A.; Aftandilian, C.; Bhat, R.; Choi, S.W.; et al. Molecular
and Phenotypic Diversity of <I>CBL</I>-Mutated Juvenile
Myelomonocytic Leukemia. Haematologica 2020, 107, 178-186,
doi:10.3324/haematol.2020.270595. https://doi.org/10.3324/haematol.2020.270595 PMid:33375775 PMCid:PMC8719097
- Perez,
B.; Mechinaud, F.; Galambrun, C.; Ben Romdhane, N.; Isidor, B.; Philip,
N.; Derain-Court, J.; Cassinat, B.; Lachenaud, J.; Kaltenbach, S.; et
al. Germline Mutations of the CBL Gene Define a New Genetic Syndrome
with Predisposition to Juvenile Myelomonocytic Leukaemia. J. Med.
Genet. 2010, 47, 686-691, doi:10.1136/jmg.2010.076836. https://doi.org/10.1136/jmg.2010.076836 PMid:20543203
- Bresolin,
S.; Zecca, M.; Flotho, C.; Trentin, L.; Zangrando, A.; Sainati, L.;
Stary, J.; de Moerloose, B.; Hasle, H.; Niemeyer, C.M.; et al. Gene
Expression-Based Classification as an Independent Predictor of Clinical
Outcome in Juvenile Myelomonocytic Leukemia. J. Clin. Oncol. Off. J.
Am. Soc. Clin. Oncol. 2010, 28, 1919-1927,
doi:10.1200/JCO.2009.24.4426. https://doi.org/10.1200/JCO.2009.24.4426 PMid:20231685
- Strullu,
M.; Caye, A.; Lachenaud, J.; Cassinat, B.; Gazal, S.; Fenneteau, O.;
Pouvreau, N.; Pereira, S.; Baumann, C.; Contet, A.; et al. Juvenile
Myelomonocytic Leukaemia and Noonan Syndrome. J. Med. Genet. 2014, 51,
689-697, doi:10.1136/jmedgenet-2014-102611. https://doi.org/10.1136/jmedgenet-2014-102611 PMid:25097206
- Hofmans,
M.; Schröder, R.; Lammens, T.; Flotho, C.; Niemeyer, C.; Van Roy, N.;
Decaluwe, W.; Philippé, J.; De Moerloose, B. Noonan Syndrome‐associated
Myeloproliferative Disorder with Somatically Acquired Monosomy 7:
Impact on Clinical Decision Making. Br. J. Haematol. 2019, 187,
doi:10.1111/bjh.16191. https://doi.org/10.1111/bjh.16191 PMid:31617209
- O'Halloran,
K.; Ritchey, A.K.; Djokic, M.; Friehling, E. Transient Juvenile
Myelomonocytic Leukemia in the Setting of PTPN11 Mutation and Noonan
Syndrome with Secondary Development of Monosomy 7: O'Halloran et Al.
Pediatr. Blood Cancer 2017, 64, e26408, doi:10.1002/pbc.26408. https://doi.org/10.1002/pbc.26408 PMid:28084675
- Hasle,
H.; Clemmensen, I.H.; Mikkelsen, M. Risks of Leukaemia and Solid
Tumours in Individuals with Down's Syndrome. The Lancet 2000, 355,
165-169, doi:10.1016/S0140-6736(99)05264-2. https://doi.org/10.1016/S0140-6736(99)05264-2 PMid:10675114
- Creutzig,
U.; Ritter, J.; Vormoor, J.; Ludwig, W.D.; Niemeyer, C.; Reinisch, I.;
Stollmann-Gibbels, B.; Zimmermann, M.; Harbott, J. Myelodysplasia and
Acute Myelogenous Leukemia in Down's Syndrome. A Report of 40 Children
of the AML-BFM Study Group. Leukemia 1996, 10, 1677-1686.
- Ravindranath,
Y.; Abella, E.; Krischer, J.; Wiley, J.; Inoue, S.; Harris, M.;
Chauvenet, A.; Alvarado, C.; Dubowy, R.; Ritchey, A. Acute Myeloid
Leukemia (AML) in Down's Syndrome Is Highly Responsive to Chemotherapy:
Experience on Pediatric Oncology Group AML Study 8498 [See Comments].
Blood 1992, 80, 2210-2214, doi:10.1182/blood.V80.9.2210.2210. https://doi.org/10.1182/blood.V80.9.2210.2210 PMid:1384797
- Lange,
B.J.; Kobrinsky, N.; Barnard, D.R.; Arthur, D.C.; Buckley, J.D.;
Howells, W.B.; Gold, S.; Sanders, J.; Neudorf, S.; Smith, F.O.; et al.
Distinctive Demography, Biology, and Outcome of Acute Myeloid Leukemia
and Myelodysplastic Syndrome in Children with Down Syndrome: Children's
Cancer Group Studies 2861 and 2891. Blood 1998, 91, 608-615.
- Hitzler,
JK; Zipursky, A. Origins of Leukaemia in Children with Down Syndrome.
Nat. Rev. Cancer 2005, 5, 11-20, doi:10.1038/nrc1525. https://doi.org/10.1038/nrc1525 PMid:15630411
- Pine,
S.R.; Guo, Q.; Yin, C.; Jayabose, S.; Druschel, C.M.; Sandoval, C.
Incidence and Clinical Implications of GATA1 Mutations in Newborns with
Down Syndrome. Blood 2007, 110, 2128-2131,
doi:10.1182/blood-2007-01-069542. https://doi.org/10.1182/blood-2007-01-069542 PMid:17576817
- Wechsler,
J.; Greene, M.; McDevitt, M.A.; Anastasi, J.; Karp, J.E.; Le Beau, MM;
Crispino, J.D. Acquired Mutations in GATA1 in the Megakaryoblastic
Leukemia of Down Syndrome. Nat. Genet. 2002, 32, 148-152,
doi:10.1038/ng955. https://doi.org/10.1038/ng955 PMid:12172547
- Yoshida,
K.; Toki, T.; Okuno, Y.; Kanezaki, R.; Shiraishi, Y.; Sato-Otsubo, A.;
Sanada, M.; Park, M.; Terui, K.; Suzuki, H.; et al. The Landscape of
Somatic Mutations in Down Syndrome-Related Myeloid Disorders. Nat.
Genet. 2013, 45, 1293-1299, doi:10.1038/ng.2759. https://doi.org/10.1038/ng.2759 PMid:24056718
- Izraeli,
S. The Acute Lymphoblastic Leukemia of Down Syndrome - Genetics and
Pathogenesis. Eur. J. Med. Genet. 2016, 59, 158-161,
doi:10.1016/j.ejmg.2015.11.010. https://doi.org/10.1016/j.ejmg.2015.11.010 PMid:26631987
- Mullighan,
C.G.; Collins-Underwood, J.R.; Phillips, L.A.A.; Loudin, M.G.; Liu, W.;
Zhang, J.; Ma, J.; Coustan-Smith, E.; Harvey, R.C.; Willman, C.L.; et
al. Rearrangement of CRLF2 in B-Progenitor- and Down
Syndrome-Associated Acute Lymphoblastic Leukemia. Nat. Genet. 2009, 41,
1243-1246, doi:10.1038/ng.469. https://doi.org/10.1038/ng.469 PMid:19838194 PMCid:PMC2783810
- Kroll,
M.; Kaupat-Bleckmann, K.; Mörickel, A.; Altenl, J.; Schewel, D.M.;
Stanullal, M.; Zimmermann, M.; Schrappe, M.; Cario, G.
Methotrexate-Associated Toxicity in Children with Down Syndrome and
Acute Lymphoblastic Leukemia during Consolidation Therapy with High
Dose Methotrexate According to ALL-BFM Treatment Regimen. Haematologica
2020, 105, 1013-1020, doi:10.3324/haematol.2019.224774. https://doi.org/10.3324/haematol.2019.224774 PMid:31371414 PMCid:PMC7109740
- Muramatsu,
H.; Sakaguchi, H.; Taga, T.; Tabuchi, K.; Adachi, S.; Inoue, M.; Kitoh,
T.; Suminoe, A.; Yabe, H.; Azuma, E.; et al. Reduced Intensity
Conditioning in Allogeneic Stem Cell Transplantation for AML with Down
Syndrome: RIC for AML With DS. Pediatr. Blood Cancer 2014, 61, 925-927,
doi:10.1002/pbc.24883. https://doi.org/10.1002/pbc.24883 PMid:24302531
- Shah,
N.; Al-Ahmari, A.; Al-Yamani, A.; Dupuis, L.; Stephens, D.; Hitzler, J.
Outcome and Toxicity of Chemotherapy for Acute Lymphoblastic Leukemia
in Children with down Syndrome: ALL and Down Syndrome Outcomes.
Pediatr. Blood Cancer 2009, 52, 14-19, doi:10.1002/pbc.21737. https://doi.org/10.1002/pbc.21737 PMid:18802938
- Nutt,
S.L.; Heavey, B.; Rolink, A.G.; Busslinger, M. Commitment to the
B-Lymphoid Lineage Depends on the Transcription Factor Pax5. Nature
1999, 401, 556-562, doi:10.1038/44076. https://doi.org/10.1038/44076 PMid:10524622
- Coyaud,
E.; Struski, S.; Prade, N.; Familiades, J.; Eichner, R.; Quelen, C.;
Bousquet, M.; Mugneret, F.; Talmant, P.; Pages, M.-P.; et al. Wide
Diversity of PAX5 Alterations in B-ALL: A Groupe Francophone de
Cytogenetique Hematologique Study. Blood 2010, 115, 3089-3097,
doi:10.1182/blood-2009-07-234229. https://doi.org/10.1182/blood-2009-07-234229 PMid:20160164
- Gu,
Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe,
J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al.
PAX5-Driven Subtypes of B-Progenitor Acute Lymphoblastic Leukemia. Nat.
Genet. 2019, 51, 296-307, doi:10.1038/s41588-018-0315-5. https://doi.org/10.1038/s41588-018-0315-5 PMid:30643249 PMCid:PMC6525306
- Shah,
S.; Schrader, K.A.; Waanders, E.; Timms, A.E.; Vijai, J.; Miething, C.;
Wechsler, J.; Yang, J.; Hayes, J.; Klein, R.J.; et al. A Recurrent
Germline PAX5 Mutation Confers Susceptibility to Pre-B Cell Acute
Lymphoblastic Leukemia. Nat. Genet. 2013, 45, 1226-1231,
doi:10.1038/ng.2754. https://doi.org/10.1038/ng.2754 PMid:24013638 PMCid:PMC3919799
- Auer,
F.; Rüschendorf, F.; Gombert, M.; Husemann, P.; Ginzel, S.; Izraeli,
S.; Harit, M.; Weintraub, M.; Weinstein, O.Y.; Lerer, I.; et al.
Inherited Susceptibility to Pre B-ALL Caused by Germline Transmission
of PAX5 c.547G>A. Leukemia 2014, 28, 1136-1138,
doi:10.1038/leu.2013.363. https://doi.org/10.1038/leu.2013.363 PMid:24287434
- Georgopoulos,
K.; Bigby, M.; Wang, J.H.; Molnar, A.; Wu, P.; Winandy, S.; Sharpe, A.
The Ikaros Gene Is Required for the Development of All Lymphoid
Lineages. Cell 1994, 79, 143-156, doi:10.1016/0092-8674(94)90407-3. https://doi.org/10.1016/0092-8674(94)90407-3 PMid:7923373
- Chen,
Q.; Shi, Y.; Chen, Y.; Ji, T.; Li, Y.; Yu, L. Multiple Functions of
Ikaros in Hematological Malignancies, Solid Tumor and Autoimmune
Diseases. Gene 2019, 684, 47-52, doi:10.1016/j.gene.2018.10.045. https://doi.org/10.1016/j.gene.2018.10.045 PMid:30352248
- Mullighan,
C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.A.; Miller, C.B.;
Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1
and Prognosis in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2009,
360, 470-480, doi:10.1056/NEJMoa0808253. https://doi.org/10.1056/NEJMoa0808253 PMid:19129520 PMCid:PMC2674612
- Zhang,
J.; McCastlain, K.; Yoshihara, H.; Xu, B.; Chang, Y.; Churchman, M.L.;
Wu, G.; Li, Y.; Wei, L.; Iacobucci, I.; et al. Deregulation of DUX4 and
ERG in Acute Lymphoblastic Leukemia. Nat. Genet. 2016, 48, 1481-1489,
doi:10.1038/ng.3691. https://doi.org/10.1038/ng.3691 PMid:27776115 PMCid:PMC5144107
- Den
Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.;
Buijs-Gladdines, JGCAM; Peters, STCJM; Van Zutven, LJCM; Beverloo,
H.B.; Van der Spek, PJ; Escherich, G.; et al. A Subtype of Childhood
Acute Lymphoblastic Leukaemia with Poor Treatment Outcome: A
Genome-Wide Classification Study. Lancet Oncol. 2009, 10, 125-134,
doi:10.1016/S1470-2045(08)70339-5. https://doi.org/10.1016/S1470-2045(08)70339-5 PMid:19138562
- Martinelli,
G.; Iacobucci, I.; Storlazzi, C.T.; Vignetti, M.; Paoloni, F.; Cilloni,
D.; Soverini, S.; Vitale, A.; Chiaretti, S.; Cimino, G.; et al. IKZF1
(Ikaros) Deletions in BCR-ABL1-Positive Acute Lymphoblastic Leukemia
Are Associated with Short Disease-Free Survival and High Rate of
Cumulative Incidence of Relapse: A GIMEMA AL WP Report. J. Clin. Oncol.
Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5202-5207,
doi:10.1200/JCO.2008.21.6408. https://doi.org/10.1200/JCO.2008.21.6408 PMid:19770381
- van
der Veer, A.; Zaliova, M.; Mottadelli, F.; De Lorenzo, P.; Te Kronnie,
G.; Harrison, C.J.; Cavé, H.; Trka, J.; Saha, V.; Schrappe, M.; et al.
IKZF1 Status as a Prognostic Feature in BCR-ABL1-Positive Childhood
ALL. Blood 2014, 123, 1691-1698, doi:10.1182/blood-2013-06-509794. https://doi.org/10.1182/blood-2013-06-509794 PMid:24366361
- Zaliova,
M.; Zimmermannova, O.; Dörge, P.; Eckert, C.; Möricke, A.; Zimmermann,
M.; Stuchly, J.; Teigler-Schlegel, A.; Meissner, B.; Koehler, R.; et
al. ERG Deletion Is Associated with CD2 and Attenuates the Negative
Impact of IKZF1 Deletion in Childhood Acute Lymphoblastic Leukemia.
Leukemia 2014, 28, 182-185, doi:10.1038/leu.2013.282. https://doi.org/10.1038/leu.2013.282 PMid:24072102
- Kuehn,
H.S.; Boisson, B.; Cunningham-Rundles, C.; Reichenbach, J.;
Stray-Pedersen, A.; Gelfand, E.W.; Maffucci, P.; Pierce, K.R.; Abbott,
J.K.; Voelkerding, K.V.; et al. Loss of B Cells in Patients with
Heterozygous Mutations in IKAROS. N. Engl. J. Med. 2016, 374,
1032-1043, doi:10.1056/NEJMoa1512234. https://doi.org/10.1056/NEJMoa1512234 PMid:26981933 PMCid:PMC4836293
- Yoshida,
N.; Sakaguchi, H.; Muramatsu, H.; Okuno, Y.; Song, C.; Dovat, S.;
Shimada, A.; Ozeki, M.; Ohnishi, H.; Teramoto, T.; et al. Germline
IKAROS Mutation Associated with Primary Immunodeficiency That
Progressed to T-Cell Acute Lymphoblastic Leukemia. Leukemia 2017, 31,
1221-1223, doi:10.1038/leu.2017.25. https://doi.org/10.1038/leu.2017.25 PMid:28096536
- Churchman,
M.L.; Qian, M.; Te Kronnie, G.; Zhang, R.; Yang, W.; Zhang, H.; Lana,
T.; Tedrick, P.; Baskin, R.; Verbist, K.; et al. Germline Genetic IKZF1
Variation and Predisposition to Childhood Acute Lymphoblastic Leukemia.
Cancer Cell 2018, 33, 937-948.e8, doi:10.1016/j.ccell.2018.03.021. https://doi.org/10.1016/j.ccell.2018.03.021 PMid:29681510 PMCid:PMC5953820
- Shahin,
T.; Mayr, D.; Shoeb, M.R.; Kuehn, H.S.; Hoeger, B.; Giuliani, S.;
Gawriyski, L.M.; Petronczki, Ö.Y.; Hadjadj, J.; Bal, S.K.; et al.
Identification of Germline Monoallelic Mutations in IKZF2 in Patients
with Immune Dysregulation. Blood Adv. 2022, 6, 2444-2451,
doi:10.1182/bloodadvances.2021006367. https://doi.org/10.1182/bloodadvances.2021006367 PMid:34920454 PMCid:PMC9006292
- Chen,
S.; Wang, W.; Lee, S.; Nafa, K.; Lee, J.; Romans, K.; Watson, P.;
Gruber, S.B.; Euhus, D.; Kinzler, K.W.; et al. Prediction of Germline
Mutations and Cancer Risk in the Lynch Syndrome. JAMA 2006, 296, 1479,
doi:10.1001/jama.296.12.1479. https://doi.org/10.1001/jama.296.12.1479 PMid:17003396 PMCid:PMC2538673
- Ripperger,
T.; Schlegelberger, B. Acute Lymphoblastic Leukemia and Lymphoma in the
Context of Constitutional Mismatch Repair Deficiency Syndrome. Eur. J.
Med. Genet. 2016, 59, 133-142, doi:10.1016/j.ejmg.2015.12.014. https://doi.org/10.1016/j.ejmg.2015.12.014 PMid:26743104
- Sarasin,
A.; Quentin, S.; Droin, N.; Sahbatou, M.; Saada, V.; Auger, N.;
Boursin, Y.; Dessen, P.; Raimbault, A.; Asnafi, V.; et al. Familial
Predisposition to TP53/Complex Karyotype MDS and Leukemia in DNA
Repair-Deficient Xeroderma Pigmentosum. Blood 2019, 133, 2718-2724,
doi:10.1182/blood-2019-01-895698. https://doi.org/10.1182/blood-2019-01-895698 PMid:30914417 PMCid:PMC6610036
- Poppe,
B.; Van Limbergen, H.; Van Roy, N.; Vandecruys, E.; De Paepe, A.;
Benoit, Y.; Speleman, F. Chromosomal Aberrations in Bloom Syndrome
Patients with Myeloid Malignancies. Cancer Genet. Cytogenet. 2001, 128,
39-42, doi:10.1016/S0165-4608(01)00392-2. https://doi.org/10.1016/S0165-4608(01)00392-2 PMid:11454428
- Dembowska-Baginska,
B.; Perek, D.; Brozyna, A.; Wakulinska, A.; Olczak-Kowalczyk, D.;
Gladkowska-Dura, M.; Grajkowska, W.; Chrzanowska, K.H. Non-Hodgkin
Lymphoma (NHL) in Children with Nijmegen Breakage Syndrome (NBS).
Pediatr. Blood Cancer 2009, 52, 186-190, doi:10.1002/pbc.21789. https://doi.org/10.1002/pbc.21789 PMid:18937313
- Rafei,
H.; DiNardo, C.D. Hereditary Myeloid Malignancies. Best Pract. Res.
Clin. Haematol. 2019, 32, 163-176, doi:10.1016/j.beha.2019.05.001. https://doi.org/10.1016/j.beha.2019.05.001 PMid:31203998
- Cunniff,
C.; Bassetti, J.A.; Ellis, N.A. Bloom's Syndrome: Clinical Spectrum,
Molecular Pathogenesis, and Cancer Predisposition. Mol. Syndromol.
2017, 8, 4-23, doi:10.1159/000452082. https://doi.org/10.1159/000452082 PMid:28232778 PMCid:PMC5260600
- Ferris,
M.A.; Smith, A.M.; Heath, S.E.; Duncavage, E.J.; Oberley, M.; Freyer,
D.; Wynn, R.; Douzgou, S.; Maris, J.M.; Reilly, A.F.; et al. DNMT3A
Overgrowth Syndrome Is Associated with the Development of Hematopoietic
Malignancies in Children and Young Adults. Blood 2022, 139, 461-464,
doi:10.1182/blood.2021014052. https://doi.org/10.1182/blood.2021014052 PMid:34788385 PMCid:PMC8777205
- Douglas,
S.P.M.; Siipola, P.; Kovanen, P.E.; Pyörälä, M.; Kakko, S.; Savolainen,
E.-R.; Salmenniemi, U.; Orte, K.; Kytölä, S.; Pitkänen, E.; et al.
ERCC6L2 Defines a Novel Entity within Inherited Acute Myeloid Leukemia.
Blood 2019, 133, 2724-2728, doi:10.1182/blood-2019-01-896233. https://doi.org/10.1182/blood-2019-01-896233 PMid:30936069
- Ma,
S.; E, C.; C, F.; A, Z.; Se, M.; As, AH; A, B.; B, L.; M, R.; T, M.; et
al. MBD4 Guards against Methylation Damage and Germ Line Deficiency
Predisposes to Clonal Hematopoiesis and Early-Onset AML. Blood 2018,
132, doi:10.1182/blood-2018-05-852566. https://doi.org/10.1182/blood-2018-05-852566 PMid:30049810 PMCid:PMC6172562
- van
der Werff Ten Bosch, J.; van den Akker, M. Genetic Predisposition and
Hematopoietic Malignancies in Children: Primary Immunodeficiency. Eur.
J. Med. Genet. 2016, 59, 647-653, doi:10.1016/j.ejmg.2016.03.001. https://doi.org/10.1016/j.ejmg.2016.03.001 PMid:26975585
[TOP]