Giovanna Cannas1,2, Solène Poutrel1 and Xavier Thomas3
1 Hospices Civils de Lyon, Department of Internal Medicine, Edouard Herriot Hospital, Lyon, France.
2
Claude Bernard University Lyon 1, Laboratoire Interuniversitaire de
Biologie de la Motricité EA7424, Equipe ‘Vascular biology and red blood
cell’, Villeurbanne, France.
3 Hospices Civils de Lyon, Hematology Department, Lyon-Sud Hospital, Pierre Bénite, France.
Corresponding
author: Xavier Thomas, M.D., Ph.D., Hospices Civils de Lyon,
Hematology Department, Lyon-Sud Hospital, Bât. 1G, 165 chemin du
Grand Revoyet, 69495 Pierre Bénite, France. Tel: +33 478862235. Fax:
+33 4 72678880. E-mail:
xavier.thomas@chu-lyon.fr
Published: February 15, 2017
Received: December 12, 2016
Accepted: January 20, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017015 DOI
10.4084/MJHID.2017.015
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
While
hydroxycarbamide (hydroxyurea, HU) has less and fewer indications in
malignant hemopathies, it represents the only widely used drug which
modifies sickle cell disease pathogenesis. Clinical experience with HU
for patients with sickle cell disease has been accumulated over the
past 25 years in Western countries. The review of the literature
provides increasing support for safety and efficacy in both children
and adults for reducing acute vaso-occlusive events including pain
episodes and acute chest syndrome. No increased incidence of leukemia
and teratogenicity was demonstrated. HU has become the standard-of-care
for sickle cell anemia but remains underused. Barriers to its use
should be identified and overcome.
|
Introduction
Hydroxycarbamide
(Hydroxyurea, HU) was first synthesized in Germany in 1869,[1] but its
potential biologic significance was not recognized until 1928.[2] It is
a simple compound of the formula, H2NCONHOH, which the =C−NHOH moiety
is responsible for its biological activity (Figure 1).
HU is a potent inhibitor of DNA synthesis. It is antimitotic and
cytotoxic depending upon the used concentration, the duration of
exposure, and the sensitivity of the organism. HU is active mainly in
the S-phase of the cell cycle. In the 1950s the drug was evaluated in a
large number of experimental tumor models and was found to have broad
anti-tumor activity against both leukemia and solid tumors.[3] Clinical
trials began in the 1960s.[4] As an antineoplastic drug, HU has some
advantages. It may be used with ambulatory patients and has relatively
few side effects, which are relieved almost immediately after
withdrawal of the drug. The drug is readily absorbed from the
gastrointestinal tract following oral administration. Peak serum
concentrations are reached in 1 to 2 hours, and the serum half-life is
about 5.5 hours. It is rapidly excreted in the urine, and it is
reported that up to 70% of the dose is excreted unchanged.[5] At
present, HU has only a limited medical use in acute leukemia,
consisting in reducing and controlling white blood cell count in
patients with hyperleukocytosis. The principal use of HU has been as a
myelosuppressive agent in the myeloproliferative syndromes. The
efficacy of HU as initial therapy for chronic myeloid leukemia (CML)
has been known for a number of years.[6] Since the introduction of
tyrosine kinase inhibitors in the treatment of CML, hydroxyurea is
essentially used in the BCR-ABL1-negative myeloproliferative neoplasms,
including polycythemia vera, essential thrombocythemia, and primary
myelofibrosis.[7] In high-risk patients with polycythemia vera or
essential thrombocythemia, HU remains the first-line cytoreductive drug
of choice, the second-line choice being represented by interferon-alpha
and busulfan.[8,9] Survival is relatively long in these diseases, and
risk of leukemic transformation low. Treatment with HU has not been
shown to modify these favorable outcomes, while controlled clinical
trials have shown increased risk of acute leukemia with the use of
chlorambucil, radiophosphorus and pipobroman, and increased risk of
fibrotic transformation with the use of anagrelide.[7] The introduction
of new drugs should, therefore, be careful. This is particularly
important when considering the use of JAK inhibitor ruxolitinib, which
was recently approved for use in these pathologies. HU also remains the
first-line drug of choice for myelofibrosis-associated splenomegaly,
while hydroxyurea-refractory splenomegaly is often managed with
ruxolitinib therapy or splenectomy.[10] In addition to its use as an
anti-cancer agent, HU has found some marginal applications in
dermatology.[11]
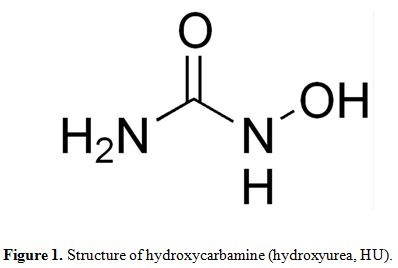 |
Figure 1. Structure of hydroxycarbamine (hydroxyurea, HU). |
While HU is an old drug that can still be used to
control essential thrombocythemia and polycythemia vera in patients
with high-risk disease, it has emerged over the last decades as the
primary disease-modifying therapy for sickle cell anemia, a
non-malignant inherited disease. The purpose of this short review is to
provide the reader a comprehensive understanding of HU and to reinforce
the fact that HU is a safe and effective medication for the treatment
of sickle cell disease.
Sickle Cell Disease: Historical Considerations
Sickle
cell anemia, first described by James B Herrick in 1910,[12] is the
first inherited disease identified at the molecular level. In 1949,
Linus Pauling confirmed an intrinsic dissimilarity in the hemoglobin
from patients with sickle cell anemia on electrophoretic mobility
patterns.[13] Because of the heterozygote state, sickle cell trait,
appeared to persist in some populations with prevalence as high as
20%-40% and the sickle cell trait allele frequency overlapped with
malarial endemicity, AC Allison hypothesized that sickle hemoglobin
(HbS) must confer a selective advantage of malarial resistance in the
carrier state.[14] A recent meta-analysis confirmed a strong protective
advantage of sickle cell trait for Plasmodium falciparum malaria,
suggesting that HbS does not protect against infection itself, but
rather to progression to clinical malaria and its childhood
associated-mortality.[15] Although not elucidated, the suggested
mechanisms involved in this epidemiologic observation comprise a
protective effect through enhanced immunity, increased clearance of
infected erythrocytes, and reduced parasite growth. In 1956, VM
Ingram discovered a single amino acid substitution in HbS.[16] The
genetic basis for the abnormal hemoglobin was a single base-pair change
(A → T) in the β-globin gene, resulting in a substitution of a valine
for glutamic acid at position 6. Structural changes promote
polymerization into long fibrils, distorting the red cell into a sickle
shape, leading to erythrocytes dehydrated, rigid and prone to
hemolysis, and so to occluding the microvasculature causing acute and
chronic tissue ischemia and injury. It took then until the 1970s for
systematic research into the laboratory screening techniques and
clinical sequelae of sickling disorders to be prioritized.[17] At that
time, only 50% of afflicted children survived into adulthood.[18] As a
result of the institution of the National Sickle Cell Anemia Control
Act, a Hemoglobinopathy Reference Laboratory was created to standardize
techniques and elaborate screening programs.[19] By the 1990s,
widespread mandatory newborn screening and the routine administration
of penicillin to prevent pneumococcal sepsis increased childhood
survival to over 90%.[20] Currently, the most common screening
techniques include sickle solubility testing, hemoglobin
electrophoresis, high-performance liquid chromatography, and
isoelectric focusing, each with their own advantages and limitations.
Recent advances in technology have also allowed for detection of sickle
cell trait from DNA through exome sequencing.[21,22] Indeed
misclassification of individuals with sickle cell trait and sickle cell
disease in early case reports led to confusing series in which sickle
cell disease complications were ascribed to individuals with sickle
cell trait.
No specific therapy was available until the 1970s when
it was recognized that patients with increased red blood cell HbF had
fewer adverse clinical events. First described as a potential therapy
for sickle-cell anemia in 1984, HU enhances the production of fetal
hemoglobin production in sickle erythrocytes.[23] The two most common
acute morbidities in sickle cell anemia are vaso-occlusive pain crises
and acute chest syndrome, corresponding to the occlusion of small
vessels in the bone marrow and lungs, respectively.[24,25] Other
pulmonary complications of sickle cell disease include pulmonary
hypertension, pulmonary artery thrombosis, and pulmonary fibrosis, with
an increased prevalence of reactive airways disease, increased
tricuspid regurgitant jet velocity, sleep-disordered breathing, and
nocturnal hypoxemia.[26] On a chronic basis, vaso-occlusion may damage
the lungs, kidney or brain accounting ultimately for most deaths in
patients with sickle cell disease.[27] Clinical studies with HU
demonstrated a decreased rate of vaso-occlusive disease and acute chest
syndrome, and an improved survival.[28] Consequently, HU became in 1998
the only US Food and Drug Administration-approved therapy for sickle
cell disease. The European Medicines Agency authorized HU in 2007 for
pediatric and adult patients with sickle cell anemia. In 2008, the
Agency for Healthcare Research and Quality published a comprehensive
review,[29] and a consensus conference on HU in the treatment of
sickle cell disease was organized by the National Institute of
Health.[30]
HU Mechanisms of Action in Sickle Cell Anemia
In
sickle cell anemia, the red cells almost contain only HbS. Only a
smaller population of red cells comes directly from immature
progenitors, which contain the fetal hemoglobin (HbF). These nearly
normal cells mitigate the damage caused by HbS.[31]
Cells with high levels of HbS lose deformability when deoxygenated,
leading to vascular obstruction and ischemia. Membrane damage shortens
the life span of the cell leading to chronic intravascular and
extravascular hemolysis. Damage red cells showed an increased adherence
to vascular endothelium leading to vaso-occlusion and proliferative
lesions involving many cells and factors underlying large-vessel
stroke.[32] Shifting hemoglobin production from HbS
to HbF represents then a major therapeutic approach to sickle cell
anemia. Low level of HbF is one of the strongest predictors of
morbidity and mortality in sickle cell disease.[27]
The cytotoxic effect of HU reduces the production of red cells
containing a high level of HbS, which tend to arise from rapidly
dividing precursors, and favors the production of cells containing a
high level of HbF.[32] The exact mechanism by which
HU induces HbF remains unclear. The increase in HbF appears to
interfere with HbS polymerization both by preventing contact between
adjacent HbS molecules and by forming mixed hybrids with HbS that have
greater solubility than HbS polymers.[33] HU may
increase HbF indirectly by killing dividing late erythroid cells,
causing recruitment of more primitive erythroid precursors which
produce high levels of HbF, or by acting directly on the primitive
precursors stimulating HbF production.[34] However,
induction of HbF was unlikely to explain all the clinical effects of
HU. Prior to any rise in HbF, sickle erythrocytes show reduced adhesion
to endothelial cells. HU reduces adhesion molecule expression on sickle
erythrocytes, including very late activation antigen-4 and CD36.[35]
Other rheological properties of sickle erythrocytes, including
erythrocyte hydration status and whole cell deformability, can be
increased by HU. HU also reduces white blood cells and platelets
reducing their roles in vascular injury. Neutrophilia has long been
identified as a marker of severity in sickle cell disease.[27]
Neutrophils release pro-inflammatory mediators involved in endothelial
damage and cytokine release, which trigger sickling, and contribute to
slow transit time via their adhesive properties and an increase in
blood viscosity.[36] The drug also produces nitric
oxide, which stimulates soluble guanylate cyclase (an enzyme containing
heme iron) resulting in the production of HbF.[37] Some of the clinical effects are mediated by nitric oxide-induced vasodilatation or reduced platelet activation.
The Use of HU in Sickle Cell Disease placebo.
HU was initially tested in anemic baboons.[38] The first patients were tested in 1984 showing a response within 72 hours after therapy with an elevated level of HbF.[23]
Subsequent prospective studies confirmed the efficacy and tolerability
of HU in this setting. Recent reviews of the literature on HU therapy
in sickle cell disease showed that HU was consistently associated with
overall increases in HbF, a reduction of vaso-occlusive crises,
decreased rates of hospitalization, and prevention of pulmonary
complications.[39,40] The benefit of HU regarding the frequency of acute clinical events was demonstrated in randomized studies (Table 1),[28,41-47]
but also in observational studies (uncontrolled longitudinal studies,
retrospective case series, or prospective cohort studies using
historical controls) (Table 2).[48-64]
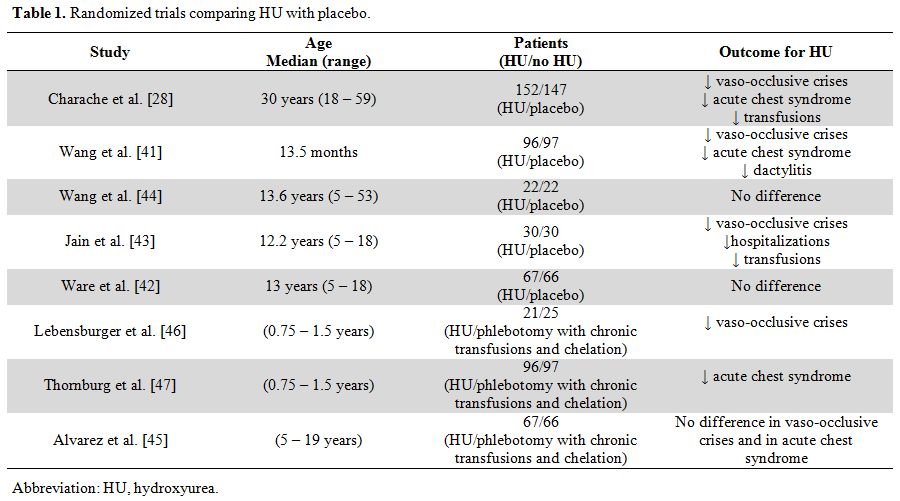 |
Table 1. Randomized trials comparing HU with placebo. |
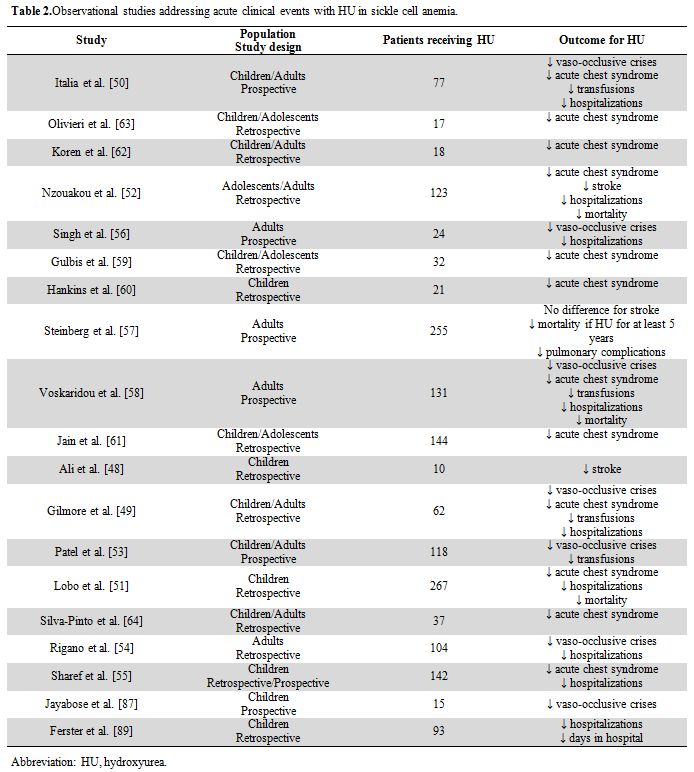 |
Table 2. Observational studies addressing acute clinical events with HU in sickle cell anemia. |
Treatment with HU in adults:
Most studies included both children and adults. Among specific studies
for adult patients, the most important was a multicenter, double-blind,
randomized controlled study that ran from 1992 to 1994 and that was
stopped early after inclusion of 299 patients, because of a significant
reduction of events in the Hu arm.[28] HU improved
the clinical course of sickle cell disease by significantly reducing
the annual rate of crises, increasing the median time to the first and
second crisis, reducing the incidence of acute chest syndrome, and
reducing transfusion requirements. Furthermore, the recommended dose of
HU was not always needed to achieve a clinical response. Among the
other randomized studies, no difference was noted in terms of frequency
of vaso-occlusive crises in three studies.[42,44,45] In one study, there were equivalent liver iron contents and similar rates of stroke in both arms.[42]
However, two of these three trials terminated earlier due to poor
accrual. In the third study (SWiTCH trial), given the low rates of
acute chest syndrome observed in the trial, the number of patients was
not sufficient to determine whether there was a true difference between
acute chest syndrome in the two arms.[45] Although
studies of various designs showed that HU decreased the occurrence of
acute chest disease, most studies provided lower-quality evidence for
such effect.[49,58,62-64]
Regarding pulmonary hypertension and tricuspid regurgitant velocity,
the lack of randomization and prospective follow-up makes
interpretation of results difficult. If most studies showed no
difference among groups[26,65-70] or even higher proportion of patients with prior exposure to HU in a group with increased tricuspid regurgitant velocity,[71] some studies tended to provide evidence of HU effect.[72,73] Evidence for primary stroke prevention was limited to observational data.[50,58]
While current evidence supports the use of chronic blood transfusions
to prevent progressive disease and especially clinical stroke, HU
represents an attractive alternative treatment option in order to avoid
indefinite blood transfusion therapy which can lead to serious
complications such as infections, iron overload, transfusion reactions,
and erythrocyte allo- and auto-antibody formation.[74]
The therapeutic switch from transfusions to HU should follow an overlap
period of dual therapy because the benefits of HU have a slow onset and
treatment should reach a stable maximum tolerated dose. After the
switch, the problem of hemosiderosis persists. Despite effective oral
chelators, the greatest challenge of serial phlebotomy in patients with
sickle cell anemia is the underlying anemia, but HU therapy at a stable
maximum tolerated dose typically raises the hemoglobin concentration,
allowing a safe procedure.[75] Before the era of HU, the average life expectancy was in the 40s.[27] HU was associated with decreased mortality in symptomatic patients compared with those receiving only short-term HU or no HU.[57]
It typically takes less than 6 months for patients to be stabilized on
a dose that defines their maximal tolerated dose. Before the maximal
tolerated dose is established, the number of cells with high HbF levels
increases.[44,50,53,56]
At 6 months, the HbF level is typically doubled, the hemoglobin level
is increased, and the absolute reticulocyte count, bilirubin level, and
lactate dehydrogenase level are reduced.[32] Patients
should receive HU therapy as a continuous treatment unless adverse
events occur. The optimal dose is still a source of debate. Dose
escalation has been suggested toward the maximum tolerated dose (MTD).
However, the stepwise approach of dose escalation generally requires
several months and patients receiving HU have variable pharmacokinetics
and pharmacodynamics.[76] Creatinine, reticulocyte
count, and body mass index are among the simplest parameters that best
predicted the HU maximum tolerated dose. It has been demonstrated a
near linear dose response to HU. The treatment dose correlated
positively with both the plasma drug concentration and the percentage
of HbF response.[77] However, the dose does not need
to be titrated to a particular HbF threshold. The dose can be escalated
simply to reach an acceptable nontoxic degree of marrow suppression
with target counts for both neutrophils and reticulocytes.[78] It has been suggested that HU may have benefits for the less common genotypes, especially HbSC or HbS/β+ thalassemia.[79]
Because the primary effect of HU is damaging DNA replication by
inhibiting ribonucleotide reductase, concerns have been raised about an
oncogenic potential, especially after prolonged use. Although fears
have been amplified by its original use as chemotherapy for chronic
myeloproliferative diseases, which could evolve to acute leukemia,
oncogenicity of HU is probably quite low or non-existent. Only a few
cases of acute leukemia have been reported, but do not appear more
frequent than in the untreated population.[80]
Similarly, the benefits and harms of HU therapy in women with sickle
cell disease during pregnancy and lactation represent a relevant issue.[79]
No clear teratogenic phenotype exists for HU, but more data should be
collected. Women with sickle cell anemia receiving HU have had
successful normal pregnancies.[81] A variety of
factors can lead to treatment failure. Poor adherence is recognized as
a common problem and seems in part related to adverse events of the
drug and inconvenience associated with monitoring.
Treatment with HU in children:
The use of HU in children brings theoretically the best satisfactions
regarding prevention of end-organ damage. However, it also carries
potential risks in terms of growth and development and remains
questionable for the risk of secondary malignancy after exposure to the
drug for long periods. Observational studies in children have noted
significant improvements in splenic uptake, glomerular filtration rate,
renal hypertrophy, the ability to concentrate urine, microalbuminuria,
and retinopathy.[82-86] As in adults studies,
beneficial results with HU were reported showing a reduction in
hospital admissions and days spent in the hospital, and potentially a
reduced frequency of acute chest syndrome.[62,87-89] Evidence on benefits of HU use in children below 5 years is that it is associated with decreased pain crises and dactylitis.[41,90] However, most studies provided lower-quality evidence for the occurrence of acute chest syndrome.[46,47,60,61] There were few pieces of evidence that HU prevents stroke or the recurrence of stroke in children.[91] As in adults, evidence for primary stroke prevention was limited to observational data.[51] HU treatment was shown to lower secondary stroke rates in children with previous stroke.[92]
Transcranial Doppler screening is used for primary stroke prevention.
Abnormal velocities are the most common indication to commence chronic
transfusion therapy in children. HU can lower transcranial Doppler
velocities[93] and its utility in this setting is under investigations.[94]
Although case reports have shown a reversal of splenic dysfunction
after HU therapy, larger studies demonstrated that HU is clearly not
enough to completely prevent the major complications of the disease.[95] Growth and development appeared to be unaffected in all studies.[95,96] HU should, therefore, be offered early and routinely as a preventive treatment for sickle cell anemia in children.
Distribution of HbF
More
than 75% of the hemoglobin of the newborn is HbF. It diminishes over a
period of several months to adult levels. HbF is becoming <2% by one
year-of-age and <1% by 2 years. In most patients with sickle cell
disease, HbF levels are increased. HbF is produced by a small number of
erythroid precursors: the F-cells. Both HbF concentration and its
distribution among erythrocytes are heritable. A correlation has been
demonstrated between the number of F-cells and the percent of HbF in
the hemolysate.[97] The concentration of HbF in each F-cell (HbF/F-cell) is changing during maturation.[98]
However, quantitative methods for measuring this amount and plotting
the distribution of HbF among F-cells are not available. The
distribution of HbF concentrations among F-cells is the most critical
element in the physiopathology of sickle cell anemia.[99]
Compound heterozygosity for HbS and gene deletion hereditary
persistence of HbF (HPFH) represents a condition in which the typical
HbF level is 30%.[100] In this setting, HbF is
distributed among all erythrocytes, each cell containing about 10 pg of
HbF. Patients with sickle cell anemia have individually characteristic
distributions of HbF/F-cell regardless of their total HbF level.[101] HU can induce HbF in most patients, but the HbF response to HU is highly variable.[102]
Higher HbF levels were associated with a reduced rate of painful
episodes, fewer leg ulcers, less osteonecrosis, and less frequent acute
chest syndromes. However, HbF level had a weak or no clear association
with priapism, urine albumin excretion, stroke and silent cerebral
infarction, systemic blood pressure, and tricuspid regurgitant
velocity.[103] HbF is unevenly
distributed when high levels are successfully induced with HU. Total
HbF and F-cell percentages are generally not good predictors of disease
severity since they provide no information on F-cells with sufficient
levels of HbF to protect against polymer-induced damage. Very few
protected F-cells are present when HbF levels are about 5%, but more
cells are possible when they reach 10%.[99] The
calculated mean HbF/F-cell in HU-treated sickle cell anemia is about 8
pg. Early starting treatment seems to retard the fall in HbF, but many
F-cells will continue to be poorly protected from polymer-induced
damage even with 20% HbF.[99]
A Clinical and Economic Problem
It
is estimated that 7% of the world population are carriers for
hemoglobin disorders. Sickle cell disease is the most important
potentially devastating, recessively inherited condition. The β-globin
gene point mutation resulting in HbS has undergone evolutionary
selection in the world because of its overwhelming malaria protective
effects. High prevalence areas include Africa, the Middle-East, and
Indian subcontinent with sickle cell trait affecting up to 300 million
individuals worldwide.[104] In Africa, one on 14 persons with sickle cell anemia is an asymptomatic carrier.[32] One in 700 newborns is affected.[105]
However, recent studies suggest that only 16% of polled individuals are
aware of their sickle cell trait status, and only 37% of parents report
having received notification of the sickle cell trait status of their
children.[106,107] Sickle cell disease represents
then an emerging global health burden in limited-resource countries, in
which the development of sickle cell disease strategies should include
sickle cell awareness, early detection, and the development of care and
treatment programs.[108-110] The main recommendation
is to educate all patients and their families about HU therapy.
Although Food and Drug administration-label recommends treatment only
for adults with sickle cell anemia severely affected with at least 3
painful crises over the prior 12 months, there are strong
recommendations to treat adults with common clinical symptoms and to
offer HU to children after age 9 months, regardless of clinical
symptoms.[111] HU is relatively cheap. Especially in
limited-resource countries without a safe and adequate blood supply, HU
may represent a clinically useful and cost-effective therapeutic
strategy for preventing cerebrovascular disease.[112]
In the United States, it was reported per year 113,000 hospitalizations
for sickle cell disease generating total hospital costs of about $488
million.[113] The average cost of HU was estimated at about $1,000 per year plus $400 per year for visits and tests.[28]
This cost was offset by reduced costs for hospitalization, emergency
room visits, and transfusions. The net savings was estimated at about
$5,000 per patient per year.[114] Beyond HU Therapy
HU
is currently the only US Food and Drug Administration-approved
medication to modify the disease course in sickle cell disease.
However, elucidation of the multiple pathophysiologic mechanisms
leading to vaso-occlusion and tissue injury in sickle cell disease is
currently resulting in the identification of new treatment modalities.[115] In addition to HU, a number of drugs have been proposed: histone deacetylase inhibitors,[116] decitabine,[117] thalidomide and related compounds,[118] pomalidomide.[119] Optimally efficient induction of HbF may require combined use of drugs.[120]
Carbon monoxide is also a potent antisickling agent that attaches to Hb
and therefore reverse HbS polymerization. Shifting the oxyhemoglobin to
the left or preventing cell dehydration can ameliorate sickling.[121]
Sanguinate is a bovine pegylated Hb product designed to reduce sickling
by delivering carbon monoxide to HbS and then carrying O2.[122]
Because adhesive cell interactions contribute to vaso-occlusion, drugs
targeting either red blood cell or leukocyte adhesion appear as
attractive therapeutic modalities. Drugs targeting selectin-mediated
adhesion are being especially investigated including the selectin
inhibitors GMI-1070 (rivipansel)[123] and the humanized monoclonal antibody SelG1.[124]
Heparin derivatives, such as sevuparin or tinzaparin, also have a
well-known ability to inhibit adhesive interactions via P-selectin.[125,126]
Targeting signaling pathways that activate adhesion molecules is
another potential therapeutic modality. This can be achieved via
beta-blockers administration[127] or through the use of MEK inhibitors[128]
that might reduce red blood cell adhesion. Poloxamer 188, a
nonspecific inhibitor of adhesion is also currently being studied.[129]
Vaso-occlusion can engender an inflammatory response typical of
hypoxia/reperfusion injury. Down-regulation of inflammatory pathways
can, therefore, represent another approach to ameliorate sickle cell
disease. Invariant NKT cells are involved in this pathogenesis. Their
activation can be down-regulated by activation of the adenosine A2A
receptor. Regadenoson is a partially selective adenosine A2A receptor
agonist. It has been proposed in the treatment of vaso-occlusive
crisis, which involves invariant NKT cells as contributors to the
inflammatory component.[130] A humanized monoclonal antibody against invariant NKT cells has also recently shown some efficacy.[131]
Because inflammatory pathways are important to both vaso-occlusion and
tissue injury, targeting inflammatory mediators, such as leukotrienes,
has also been proposed as a promising approach for the development of
novel therapies in sickle cell disease.[132] Intravenous γ globulin infusion can also reduce inflammation via inhibition of neutrophil adhesion.[133] Statins that decrease endothelial inflammation have also been studied in sickle cell disease.[134]
Drugs that increase HbF levels are the archetypal antisickling agents,
because HbF interferes with polymerization of HbS, thereby lessening
hemolytic rate and resulting in total Hb levels seen with HU therapy.
The interference lengthens the delay time, allowing cells to avoid
getting stuck in the microvasculature, even if hemolysis does not
happen. Despite promising results, high mortality rates in patients
older than 16 years and a paucity of suitable HLA-identical donors have
limited the implementation of allogeneic stem cell transplantation in
this patient population.[135] In the future,
correction of the β-globin gene may be the ideal approach to curing
sickle cell disease. However, there are still many concerns regarding
this approach.Despite
the development of these many new treatment modalities and the
promising results of the initial trials, HU remains a well-tolerated,
safe, cheap, and efficacious for most patients with sickle cell
disease, and should currently be considered standard-of-care for this
disease. Conclusions
HU
is a remarkably effective drug for a large proportion of patients with
sickle cell disease and appears to be the best currently available
treatment option in this setting. Treatment is indicated for patients
with “frequent pain episodes, history of acute chest syndrome, other
severe vaso-occlusive events, or severe symptomatic anemia”.[32]
Treatment endpoints remain “less pain and improved well-being,
increased HbF to 15%-20%, increased hemoglobin level, and acceptable
myelotoxicity”.[32] However, studies regarding a
better understanding of HU effects, the ability to predict individual
response, and the clinical applications for modifying disease effects
are still ongoing. Two decades after the approval of HU, most patients
with sickle cell disease are suboptimally treated with it, or not
treated at all since this disease has continued to be treated with
analgesics for pain relief. HU remains underutilized for a variety of
reasons. It is likely that optimal therapy will only be achieved with a
multi-targeted approach. However, any of the new therapies may be
similarly underused, which may be the most difficult problem. HU is
currently prescribed only sparingly and therefore has only limited
effectiveness. Early initiation and broader use of HU should alter the
natural history of sickle cell anemia. HU should be extended to
low-resource settings, where the burden of the disease and the need for
such a drug is the greatest. References
- Dresler WFC, Stein R. Über den hydroxylharnstoff. Justus Liebigs Ann Chem 1869; 150:242-52. https://doi.org/10.1002/jlac.18691500212

- Rosenthal
F, Wislicki L, Koller L. Über die beziehungen von schwersten blutgiften
zu abbauprodukten des eiweisses: ein beitrag zum entstehungmechanismus
der pernizosen anemie. KlinWochenschr 1928; 7:972-7. https://doi.org/10.1007/BF01716922 .
- Stearns B, Losee KA, Bernstein J. Hydroxyurea: a new type of potential antitumor agent. J Med Chem 1963; 6:201. https://doi.org/10.1021/jm00338a026 PMid:14188794
- Fishbein
WN, Carbone PP, Freireich EJ, et al. Clinical trials of hydroxyurea in
patients with cancer and leukemia.Clin Pharmacol Ther 1964; 5:574-80. https://doi.org/10.1002/cpt196455574
- Becloff GL. Pharmacological, metabolic and clinical experience with hydroxyurea. Clin Trials J 1967; 4:873-83.
- Kennedy BJ. Hydroxyurea therapy in chronic myelogenous leukemia.Cancer 1972; 29:1052-6. https://doi.org/10.1002/1097-0142(197204)29:4<1052::AID-CNCR2820290454>3.0.CO;2-7
- Tefferi A, Pardanani A. Myeloproliferative neoplasms. A contemporary review. JAMA Oncol 2015; 1:97-105. https://doi.org/10.1001/jamaoncol.2015.89 PMid:26182311
- Cortelazzo
S, Finazzi G, Specchia G, et al. Hydroxyurea for patients with
essential thrombocythemia and a high risk of thrombosis. N Engl J Med
1995; 332:1132-6. https://doi.org/10.1056/NEJM199504273321704 PMid:7700286
- Fruchtman
SM, Mack K, Kaplan ME, et al. From efficacy to safety: a Polycytemia
Vera Study Group report on hydroxyurea in patients with polycytemia
vera. Semin Hematol 1997; 34:17-23. PMid:9025158
- Tefferi A. Primary myelofibrosis:2014 update on diagnosis, risk-stratification, and management. Am J Hematol 2014; 89:915-25. https://doi.org/10.1002/ajh.23703 PMid:25124313
- Leavell UW, Yarbro JW. Hydroxyurea. A new treatment for psoriasis. Arch Dermatol 1970; 102:144-50. https://doi.org/10.1001/archderm.1970.04000080016003
- Herrick
JB. Peculiar elongated and sickle-shaped red blood corpuscules in a
case of severe anemia. Arch Intern Med 1910; 6:517-21. https://doi.org/10.1001/archinte.1910.00050330050003
- Pauling L, Itano HA.Sickle cell anemia, a molecular disease. Science 1949; 109:443. https://doi.org/10.1126/science.110.2865.543
- Allison
AC.The distribution of the sickle-cell trait in East Africa and
elsewhere, and its apparent relationship to the incidence of
subtertianmalaria.Trans R SocTrop Med Hyg 1954; 48:312-8. https://doi.org/10.1016/0035-9203(54)90101-7
- Taylor
SM, Parobeck CM, Fairhurst RM. Haemoglobinopathies and the clinical
epidemiology of malaria: a systematic review and meta-analysis. Lancet
Infect Dis 2012; 12:457-68. https://doi.org/10.1016/S1473-3099(12)70055-5
- Ingram
VM. A specific chemical difference between the globins of normal human
and sickle-cell anaemiahaemoglobin.Nature 1956; 178:792-4. https://doi.org/10.1038/178792a0 PMid:13369537
- Schmidt
RM, Brosious EM. Evaluation of proficiency in the performance of tests
for abnormal hemoglobins.Am J ClinPathol 1974;62:664-9. https://doi.org/10.1093/ajcp/62.5.664 PMid:4413315
- Scott RB. Health care priority and sickle cell anemia. JAMA 1970; 214:731-4. https://doi.org/10.1001/jama.1970.03180040039008 PMid:5536114
- Benson
JM, Therrell BL Jr. History and current status of newborn screening for
hemoglobinopathies. SeminPerinatol 2010; 34: 134-44. https://doi.org/10.1053/j.semperi.2009.12.006 PMid:20207263
- Quinn
CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children
and adolescents with sickle cell disease. Blood 2010; 115:3447-52. https://doi.org/10.1182/blood-2009-07-233700 PMid:20194891 PMCid:PMC2867259
- Naik
RP, Derebail VK, Grams ME, et al. Association of sickle cell trait with
chronic kidney disease and albuminuria in African Americans. JAMA 2014;
312:2115-25. https://doi.org/10.1001/jama.2014.15063 PMid:25393378 PMCid:PMC4356116
- Auer
PL, Johnsen JM, Johnson AD, et al. Imputation of exome sequence
variants into population-based samples and blood-cell-trait associated
loci in African Americans: NHLBI GO Exome Sequencing Project. Am J Hum
Genet 2012; 91:794-808. https://doi.org/10.1016/j.ajhg.2012.08.031 PMid:23103231 PMCid:PMC3487117
- Platt
OS, Orkin SH, Dover G, et al. Hydroxyurea enhances fetal hemoglobin
production in sickle cell anemia. J Clin Invest 1984; 74 :652-6. https://doi.org/10.1172/JCI111464 PMid:6205021 PMCid:PMC370519
- Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease: rates and risk factors. N Engl J Med 1991; 325:11-6. https://doi.org/10.1056/NEJM199107043250103 PMid:1710777
- Castro
O, Brambilla DJ, Thorington BD, et al. The acute chest syndrome in
sickle cell disease: incidence and risk factors.The Cooperative Study
of Sickle Cell Disease.Blood 1994; 84:643-9. PMid:7517723
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med 2008; 359:2254-65. https://doi.org/10.1056/NEJMra0804411 PMid:19020327
- Platt
OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease:
life expectancy and risk factors for early death. N Engl J Med 1994;
330:1639-44. https://doi.org/10.1056/NEJM199406093302303 PMid:7993409
- Charache
S, Terrin ML, Moore RD, et al. Effects of hydroxyurea on the frequency
of painful crises in sickle cell anemia. N Engl J Med 1995;
332:1317-22. https://doi.org/10.1056/NEJM199505183322001 PMid:7715639
- Segal
JB, Strouse JJ, Beach MC, et al. Hydroxyurea for the treatment of
sickle cell disease. Evid Rep Technol Assess 2008; 165:1-95.
- Brawley
OW, Cornelius LJ, Edwards LR, et al. National Institutes of Health
Consensus Development Conference statement: hydroxyurea treatment for
sickle cell disease. Ann Intern Med 2008; 148:932-8. https://doi.org/10.7326/0003-4819-148-12-200806170-00220 PMid:18458271
- Franco
RS, Yasin Z, Palascak MB, et al. The effect of fetal hemoglobin on the
survival characteristics of sickle cells.Blood 2006; 108:1073-6. https://doi.org/10.1182/blood-2005-09-008318 PMid:16861353 PMCid:PMC1895865
- Platt OS.Hydroxyurea for the treatmet of sickle cell anemia. N Engl J Med 2008; 358:1362-9. https://doi.org/10.1056/NEJMct0708272 PMid:18367739
- Halsey C, Roberts IAG. The role of hydroxyurea in sickle cell disease. Br J Haematol 2003; 120:177-86. https://doi.org/10.1046/j.1365-2141.2003.03849.x PMid:12542474
- Kolata G. Globin gene studies create a puzzle. Science 1984; 223:470-1. https://doi.org/10.1126/science.6197757 PMid:6197757
- Styles
LA, Lubin B, Vichinsky E, et al. Decrease of very late activation
antigen-4 and CD36 on reticulocytes in sickle cell patients treated
with hydroxyurea. Blood 1997; 89:2554-9. PMid:9116302
- Nogochi CT, Schechter AN, Rodgers GP. Sickle cell disease pathophysiology. Bailliere's Clinical Haematology 1993; 6:57-91. https://doi.org/10.1016/S0950-3536(05)80066-6
- Cokic
VP, Smith RD, Beleslin-Cokic BB, et al. Hydroxyurea induces fetal
hemoglobin by the nitric oxide-dependent activation of soluble
guanylylcyclase. J Clin Invest 2003; 111:231-9. https://doi.org/10.1172/JCI200316672 PMid:12531879 PMCid:PMC151872
- Letvin
NL, Linch DC, Beardsley GP, McIntyre KW, Nathan DG. Augmentation of
fetal-hemoglobin production in anemic monkeys by hydroxyurea. N Engl J
Med 1984; 310:869-73. https://doi.org/10.1056/NEJM198404053101401 PMid:6199670
- Wong
TE, Brandow AM, Lim W, Lottenberg R. Update on the use of hydroxyurea
therapy in sickle cell disease. Blood 2014; 124:3850-7. https://doi.org/10.1182/blood-2014-08-435768 PMid:25287707 PMCid:PMC4271176
- Buckner
TW, Ataga KI. Does hydroxyurea prevent pulmonary complications of
sickle cell disease? Hematology (Educational Sessions of the American
Society of Hematology) 2014; 432-7. https://doi.org/10.1182/asheducation-2014.1.432 PMid:25696890
- Wang
WC, Ware RE, Miller ST, et al. Hydroxycarbamine in very young children
with sickle-cell anaemia: a multicentre, randomized, controlled trial
(BABY HUG). Lancet 2011; 377:1663-72. https://doi.org/10.1016/S0140-6736(11)60355-3
- Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood 2012; 119:3925-32. https://doi.org/10.1182/blood-2011-11-392340 PMid:22318199 PMCid:PMC3350359
- Jain
DL, Sarathi V, Desai S, et al. Low fixed-dose hydroxyurea in severely
affected Indian children with sickle cell disease. Hemoglobin 2012;
36:323-32. https://doi.org/10.3109/03630269.2012.697948 PMid:22734586
- Wang
W, Brugnara C, Snyder C, et al. The effects of hydroxycarbamine and
magnesium on haemoglobin SC disease: results of the multicentre CHAMPS
trial. Br J Haematol 2011; 152:771-6. https://doi.org/10.1111/j.1365-2141.2010.08523.x PMid:21275961
- Alvarez
O, Yovetich NA, Scott JP, et al. Pain and other non-neurological
adverse events in children with sickle cell anemia and previous stroke
who received hydroxyurea and phlebotomy or chronic transfusions and
chelation: results from the SWiTCH clinical trial. Am J Hematol 2013;
88:932-8. https://doi.org/10.1002/ajh.23547 PMid:23861242 PMCid:PMC4631259
- Lebensburger
JD, Miller ST, Howard TH, et al. Influence of severity of anemia on
clinical findings in infants with sickle cell anemia: analyses from the
BABY HUG study. Pediatr Blood Cancer 2012; 59:675-8. https://doi.org/10.1002/pbc.24037 PMid:22190441 PMCid:PMC3337342
- Thornburg CD, Files BA, Luo Z, et al. Impact of hydroxyurea on clinical events in the BABY HUG trial. Blood 2012; 120:4304-10. https://doi.org/10.1182/blood-2012-03-419879 PMid:22915643 PMCid:PMC3507142
- Ali
SB, Moosang M, King L, et al. Stroke recurrence in children with sickle
cell disease treated with hydroxyurea following first clinical stroke.
Am J Hematol 2011; 86:846-50. https://doi.org/10.1002/ajh.22142 PMid:21898530
- Gilmore
A, Cho G, Howard J, et al. Feasibility and benefit of hydroxycarbamide
as a long-term treatment for sickle cell disease patients: results from
the North West London Sickle Cell Disease Registry. Am J Hematol 2011;
86:958-61. https://doi.org/10.1002/ajh.22146 PMid:21948113
- Italia
K, Jain D, Gattani S, et al. Hydroxyurea in sickle cell disease – a
study of clinic-pharmacological efficacy in the Indian haplotype. Blood
Cells Mol Dis 2009; 42:25-31. https://doi.org/10.1016/j.bcmd.2008.08.003 PMid:18954999
- Lobo
CL, Pinto JF, Nascimento EM, et al.The effect of hydroxycarbamine
therapy on survival of children with sickle cell disease. Br J Haematol
2013; 161:852-60. https://doi.org/10.1111/bjh.12323 PMid:23590693
- Nzouakou
R, Bachir D, Lavaud A, et al. Clinical follow-up of hydroxyurea-treated
adults with sickle cell disease. Acta Haematol 2011; 125:145-52. https://doi.org/10.1159/000322248
- Patel
DK, Mashon RS, Patel S, et al. Low dose hydroxyurea is effective in
reducing the incidence of painful crisis and frequency of blood
transfusion in sickle cell anemia patients from eastern India.
Hemoglobin 2012; 36:409-20. https://doi.org/10.3109/03630269.2012.709897 PMid:22881992
- Rigano
P, Pecoraro A, Calvaruso G, et al. Cerebrovascular events in sickle
cell-beta thalassemia treated with hydroxyurea: a single center
prospective survey in adult Italians. Am J Hematol 2013; 88:E261-4. https://doi.org/10.1002/ajh.23531 PMid:23828131
- Sharef
SW, Al-Hajri M, Beshlawi I, et al. Optimizing hydroxyurea use in
children with sickle cell disease: low dose regimen is effective. Eur J
Haematol 2013; 90:519-24. https://doi.org/10.1111/ejh.12103 PMid:23489171
- Singh
H, Dulhani N, Kumar BN, et al. Effective control of sickle cell disease
with hydroxyurea therapy. Indian J Pharmacol 2010; 42:32-5. https://doi.org/10.4103/0253-7613.62409 PMid:20606834 PMCid:PMC2885637
- Steinberg
MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term
use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J
Hematol 2010; 85:403-8. https://doi.org/10.1002/ajh.21699
- Voskaridou
E, Christoulas D, Bilalis A, et al. The effect of prolonged
administration of hydroxyurea on morbidity and mortality in adult
patients with sickle cell syndromes: results of a 17-year,
single-center trial (LaSHS). Blood 2010; 115:2354-63. https://doi.org/10.1182/blood-2009-05-221333 PMid:19903897
- Gulbis
B, Haberman D, Dufour D, et al. Hydroxyurea for sickle cell disease in
children and for prevention of cerebrovascular events: the Belgian
experience. Blood 2005; 105:2685-90. https://doi.org/10.1182/blood-2004-07-2704 PMid:15604217
- Hankins
JS, Ware RE, Rogers ZR, et al. Long-term hydroxyurea therapy for
infants with sickle cell anemia: the HUSOFT extension study. Blood
2005; 106:2269-75. https://doi.org/10.1182/blood-2004-12-4973 PMid:16172253 PMCid:PMC1895275
- Jain
DL, Apte M, Colah R, et al. Efficacy of fixed low dose hydroxyurea in
Indian children with sickle cell anemia: a single centre experience.
Indian Pediatr 2013; 50:929-33. https://doi.org/10.1007/s13312-013-0264-0 PMid:23798623
- Koren
A, Segal-Kupershmit D, Zalman L, et al. Effect of hydroxyurea in sickle
cell anemia: a clinical trial in children and teenagers with severe
sickle cell beta-thalassemia. Pediatr Hematol Oncol 1999; 16:221-32. https://doi.org/10.1080/088800199277272 PMid:10326220
- Olivieri
NF, Vichinsky EP. Hydroxyurea in children with sickle cell disease:
impact on splenic function and compliance with therapy. J Pediatr
Hematol Oncol 1998; 20:26-31. https://doi.org/10.1097/00043426-199801000-00004
- Silva-Pinto
AC, Angulo IL, Brunetta DM, et al. Clinical and hematological effects
of hydroxyurea therapy in sickle cell patients: a single-center
experience in Brazil. Sao Paulo Med J 2013; 131:238-43. https://doi.org/10.1590/1516-3180.2013.1314467 PMid:24141294
- Desai
PC, May RC, Jones SK, et al. Longitudinal study of
echocardiography-derived tricuspid regurgitant jet velocity in sickle
cell disease. Br J Haematol 2013; 162:836-41. https://doi.org/10.1111/bjh.12453 PMid:23829561 PMCid:PMC3759564
- Gordeuk
VR, Campbell A, Rana S, et al. Relationship of erythropoietin, fetal
hemoglobin, and hydroxyurea treatment to tricuspid regurgitation
velocity in children with sickle cell disease. Blood 2009; 114:4639-44.
https://doi.org/10.1182/blood-2009-04-218040 PMid:19724057 PMCid:PMC2780300
- Pashankar
FD, Carbonella J, Bazzy-Asaad A, et al. Prevalence and risk factors of
elevated pulmonary artery pressures in children with sickle cell
disease. Pediatrics 2008; 121:777-82. https://doi.org/10.1542/peds.2007-0730 PMid:18381543
- Voskaridou
E, Tsetsos G, Tsoutsias A, et al. Pulmonary hypertension in patients
with sickle cell/beta thalassemia: incidence and correlation with serum
N-terminal pro-brain natriuretic peptide concentrations. Haematologica
2007; 92:738-43. https://doi.org/10.3324/haematol.11136 PMid:17550845
- Fonseca
GH, Souza R, Salemi VM, et al. Pulmonary hypertension diagnosed by
right heart catheterization in sickle cell disease. EurRespir J 2012;
39:112-8. https://doi.org/10.1183/09031936.00134410 PMid:21778170
- Parent
F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary
hypertension in sickle cell disease. N Engl J Med 2011; 365:44-53. https://doi.org/10.1056/NEJMoa1005565 PMid:21732836
- Dahoui
HA, Hayek MN, Nietert PJ, et al. Pulmonary hypertension in children and
young adults with sickle cell disease: evidence for familial
clustering. Pediatr Blood Cancer 2010; 54:398-402. https://doi.org/10.1002/pbc.22306 PMid:19827138
- Ataga
KI, Moore CG, Jones S, et al. Pulmonary hypertension in patients with
sickle cell disease: a longitudinal study. Br J Haematol 2006;
134:109-15. https://doi.org/10.1111/j.1365-2141.2006.06110.x PMid:16803576
- De
Castro LM, Jonassaint JC, Graham FL, et al. Pulmonary hypertension
associated with sickle cell disease: clinical and laboratory endpoints
and disease outcomes. Am J Hematol 2008; 83:19-25. https://doi.org/10.1002/ajh.21058 PMid:17724699
- Chou
ST, Jackson T, Vege S, et al. High prevalence of red blood cell
alloimmunization in sickle cell disease despite transfusion from
Rh-matched minority donors. Blood 2013; 122:1062-71. https://doi.org/10.1182/blood-2013-03-490623 PMid:23723452
- Aygun
B, Mortier NA, Kesler K, et al. Therapeutic phlebotomy is safe in
children with sickle cell anaemia and can be effective treatment for
transfusional iron overload. Br J Haematol 2015; 169:262-6. https://doi.org/10.1111/bjh.13280 PMid:25612463 PMCid:PMC4631316
- Ware
RE, Despotovic JM, Mortier NA, et al. Pharmacokinetics,
pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for
children with sickle cell anemia. Blood 2011; 118:4985-91. https://doi.org/10.1182/blood-2011-07-364190 PMid:21876119 PMCid:PMC3208303
- Charache
S, Dover GJ, Moore RD, et al. Hydroxyurea. Effects on hemoglobin F
production in patients with sickle cell anemia.Blood 1992; 79:2555-65.
PMid:1375104
- Ware
RE. Optimizing hydroxyurea therapy for sickle cell anemia. Hematology
(Educational Sessions of the American Society of Hematology) 2015;
436-43. https://doi.org/10.1182/asheducation-2015.1.436 PMid:26637755
- Savage
WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of
sickle cell disease: a summary of needed research. Am J Hematol 2015;
90:273-5. https://doi.org/10.1002/ajh.23945 PMid:25639238
- Flanagan
JM, Howard TA, Mortier N, et al. Assessment of genotoxicity associated
with hydroxyurea therapy in children with sickle cell anemia. Mutat Res
2010; 698:38-42. https://doi.org/10.1016/j.mrgentox.2010.03.001 PMid:20230905
- Ballas
SK, McCarthy WF, Guo N, et al. Exposure to hydroxyurea and pregnancy
outcomes in patients with sickle cell anemia. J Natl Med Assoc 2009;
101:1046-51. https://doi.org/10.1016/S0027-9684(15)31072-5
- Alvarez
O, Miller ST, Wang WC, et al. Effect of hydroxyurea treatment on renal
function parameters: results from the multicenter placebo-controlled
BABY HUG clinical trial for infants with sickle cell anemia. Pediatr
Blood Cancer 2012; 59:668-74. https://doi.org/10.1002/pbc.24100 PMid:22294512 PMCid:PMC3396762
- Hankins
JS, Helton KJ, McCarville MB, et al. Preservation of spleen and brain
function in children with sickle cell anemia treated with hydroxyurea.
Pediatr Blood Cancer 2008; 50:293-7. https://doi.org/10.1002/pbc.21271 PMid:17554794
- Aygun
B, Mortier NA, Smeltzer MP, et al. Hydroxyurea treatment decreases
glomerular hyperfiltration in children with sickle cell anemia. Am J
Hematol 2013; 88:116-9. https://doi.org/10.1002/ajh.23365 PMid:23255310 PMCid:PMC4673980
- Estepp
JH, Smeltzer MP, Wang WC, et al. Protection from sickle cell
retinopathy is associated with elevated HbF levels and hydroxycarbamine
use in children. Br J Haematol 2013; 161:402-5. https://doi.org/10.1111/bjh.12238 PMid:23384083
- McKie
KT, Hanevold CD, Hernandez C, et al. Prevalence, prevention, and
treatment of microalbuminuria and proteinuria in children with sickle
cell disease. J Pediatr Hematol Oncol 2007; 29:140-4. https://doi.org/10.1097/mph.0b013e3180335081
- Jayabose
S, Tugal O, Sandoval C, et al. Clinical and hematologic effects of
hydroxyurea in children with sickle cell anemia. J Pediatr 1996;
129:559-65. https://doi.org/10.1016/S0022-3476(96)70121-X
- Ferster
A, Vermylen C, Cornu G, et al. Hydroxyurea for treatment of severe
sickle cell anemia : a pediatric clinical trial. Blood 1996; 88:1960-4.
PMid:8822914
- Ferster
A, Tahriri P, Vermylen C, et al. Five years of experience with
hydroxyurea in children and young adults with sickle cell disease.
Blood 2001; 97:3628-32. https://doi.org/10.1182/blood.V97.11.3628 PMid:11369660
- Mulaku
M, Opiyo N, Karumbi J, et al. Evidence review of hydroxyurea for the
prevention of sickle cell complications in low-income countries. Arch
Dis Child 2013; 98:908-14. https://doi.org/10.1136/archdischild-2012-302387 PMid:23995076 PMCid:PMC3812872
- Ware
RE, Zimmerman SA, Schultz WH. Hydroxyurea as an alternative to blood to
blood transfusions for the prevention of recurrent stroke in children
with sickle cell disease. Blood 1999; 94:3022-6.
PMid:10556185
- Ware
RE, Zimmerman SA, Sylvestre PB, et al. Prevention of secondary stroke
and resolution of transfusional iron overload in children with sickle
cell anemia using hydroxyurea and phlebotomy. J Pediatr 2004;
145:346-52. https://doi.org/10.1016/j.jpeds.2004.04.058 PMid:15343189
- Zimmerman
SA, Schultz WH, Burgett S, et al. Hydroxyurea therapy lowers
transcranial Doppler flow velocities in children with sickle cell
anemia. Blood 2007; 110:1043-7. https://doi.org/10.1182/blood-2006-11-057893 PMid:17429008
- Hankins
JS, McCarville MB, Rankine-Mullings A, et al. Prevention of conversion
to abnormal TCD with hydroxyurea in sickle cell anemia: A phase III
international randomized clinical trial. Am J Hematol 2015;
90:1099-105. https://doi.org/10.1002/ajh.24198 PMid:26414435 PMCid:PMC4715740
- Wang
WC, Wynn LW, Rogers ZR, et al. A two-year pilot trial of hydroxyurea in
very young children with sickle-cell anemia J Pediatr 2001; 139:790-6. https://doi.org/10.1067/mpd.2001.119590 PMid:11743503
- Wang
WC, Helms RW, Lynn HS, et al. Effects of hydroxyurea on growth in
children with sickle cell anemia: Results of the HUG-KIDS study. J
Pediatr 2002; 140:225-9. https://doi.org/10.1067/mpd.2002.121383 PMid:11865275
- Mundee
Y, Bigelow NC, Davis BH, Porter JB. Flow cytometric method for
simultaneous assay of foetal haemoglobin containing red cells,
reticulocytes and foetal haemoglobin containing reticulocytes. Clin Lab
Haematol 2001; 23:149-54. https://doi.org/10.1046/j.1365-2257.2001.00344.x PMid:11553054
- Dover
GJ, Boyer SH. Quantitation of hemoglobins within individual red cells:
asynchronous biosynthesis of fetal and adult hemoglobin during
erythroid maturation in normal subjects. Blood 1980; 56:1082-91.
PMid:6159933
- Steinberg MH, Chui DHK, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood 2014; 123:481-5. https://doi.org/10.1182/blood-2013-09-528067 PMid:24222332
- Ngo
DA, Aygun B, Akinsheye I, et al. Fetal haemoglobin levels and
haematological characteristics of compound heterozygotes for
haemoglobin S and deletional hereditary persistence of fetal
haemoglobin. Br J Haematol 2012; 156:259-64. https://doi.org/10.1111/j.1365-2141.2011.08916.x PMid:22017641
- Horiuchi
K, Osterhout ML, Kamma H, et al. Estimation of fetal hemoglobin levels
in individual red cells via fluorescence image cytometry. Cytometry
1995; 20:261-7. https://doi.org/10.1002/cyto.990200310 PMid:7587712
- Steinberg
MH, Lu ZH, Barton FB, et al. Multicenter study of hydroxyurea. Fetal
hemoglobin in sickle cell anemia: determinants of response to
hydroxyurea. Blood 1997; 89:1078-88. PMid:9028341
- Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol 2012; 87:795-803. https://doi.org/10.1002/ajh.23232 PMid:22641398 PMCid:PMC4562292
- Elion
J, Berg PE, Lapoumeroulie C, et al. DNA sequencevariation in a negative
control region 5' to the beta-globin gene correlates with the
phenotypic expression of the beta S mutation. Blood 1992; 79:787-92.
PMid:1346253
- Lorey
FW, Arnopp J, Cunningham GC. Distribution of hemoglobinopathy variants
by ethnicity in a multiethnic state.Genet Epidemiol 1996; 13:501-12. https://doi.org/10.1002/(SICI)1098-2272(1996)13:5<501::AID-GEPI6>3.0.CO;2-4
- Treadwell
MJ, McClough L, Vichinsky E. Using qualitative and quantitative
strategies to evaluate knowledge and perceptions about sickle cell
disease and sickle cell trait. J Natl Med Assoc 2006; 98:704-10.
PMid:16749645 PMCid:PMC2569269
- Kavanagh
PL, Wang CJ, Therrell BL, et al. Communication of positive newborn
screening results for sickle cell disease and sickle cell trait:
variation across states. Am J Med Genet C Semin Med Genet 2008;
148C:15-22. https://doi.org/10.1002/ajmg.c.30160 PMid:18200513
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood 2010; 115:4331-6. https://doi.org/10.1182/blood-2010-01-251348 PMid:20233970 PMCid:PMC2881491
- Piel
FB, Hay SI, Gupta S, et al. Global burden of sickle cell anaemia in
children under five, 2010-2050: modeling based on demographics, excess
mortality, and interventions. PLoS Med 2013; 10:e1001484. https://doi.org/10.1371/journal.pmed.1001484 PMid:23874164 PMCid:PMC3712914
- World
Health Organization. Sickle-cell disease: a strategy for the WHO
African Region. Report AFR/RC60/8. Geneva, Switzerland: World Health
Organization; 2010.
- Yawn
BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell
disease: summary of the 2014 evidence-based report by expert panel
members. JAMA 2014; 12:1033-48. https://doi.org/10.1001/jama.2014.10517 PMid:25203083
- Cunningham-Myrie
C, Abdulkadri A, Waugh A, et al. Hydroxyurea use in prevention of
stroke recurrence in children with sickle cell disease in a developing
country: a cost effectiveness analysis. Pediatr Blood Cancer 2015;
62:1862-4. https://doi.org/10.1002/pbc.25563 PMid:25929458
- Steiner
CA, Miller JL. Sickle cell disease patients in U.S. hospitals,
2004.Statistical brief. No.21. Rockville, Md: Agency for Healthcare
Research and Quality, December 2006.
- Moore RD, Charache S, Terrin ML, et al. Cost-effectiveness of hydroxyurea in sickle cell anemia. Am J Hematol 2000; 64:26-31. https://doi.org/10.1002/(SICI)1096-8652(200005)64:1<26::AID-AJH5>3.0.CO;2-F
- Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood 2016; 127:810-9. https://doi.org/10.1182/blood-2015-09-618553 PMid:26758919
- Atweh
GF, Sutton M, Nassif I, et al. Sustained induction of fetal hemoglobin
by pulse butyrate therapy in sickle cell disease. Blood 1999;
93:1790-7. PMid:10068649 PMCid:PMC4269326
- Saunthararajah
Y, Molokie R, Saraf S, et al. Clinical effectiveness of decitabine in
severe sickle cell disease. Br J Haematol 2008; 141:126-9. https://doi.org/10.1111/j.1365-2141.2008.07027.x PMid:18324975
- Dos
Santos JL, Lanaro C, Lima LM, et al. Design, synthesis, and
pharmacological evaluation of novel hydrid compounds to treat sickle
cell disease symptoms. J Med Chem 2011; 54:5811-9. https://doi.org/10.1021/jm200531f PMid:21766854
- Meiler
SE, Wade M, Kutlar F, et al. Pomalidomide augments fetal hemoglobin
production without the myelosuppressive effects of hydroxyurea in
transgenic sickle cell mice. Blood 2011; 118:248-58. https://doi.org/10.1182/blood-2010-11-319137 PMid:21536862 PMCid:PMC3148160
- Fard
AD, Hosseini SA, Shahjahani M, et al. Evaluation of novel fetal
hemoglobin inducer drugs in treatment of ß-hemoglobinopathy disorders.
Int J Hematol Oncol Stem Cell Res 2013; 7:47-54. PMid:24505535
PMCid:PMC3913144
- Ataga
KI, Stocker J. Senicapoc (ICA-17043): a potential therapy for the
prevention and treatment of hemolysis-associated complications in
sickle cell anemia. Expert Opin Investig Drugs 2009; 18:231-9. https://doi.org/10.1517/13543780802708011 PMid:19236269
- Misra
H, Lickliter J, Kazo F, Abuchowski A. PEGylated carboxyhemoglobin
bovine (SANGUINATE): result of a phase I clinical trial. Artif Organs
2014; 38:702-7. https://doi.org/10.1111/aor.12341 PMid:25113835
- Chang
J, Patton JT, Sarkar A, et al. GMI-1070, a novel pan-selectin
antagonist, reverses acute vascular occlusions in sickle cell mice.
Blood 2010; 116:1779-86. https://doi.org/10.1182/blood-2009-12-260513 PMid:20508165 PMCid:PMC2947397
- Mandarino
D, Kawar Z, Alvarez R, et al. Placebo-controlled, double-blind,
first-in-human, ascending single dose, healthy subject study of
intravenous-administered SelG1, a humanized anti-P-selectin antibody in
development for sickle cell disease. Blood 2013; 122:abstract 970.
- Batchvarova
M, Shan S, Zennadi R, et al. Sevuparin reduces adhesion of both sickle
red cells and leukocytes to endothelial cells in vitro and inhibits
vaso-occlusion in vivo. Blood 2013;122:abstract 182.
- Qari
MH, Aljaouni SK, Alardawi MS, et al. Reduction of painful
vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a
double-blind randomized trial. Thromb Haemost 2007; 98:392-6. https://doi.org/10.1160/th06-12-0718
- De
Castro LM, Zennadi R, Jonassaint JC, et al. Effect of propranolol as
anti-adhesive therapy in sickle cell disease. Clin Transl Sci 2012;
5:437-44. https://doi.org/10.1111/cts.12005 PMid:23253664 PMCid:PMC3762678
- Zennadi
R. MEK inhibitors, novel anti-adhesive molecules, reduce sickle red
blood cell adhesion in vitro and in vivo, and vasoocclusion in vivo.
PLoS One 2014; 9:e110306. .
- Cheung
AT, Chan MS, Ramanujam S, et al. Effects of poloxamer 188 treatment on
sickle cell vaso-occlusive crisis: computer-assisted intravital
microscopy study. J Investig Med 2004; 52:402-6. https://doi.org/10.1136/jim-52-06-35 PMid:15612454
- Field
JJ, Lin G, Okam MM, et al. Sickle cell vaso-occlusion causes activation
of iNKT cells that is decreased by the adenosine A2A receptor agonist
ragadenoson. Blood 2013; 121:3329-34. https://doi.org/10.1182/blood-2012-11-465963 PMid:23377438 PMCid:PMC3637009
- Field
JJ, Ataga KI, Majerus E, et al. A phase I single ascending dose study
of NKTT120 in stable adult sickle cell patients. Blood 2013;
122:abstract 977.
- Knight-Perry
J, DeBaun MR, Strunk RC, Field JJ. Leukotriene pathway in sickle cell
disease: a potential target for directed therapy. Expert Rev Hematol
2009; 2:57-68. https://doi.org/10.1586/17474086.2.1.57 PMid:21082995
- Chang
J, Shi PA, Chiang EY, Frenette PS. Intravenous immunoglobulins reverse
acute vaso-occlusive crises in sickle cell mice through rapid
inhibition of neutrophil adhesion. Blood 2008; 111:915-23. https://doi.org/10.1182/blood-2007-04-084061 PMid:17932253 PMCid:PMC2200843
- Hoppe
C, Kuypers F, Larkin S, et al. A pilot study of the short-term use of
simvastatin in sickle cell disease: effects on markers of vascular
dysfunction. Br J Haematol 2011; 153:655-63. https://doi.org/10.1111/j.1365-2141.2010.08480.x PMid:21477202 PMCid:PMC3601917
- Bhatia
M, Walters MC. Hematopoietic cell transplantation for thalassemia and
sickle cell disease: past, present and future. Bone Marrow Transplant
2008; 41:109-17. https://doi.org/10.1038/sj.bmt.1705943 PMid:18059330
[TOP]