The Frequency of Adrenal Insufficiency in Adolescents and Young
Adults with Thalassemia Major versus Thalassemia Intermedia in Iran
Sara Matin1, Masoud Ghanei Jahromi2, Zohreh Karemizadeh3, Sezaneh Haghpanah4,Vincenzo De Sanctis5, Ashraf Soliman6, Javad Dehbozorgian4, Zahra Majd1, Narges Rezaei4 and Mehran Karimi4*
1 Department of Pediatrics, Shiraz University of Medical Sciences, Shiraz, Iran
2 Department of Anesthesiology, Jahrom University of Medical Science, Jahrom, Iran
3 Metabolic and Endocrinology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
4 Hematology Research Center, Shiraz University of Medical Science, Shiraz, Iran.
5 Pediatric and Adolescent Outpatient Clinic, Quisisana Private Accredited Hospital, Ferrara (Italy)
6 Department of Pediatrics, Alexandria University Children’s Hospital, Alexandria, Egypt.
Corresponding author: Mehran
Karimi, Professor of Pediatrics Hematology and Oncology, Hematology
Research Center, Shiraz University of Medical Sciences, Shiraz, Iran.
Tel. and Fax: 0098-7116473239, E-mail:
karimim@sums.ac.ir
Published: January 1, 2015
Received: September 24, 2014
Accepted: November 13, 2014
Mediter J Hematol Infect Dis 2015, 7(1): e2015005, DOI
10.4084/MJHID.2015.005
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background. Endocrine
dysfunction is not uncommon complication in patients with
transfusion-dependent thalassemia and is thought to occur as a
consequence of excessive iron overload. The primary objective of this
study is to determine the frequency of adrenal insufficiency in
patients with thalassemia major and thalassemia intermediate.
Methods. This
cross-sectional study was done at the Shiraz University of Medical
Sciences, Shiraz, Southern Iran, in 2013. One hundred and ninety
patients were divided into two groups; thalassemia major(TM) and
thalassemia intermediate (TI) groups. We measured 8 AM serum cortisol,
ACTH and ferritin concentrations in all patients.
Results. The
mean age of the TM and TI group were 22.5±5.7 and 23.8±6 years,
respectively. 90 patients (47.4%) were splenectomized, 34 (36.2%) with
TM and 56 (58.2%) with TI (p :<0.001). The median and interquartile
range of serum ferritin levels were 2184±3700 ng/ml and 437±443ng/ml in
TM and TI respectively (p< 0.001). Three patients with TM (1.6%) had
low basal cortisol and ACTH levels. However, their cortisol response to
ACTH stimulation was normal.
Conclusions. Low basal
concentrations of cortisol and ACTH occurred in 1.6% of our adolescents
young adult patients with TM suggesting a central defect in cortisol
secretion at the basal state. However, cortisol response to standard –
dose ACTH was normal in all patients with TM and TI. |
Introduction
β-thalassemias are a group of hereditary blood disorders
characterized by anomalies in the synthesis of the beta chains of
hemoglobin resulting in variable phenotypes, ranging from severe anemia
to clinically asymptomatic individuals. Three main forms have been
described: thalassemia major(TM) thalassemia intermedia (TI) and
thalassemia minor. Individuals with TM usually present within the first
two years of life with severe anemia, requiring regular red blood cell
(RBC) transfusions.[1]
The life expectancy of
children and adolescents with TM and TI has markedly increased, due to
improvement of the quality and techniques of blood transfusion and new
development of effective oral iron chelators.[1]
However, TI patients still develop iron overload, despite the lack of
need for blood transfusions, because of increased intestinal iron
absorption.[2-5] Moreover, various endocrine
abnormalities have been described in patients with TM and TI and most
reports incriminate iron overload as an important factor in the
development of target-organ dysfunction.[6,7]
Iron
overload is toxic to parenchymal cells because it generates free
radicals and induces oxidative stress causing damage to biomolecules,
including lipids, proteins, and DNA.[1,3]
Therefore,
regular surveillance of these patients is essential for early detection
and management of possible complications, such as: heart failure and
arrhythmias, chronic liver diseases, endocrine problems (hypogonadism,
hypothyroidism, diabetes mellitus, hypoparathyroidism and adrenal
insufficiency), growth failure, osteoporosis and thrombophilia.[1,6]
In TM, the prevalence of adrenal insufficiency (AI) is variable because
of the variable degree and duration of iron overload and not
standardized cutoff cortisol values for diagnosing cortisol deficiency.
In addition, the prevalence of AI has not been investigated well in
adolescent patients with TI. Although, most studies have revealed
intact pituitary adrenal axis in TM, several recent studies reported a
significant prevalence of subclinical “biochemical” AI, ranging from
18-45% in these patients.[8-15] AI is either primary,
due to deposition of excess iron in the adrenal gland or secondary due
to the toxic effects of iron in the pituitary gland.[16-18]
The combined measurement of early morning serum cortisol and plasma
ACTH separates patients with primary adrenal insufficiency from healthy
individuals and from those with secondary disease.[19]
While the diagnosis of overt adrenal failure is generally
straightforward, the identification of asymptomatic patients with
subtle dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis is
still a diagnostic challenge.
We assessed basal and stimulated
cortisol secretion in relation to the degree of iron overload in 190
consecutive adolescent patients with TM and TI.
Subjects and Methods
This
study included all patients who were referred to the thalassemia
clinic, Hospital and Outpatient Clinic, affiliated with the Shiraz
University of Medical Sciences. The patients divided into two groups
according to their diagnosis: TM and TI. All patients with TM were on
regular blood transfusion every 2-4 weeks. TI patients were transfusion
independent. The diagnosis of thalassemia was based on complete blood count (CBC), Hb electrophoresis and clinical history. Inclusion criteria were all TM and TI patients aged 10 years and above. Exclusion
criteria included: history of any infection or stress such as surgery
in the past four weeks, congestive heart failure, thyroid dysfunction,
hepatic impairment, uncontrolled diabetes mellitus, and/or taking any
medications that may have an effect on adrenal function.
Four-hundred-ninety TM and 196 TI patients over 10 years old were
eligible. From these patients, 190 consecutive patients were enrolled
in our study. A venous blood sample was collected from all patients at 8 AM for measuring serum ferritin, cortisol, and ACTH concentrations. Assessment of adrenal function.
A 2.5 mL sample of fasting venous blood withdrawn from the participants
was added to CBC tube contained EDTA anti-coagulant. Plasma,
centrifuged immediately, was preserved in -20°C in the freezer, then
carried by cold box to the laboratory to test ACTH level. Furthermore,
3 mL clot blood was also withdrawn from each patient. The serum was
immediately separated without hemolysis, preserved in -20°C in
the freezer and then, carried by cold box to the laboratory to test
cortisol level. The
ACTH and cortisol levels were measured by use of an
electrochemiluminescence immunoassay (ECLIA) method (Radim Diagnostics,
Pomezia- Rome, Italy). Normal range of cortisol at 7-10 A.M. was
6.2-19.4 µg/dL and 2.3-11.9 µg/dL at afternoon, while that for
corticotropin (ACTH) was 7.2- 63.3 pg /mL.Differentiation between primary and secondary adrenal insufficiencies. Analysis of the results was considered as follows:1.A low plasma cortisol levels (< 6.2 µg/dL – 10th percentile), measured between 8.00-9.00 am, in the face of high ACTH levels (i.e. > 100 pg/mL) suggested primary AI.[19,20]2.Inappropriately normal or low ACTH levels (< 7.2 pg/mL) , in the presence of low cortisol level (< 6.2 µg/dL – 10th percentile), suggested secondary AI.[21]3.Failure
to increase cortisol levels above 18 μg/dL at 30-60 minutes post
corticotropin intravenous (i.v.) stimulation test indicated adrenal
insufficiency.[22]Standard dose ACTH stimulation test.
The ACTH stimulating test was performed with 250 μg synthetic ACTH
1–24, cosyntropin, tetracosactin, Synacthen, as i.v. bolus followed by
measurement of serum cortisol 30 and 60 min after the injection (CINACT
ampoule). Fasting serum ferritin was measured by Electro Fluorescent Assay (ELFA) method, Mini Vidas machine (bio Merieux SA, France). Statistical analyses.
Statistical analyses were performed with SPSS Software (SPSS: An IBM
Company, version 17.0, IBM Corporation, Armonk, NY, USA). Test of
normality was performed by Shapiro-Wilk Test for serum ferritin, ACTH,
and cortisol levels. Mann-Whitney test was used for comparison of serum
ferritin, ACTH, and cortisol levels between TM and TI patients. Student
t-test was used for comparison of age between the two groups.
Comparison of qualitative data was done by Chi-square test. A p value
<0 .05 was considered as statistically significant.Ethical aspects. The study was performed in accordance with provisions of the Declaration of Helsinki
and Good Clinical Practice guidelines and was approved by Medical
Ethics Committee of the Shiraz University of Medical Sciences. Written
informed consent was obtained from the patients or their parents. Results
Ninety-four TM and ninety-six TI patients were investigated. The
mean age of the TM and TI group were 22.5±5.7 and 23.8±6 years,
respectively. No significant age difference was found between the two
groups (p:0.15).
In the TM group, 55 subjects (58.5%) were females
and 39 males (41.5%). In the TI group, 41 (42.7%) were women. All
patients with TM have been on iron chelators with deferoxamine (DFO)
(70%), combined DFO and deferiprone (DFP) (10%) or deferasirox (DFX)
(20%).
The compliance to DFO and DFX iron chelators were fair
and good, respectively. Among the study subjects, 90 (47.4%) had
splenectomy, 34 (36.2%) with TM and 56 (58.2%) with TI, respectively
(p:0.001). The median and interquartile range of serum ferritin levels
in TM and TI groups were 2184±3700 ng/ml and 437±443 ng/ml respectively
(p< 0.001). (Table 1)
In general, an early morning (8 am) plasma cortisol level lower than 6.2 µg/dL – (10th
percentile) is suggestive for primary AI, whereas a value higher than
15 μg/dL makes the diagnosis highly unlikely. Therefore, we performed
an ACTH stimulation test only in 3 patients (2 males and a female;
1.6%) with low basal levels of cortisol (Table 2).
Patient
1 was on treatment with DFO and had dilated cardiomyopathy with normal
left ventricular ejection fraction (LVEF). Patient 2 has short stature
and pubertal delay with mild systolic and diastolic cardiac
dysfunction. He has been on iron chelation therapy with DFO plus DFP
and L-thyroxine therapy for primary hypothyroidism. Patient 3, while on
treatment with DFX, presented with diabetes mellitus, dilated
cardiomyopathy and normal LVEF. None of the 3 TM patients were on sex
hormone replacement therapy or had received corticosteroids treatment.
Patients 2 and 3 had poor compliance with iron chelation therapy.
The plasma cortisol response at 60 minutes post ACTH injection should reach ≥ 18 μg /dl in normal people.[22] In our TM patients, the cortisol responses to ACTH stimulation test resulted in normal range (Table 2).
All patients with TI had normal basal serum ACTH and cortisol concentrations.
The median of ACTH concentrations did not differ significantly between patients with TM and TI (p: 0.072, Table 1). The median serum ferritin and basal serum cortisol levels were significantly higher in TM versus TI patients (p<0.001, Table 1).
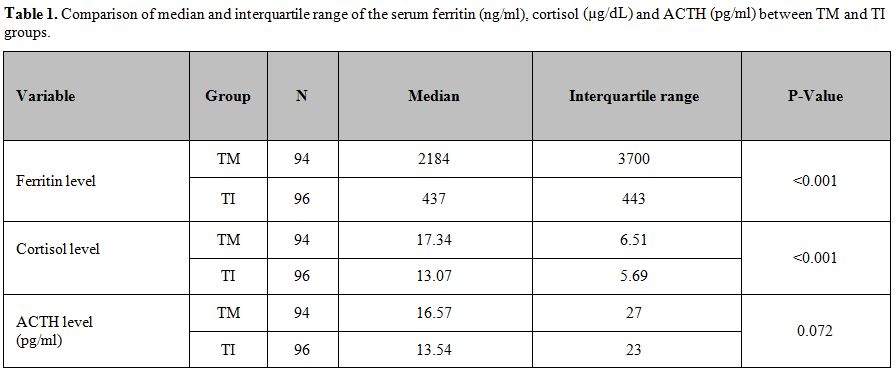 |
Table 1. Comparison of median and
interquartile range of the serum ferritin (ng/ml), cortisol (µg/dL) and
ACTH (pg/ml) between TM and TI groups. |
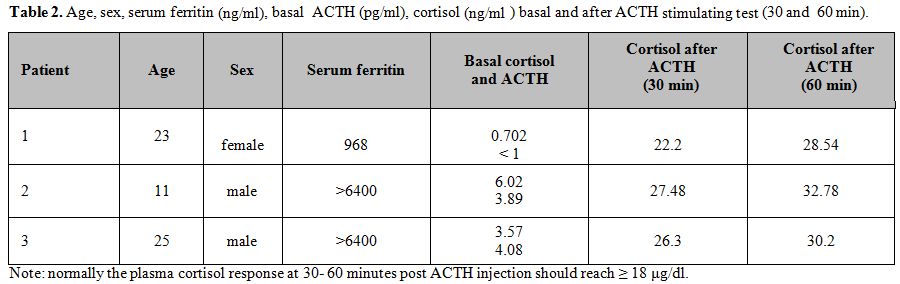 |
Table 2. Age, sex, serum ferritin (ng/ml),
basal ACTH (pg/ml), cortisol (ng/ml ) basal and after ACTH
stimulating test (30 and 60 min). |
Discussion
A
large body of evidence has emerged linking severe iron overload with
increased vulnerability to endocrine dysfunction in patients with
thalassemia.[1,2] Thalassemia patients, requiring
continuous blood transfusion, suffer from iron overload, with a
resultant increase in free non–transferrin-bound iron (NTBI) , and iron
accumulation in vital organs. In fact, the NTBI is rapidly taken up by
liver and other tissues. A particular portion of NTBI is the chelatable
labile plasma iron (LPI), which is not found in healthy individuals.
The LPI is the most toxic component; its toxicity is due to an high
reduction-oxidation (redox) potential, that generates oxygen-free
radicals such as superoxide anions, which damages DNA, proteins, and
membrane lipids in the cells.[23] Another source of
iron accumulation results from increased duodenal iron absorption due
to decreased expression of hepcidin, the central regulator of iron
homeostasis.[24,25] Iron has a marked affinity for the different endocrine glands.[26-29] The pituitary, thyroid and parathyroid gland, as well as the endocrine pancreas, are variably affected in these patients.[30-32]
It appears that the hypothalamic pituitary adrenal axis is the least
affected among the others in thalassemic patients. However, because of
the histological and imaging evidence of iron deposits in the adrenal
cortex[33,34] the potential risk of AI in these patients carries a significant risk that requires early diagnosis. Adrenal
insufficiency can be primary or secondary. Primary AI occurs when the
adrenal glands are damaged and cannot produce enough of the adrenal
hormone cortisol. The adrenal hormone aldosterone may also be lacking.[17]
Secondary adrenal insufficiency occurs when the pituitary gland fails
to produce enough adrenocorticotropin (ACTH). If ACTH output is too
low, cortisol production drops. Eventually, the adrenal glands can
shrink due to lack of ACTH stimulation.Our
study showed that all adolescents and young adults with TI had normal
ACTH- the cortisol axis. Only in three patients with TM the cortisol
and ACTH concentrations were low. However, their cortisol response to
250 μg synthetic ACTH was normal.Different
cut-offs for normal serum cortisol secretion have been proposed, and
the most reliable criterium appear to be a cortisol peak greater than
18–20 μg/dl after ACTH stimulation to exclude AI.[35]
However, this test cannot be able to detect recent onset or mild forms
of secondary AI. However, a normal cortisol response does not exclude
secondary AI, because the adrenal glands, which have not yet undergone
significant atrophy, can still respond to high dose ACTH stimulation.[36]It
is well known that the sensitivity of ACTH stimulation test to pick up
mild adrenal insufficiency improves when using the low-dose of
cosyntropin (1 μg ACTH given intravenously); but, this may result in a
higher false-positive rate. In addition, the lack of a commercially
available 1-μg dose may represent another potential error. Although
250 mcg ACTH test (owing to a massive dose of ACTH) is not sensitive
for diagnosis of partial secondary adrenal insufficiency, the basal
cortisol value during the standard-dose test has in most clinical
situations a diagnostic accuracy close to that of a low-dose of ACTH
test.[37,38] Our
3 TM patients with low basal secretion of cortisol and ACTH (secondary
AI) were asymptomatic. It is well known that the generalized weakness;
tiredness and fatigability are, in the early phase of the
adrenocortical insufficiency, transient and appearing only after
increased physical or psychical stress. They become gradually more
intensive and in more advanced stages of chronic adrenocortical
insufficiency.[21,36]A
standard-dose of 250 μg cosyntropin is useful for excluding primary AI
in those with low cortisol level during screening. A cortisol response
higher than 18 μg/dL at 30 minutes after a standard-dose ACTH confirms
adequate cortisol secretion. Most individuals with normal adrenal
function achieve much higher cortisol levels at 60 minutes after
cosyntropin injection. It
should be also noted that a large variety of total cortisol assays is
commercially available with considerable differences in specificity,
sensitivity, accuracy, precision and reproducibility of many of these
assays. Some of these assays appear to overestimate or underestimate
actual cortisol levels and, as such, hamper the correct diagnosis of
relative AI.[39]These
findings raise several interesting, relevant issues. Hematologists need
to be more vigilant about endocrine complications in thalassemia.
Whenever possible, cases of AI should be interpreted and managed in
consultation with a pediatric or adult endocrinologist, because adrenal
insufficiency usually is not in thalassemia an acute event but results
from gradual decline in pituitary function, due to iron overload.Deficiency
of cortisol level (secondary AI) with intact
renin-angiotensin-aldosterone system can cause adrenal crisis if it is
severe or patients are in acute illness. Therefore,
we recommend a systemic testing of the adrenal function prior
infection, trauma, surgical intervention or other stress and at regular
yearly interval in TM patients with iron overload especially in those
with other endocrinopathies. For
patients with proven AI, family education and stress steroids during
times of illness, injury or surgery are imperative help reduce the
morbidity and mortality associated with this serious complication.
Recovery from other endocrinopathies may be possible in thalassemia by
using intensive iron chelation therapy; however, this issue has not
been studied in cases of AI. Conclusion
Our findings support the small prevalence of AI in adolescents and
young adults with TM. All patients with TI have normal basal cortisol
and ACTH secretion.
An early morning (8 am) plasma cortisol
level lower the 10° percentile is suggestive for adrenal insufficiency,
whereas a value higher than 15 μg/dL makes the diagnosis highly
unlikely. Cortisol levels of intermediate range may be seen in patients
with primary, secondary or tertiary adrenal insufficiency. In those
cases and in patients with low basal cortisol and standard response to
ACTH a strict collaboration with the endocrinologist is needed.
References
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010 May 21;5:11. doi: 10.1186/1750-1172-5-11 http://dx.doi.org/10.1186/1750-1172-5-11

- Musallam
KM, Cappellini MD, Taher AT. Iron overload in ß-thalassemia intermedia:
an emerging concern. Curr Opin Hematol. 2013;20:187-92. http://dx.doi.org/10.1097/MOH.0b013e32835f5a5c PMid:23426199
- Musallam
KM, Taher AT, Rachmilewitz EA. ß-thalassemia intermedia: a clinical
perspective. Cold Spring Harb Perspect Med. 2012;2 :a013482. doi:
10.1101/cshperspect.a013482 http://dx.doi.org/10.1101/cshperspect.a013482
- Tanno
T, Miller JL. Iron loading and overloading due to ineffective
erythropoiesis. Adv Hematol. 2010;2010:358283. doi:
10.1155/2010/358283. Epub 2010 May 11. http://dx.doi.org/10.1155/2010/358283
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell 2004; 117: 285-97 http://dx.doi.org/10.1016/S0092-8674(04)00343-5
- Multicentre
study on prevalence of endocrine complications in thalassaemia major.
Italian working group on endocrine complications in non-endocrine
diseases. Clin Endocrinol (Oxf) 1995;42:581–6 http://dx.doi.org/10.1111/j.1365-2265.1995.tb02683.x
- Belhoul
KM, Bakir ML, Saned MS, Kadhim AM, Musallam KM, Taher AT. Serum
ferritin levels and endocrinopathy in medically treated patients with ß
thalassemia major.Ann Hematol. 2012 ;91:1107-14. http://dx.doi.org/10.1007/s00277-012-1412-7 PMid:22281991
- Sklar
CA, Lew LQ, Yoon DJ, David R. Adrenal function in thalassemia major
following long-term treatment with multiple transfusions and chelation
therapy. Evidence for dissociation of cortisol and adrenal androgen
secretion. Am J Dis Child. 1987;141:327–30 http://dx.doi.org/10.1001/archpedi.1987.04460030105036 PMid:3028128
- Jaruratanasirikul
S, Tanchotikul S, Wongcharnchailert M, Laosombat V, Sangsupavanich P,
Leetanaporn K. A low dose adrenocorticotropin test (1 microg ACTH) for
the evaluation of adrenal function in children with B-thalassemia
receiving hypertransfusion with suboptimal iron-chelating therapy. J
Pediatr Endocrinol Metab. 2007;20:1183–8. http://dx.doi.org/10.1515/JPEM.2007.20.11.1183 PMid:18183789
- Srivatsa
A, Marwaha RK, Muraldharan R, Trehan A. Assessment of adrenal endocrine
function in Asian thalassemics. Indian Pediatr. 2005;42:31–5.
PMid:15695855
- Mehrvar
A, Azarkeivan A, Faranoush M, Mehrvar N, Saberinedjad J, Ghorbani R,
Vossough P. Endocrinopathies in patients with transfusion-dependent
beta-thalassemia. Pediatr Hematol Oncol. 2008;25:187-94. http://dx.doi.org/10.1080/08880010801938207 PMid:18432501
- Vogiatzi
MG, Macklin EA, Trachtenberg FL, Fung EB, Cheung AM, Vichinsky E,
Olivieri N, Kirby M, Kwiatkowski JL, Cunningham M, Holm IA, Fleisher M,
Grady RW, Peterson CM, Giardina PJ; Thalassemia Clinical Research
Network. Differences in the prevalence of growth, endocrine and vitamin
D abnormalities among the various thalassaemia syndromes in North
America. Br J Haematol. 2009 ;146:546-56 http://dx.doi.org/10.1111/j.1365-2141.2009.07793.x PMid:19604241 PMCid:PMC2798591
- Scacchi
M, Danesi L, Cattaneo A, Valassi E, Pecori Giraldi F, Radaelli P,
Ambrogio A, D'Angelo E, Mirra N, Zanaboni L, Cappellini MD, Cavagnini
F. The pituitary-adrenal axis in adult thalassaemic patients. Eur J
Endocrinol. 2010 ;162:43-8 http://dx.doi.org/10.1530/EJE-09-0646 PMid:19820036
- Elsedfy
HH, El Kholy M, Tarif R, Hamed A, Elalfy M. Adrenal function in
thalassemia major adolescents. Pediatr Endocrinol Rev. 2011;8 (Suppl 2)
:295-9. PMid:21705981
- Soliman
AT, Yassin M, Majuid NM, Sabt A, Abdulrahman MO, De Sanctis V. Cortisol
response to low dose versus standard dose (back-to-back)
adrenocorticotrophic stimulation tests in children and young adults
with thalassemia major. Indian J Endocrinol Metab. 2013;17:1046-52. http://dx.doi.org/10.4103/2230-8210.122620 PMid:24381882 PMCid:PMC3872683
- Costin G, Kogut MD, Hyman CB, Ortega JA. Endocrine abnormalities in thalassemia major. Am J Dis Child. 1979;133:497-502. http://dx.doi.org/10.1001/archpedi.1979.02130050041009 PMid:433875
- Pasqualetti
P, Colantonio D, Collacciani A, Casale R, Natali G. Circadian pattern
of circulating plasma ACTH, cortisol, and aldosterone in patients with
beta-thalassemia. Acta Endocrinol (Copenh). 1990 Aug;123(2):174-8.
- Pasqualetti
P, Collacciani A, Colantonio D, Casale R, Natali G. Circadian rhythm of
pituitary-adrenal axis in thalassemia. Recenti Prog Med. 1990
Mar;81(3):200-1 PMid:2359871
- Wilkinson
CW, Pagulayan KF, Petrie EC, Mayer CL, Colasurdo EA, Shofer JB, Hart
KL, Hoff D, Tarabochia MA, Peskind ER. High prevalence of chronic
pituitary and target-organ hormone abnormalities after blast-related
mild traumatic brain injury. Front Neurol. 2012 Feb 7;3:11. doi:
10.3389/fneur.2012.00011. eCollection 2012. http://dx.doi.org/10.3389/fneur.2012.00011
- Oelkers
W, Diederich S, Bahr V. Diagnosis and therapy surveillance in Addison's
disease: rapid adrenocorticotropin (ACTH) test and measurement of
plasma ACTH, renin activity, and aldosterone. J Clin Endocrinol Metab
1992; 75: 259–64. PMid:1320051
- Siafarikas A. Addison disease.Diagnosis and initial management. Aust Fam Physician. 2010;39: 834-37 PMid:21301655
- Jacobson L. Hypothalamic-pituitary-adrenocortical axis regulation. Endocrinol Metab Clin North Am 2005, 34:271-92. http://dx.doi.org/10.1016/j.ecl.2005.01.003 PMid:15850842
- Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9-17. http://dx.doi.org/10.1038/nchembio.1416 PMid:24346035
- Schmidt
PJ, Fleming MD. Modulation of hepcidin as therapy for primary and
secondary iron overload disorders: preclinical models and approaches.
Hematol Oncol Clin North Am. 2014 ;28:387-401 http://dx.doi.org/10.1016/j.hoc.2013.11.004 PMid:24589273
- Chauhan
R, Sharma S, Chandra J. What regulates hepcidin in poly-transfused
ß-Thalassemia Major: Erythroid drive or store drive? Indian J Pathol
Microbiol. 2014;57:39-42 http://dx.doi.org/10.4103/0377-4929.130891 PMid:24739829
- Kishimoto
M, Endo H, Hagiwara S, Miwa A, Noda M. Immunohistochemical findings in
the pancreatic islets of a patient with transfusional iron overload and
diabetes: case report.J Med Invest. 2010;57:345-9. http://dx.doi.org/10.2152/jmi.57.345 PMid:20847537
- Matsushima
S, Tsuchiya N, Fujisawa-Imura K, Fujisawa-Imura K, Hoshimoto M, Takasu
N, Torii M, Ozaki K, Narana I, Kotani T. Ultrastructural and
morphometrical evaluation of the parathyroid gland in
iron-lactate-overloaded rats.Toxicol Pathol. 2005;33:533-9 http://dx.doi.org/10.1080/01926230591034438 PMid:16048848
- Lu
JP, Hayashi K. Selective iron deposition in pancreatic islet B cells of
transfusional iron-overloaded autopsy cases. Pathol Int. 1994;44:194-9.
http://dx.doi.org/10.1111/j.1440-1827.1994.tb02592.x
- Iancu
TC, Ward RJ, Peters TJ. Ultrastructural changes in the pancreas of
carbonyl iron-fed rats.J Pediatr Gastroenterol Nutr. 1990;10:95-101. http://dx.doi.org/10.1097/00005176-199001000-00018 PMid:2182817
- Argyropoulou
MI, Kiortsis DN, Metafratzi Z, Bitsis S, Tsatoulis A, Efremidis SC.
Pituitary gland height evaluated by MR in patients with
beta-thalassemia major: a marker of pituitary gland
function.Neuroradiology. 2001;43:1056-8. http://dx.doi.org/10.1007/s002340100634 PMid:11792043
- Wood
JC, Noetzl L, Hyderi A, Joukar M, Coates T, Mittelman S. Predicting
pituitary iron and endocrine dysfunction.Ann N Y Acad Sci.
2010;1202:123-8 http://dx.doi.org/10.1111/j.1749-6632.2010.05545.x PMid:20712782
- Noetzli
LJ, Panigrahy A, Mittelman SD, Hyderi A, Dongelyan A, Coates TD, Wood
JC. Pituitary iron and volume predict hypogonadism in transfusional
iron overload. Am J Hematol. 2012 ;87:167-71 http://dx.doi.org/10.1002/ajh.22247 PMid:22213195
- Bhamarapravati
N, Na-Nakorn S, Wasi P, Tuchinda S. Pathology of abnormal hemoglobin
diseases seen in Thailand. I. Pathology of beta-thalassemia hemoglobin
E disease. Am J Clin Pathol. 1967;47:745-58 PMid:4225872
- Drakonaki
E, Papakonstantinou O, Maris T, Vasiliadou A, Papadakis A,
Gourtsoyiannis N. Adrenal glands in beta-thalassemia major: magnetic
resonance (MR) imaging features and correlation with iron stores. Eur
Radiol. 2005;15:2462-8. http://dx.doi.org/10.1007/s00330-005-2855-1 PMid:16086182
- Hockings
GI, Nye EJ, Grice JE, Jackson RV. Short synacthen test versus insulin
stress test for the assessment of hypothalamo-pituitary axis:
controversy revisited again. Clin Endocrinol (Oxf). 1997; 46:775–6
- Dorin RI, Qualls CR, Crapo LM.Diagnosis of adrenal insufficiency. Ann Intern Med.2003; 139:194–204 http://dx.doi.org/10.7326/0003-4819-139-3-200308050-00017 PMid:12899587
- Mayenknecht
J, Diederich S, Bahr V, Plöckinger U, Oelkers W.Comparison of low and
high dose corticotropin stimulation tests in patients with pituitary
disease. J Clin Endocrinol Metab 1998;83:1558-62. http://dx.doi.org/10.1210/jcem.83.5.4831 PMid:9589655
- Annane
D, Sebille V, Charpentier C, Bollaert PE, François B, Korach JM,
Capellier G, Cohen Y, Azoulay E, Troché G, Chaumet-Riffaud P,
Bellissant E.. Effect of treatment with low doses of hydrocortisone and
fludrocortisone on mortality in patients with septic shock. JAMA
2002;288:862-71. http://dx.doi.org/10.1001/jama.288.7.862 PMid:12186604
- Cohen
J, Ward G, Prins J, Jones M, Venkatesh B. Variability of cortisol
assays can confound the diagnosis of adrenal insufficiency in the
critically ill population. Intensive Care Med 2006; 32: 1901–5 http://dx.doi.org/10.1007/s00134-006-0389-x PMid:17019540
[TOP]