Received: September 12, 2014
Accepted: December 12, 2014
Mediterr J Hematol Infect Dis 2015, 7(1): e2015010, DOI 10.4084/MJHID.2015.010
This article is available on PDF format at:

Jonathan Braue1, Vagishwari Murugesan2, Steven Holland3, Nishit Patel4, Eknath Naik5, Jennifer Leiding6, Abraham Tareq Yacoub7, Carlos N Prieto-Granada8 and John Norman Greene9
1 Morsani College of Medicine, University of South Florida, Tampa, Florida
2 Division of Infectious Diseases, Moffitt Cancer Center, Tampa, Florida
3
Laboratory of Clinical Infectious Diseases, National Institute of
Allergy and Infectious Diseases, National Institutes of Health,
Bethesda, Maryland
4 Department of Dermatology and Cutaneous Surgery, Morsani College of Medicine, University of South Florida, Tampa, Florida
5
Division of Infectious Disease and International Medicine, Morsani
College of Medicine, University of South Florida, Tampa, Florida
6
Department of Pediatrics, Division of Allergy, Immunology, and
Rheumatology, Morsani College of Medicine, University of South Florida,
Tampa, Florida
7 Division of Infectious Diseases, Moffitt Cancer Center, Tampa Florida
8 Department of Dermatology, Morsani College of Medicine, University of South Florida, Tampa, Florida
9 Chief, Division of Infectious Diseases, Moffitt Cancer Centre, Tampa
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract NF-κB essential modulator (NEMO) is a
kinase integral to the macrophage TNF-α pathway, which leads to the
intracellular destruction of Mycobacteria species. Defects in the NEMO
pathway result in spectrum of diseases, including but not limited to
ectodermal dysplasia, Mendelian susceptibility to mycobacterial
diseases, and incontinentia pigmenti. In addition, paucity of NEMO can
lead to the inability to mount a proper immune response against
opportunistic pyogenic and mycobacterial infections, leading to
dissemination to various organ systems. This manuscript will discuss
the numerous clinical manifestations of NEMO deficiency, the
differential diagnosis of atypical mycobacterial infections in
immunocompetent adults, and feature a case report of rare isolated
susceptibility to disseminated atypical mycobacteria due to a mutation
in the first exon of the NEMO gene. |
Introduction
In 1991, Nedorost et al. described two brothers, each affected with
rosacea-like lesions discovered to stem from cutaneous infection with
Mycobacterium avium-intracellulare (MAI).1 Neither brother had reason
for acquired immunoincompetence, so it was postulated that they
potentially harbored a genetic defect in the pathway responsible for
the destruction of mycobacterial species.[1] Though,
this defect remained to be uncovered. Then, in 1996 the occurrence of a
theoretically X-linked recessive susceptibility to mycobacterial
infection was reported.[2] The affected patients were
maternally related members of the family revealed to have defective
IL-12 production, which is necessary to defend against mycobacteria.[2] Later that same year, the genetic etiology of Mendelian susceptibility to mycobacterial diseases (MSMD) was first accounted.[3]
MSMD, a congenital syndrome predisposing affected individuals to
clinical disease with low virulence non-tuberculous mycobacteria, was
first shown to result from germline mutations in the autosomal gene
responsible for the production of the IFN-γ receptor ligand-binding
chain.[3] Ensuing studies demonstrated five other autosomal genes
involved in IL-12-dependent, IFN-γ-mediated immunity resulting in MSMD.[4]
However, the genetic mutation causing the X-linked pattern observed in
previous cases of familial mycobacterial infections remained elusive.
NF-κB
transcription factor is vital to the innate immune response to numerous
pathogens including mycobacteria. Two proteins known as NF-κB inhibitor
protein A and B control the immunomodulatory functions of NF-κB. In
turn, these proteins are regulated by IκB kinase alpha, beta, and gamma
otherwise known as NEMO. The gene encoding NEMO, located on the X
chromosome, is activated by TNF-α binding to its receptor leading to
downstream destruction of intracellular bacteria via the NF-κB
pathway.[5,6] In early 2000, mouse model studies linked NEMO deficiency
to the human genodermatosis incontinentia pigmenti.[6,7] Shortly
thereafter, novel human mutations in the NEMO gene were shown to lead
to the X-linked disorders, ectodermal dysplasia, and incontinentia
pigmenti.[8] Susceptibility to pyogenic and mycobacterial infections
were known to be a part of the spectrum of these diseases, and in 2004,
Orange et al reported data comprising of seven boys with mutations
leading to NEMO immunodeficiency, most of which suffered from
mycobacterial illness.[9] Also, in 2006, a novel mutation in NEMO was
reported as a cause of MSMD.[6] Thus, primary immunodeficiency in the
NEMO protein contributes further to the differential diagnosis in
patients with unexplained susceptibility to atypical mycobacteria.
Case Report
A 37-year-old Caucasian man presented to our institution for
evaluation of several multifocal chronic granulomatous skin lesions. He
had a childhood history of asthma and pneumonia, as well as recurrent
sinus infections requiring sinus surgery as an adult. Family history
revealed a male cousin who was recently hospitalized for a disseminated
Mycobacterium avium-intracellulare infection involving his liver. The patient denied any recent travel outside the country.
The
skin lesions originally erupted seven years prior, starting as a
non-painful, non-pruritic plaque on the right shoulder that remained
persistent and eventually spread to all four extremities. Sarcoidosis
was diagnosed at initial presentation and treated with corticosteroids
and hydroxychloroquine. Despite treatment and the initial response,
skin lesions remained persistent, and thus subsequent skin biopsies
were performed. Results yielded numerous acid-fast bacilli with
granulomas. A tentative diagnosis of leprosy was given, and treatment
with rifampin, clarithromycin, and minocycline ensued. Unfortunately,
this regimen failed to yield any significant degree of improvement.
Moreover, taper from the steroid treatment initially given for
sarcoidosis was attempted, only to produce worsening of the skin
lesions and development of high-grade fevers up to 105.0 ° F requiring
hospitalization.
Due to pancytopenia, persistent fevers, and
concern for disseminated infection or malignancy, a bone marrow biopsy
was performed. Analysis of the biopsy demonstrated granulomas and
acid-fast bacilli; culture isolates yielded Mycobacterium avium-intracellulare (MAI), rather than the preconceived Mycobacterium leprae.
Based on these findings, antimicrobial therapy was changed to
clarithromycin, ethambutol, and rifampin. Corticosteroids at a dose of
16 mg daily were also continued to prevent rebound exacerbation of the
skin lesions.
Despite antibiotic therapy for six months, fevers
persisted, and skin lesions advanced with new nodular masses developing
quickly over the period of a week. Multiple ulcerations with overlying
eschars on the right forearm and torso persisted, and large
erythematous nodules with central low-grade ulceration formed on the
right lower extremity. In addition, enlarged para-aortic lymph nodes
concerning for granulomatous disease were visualized on CT scan.
A repeat skin biopsy showed a persistent MAI infection resistant to
clarithromycin, ethambutol, and moxifloxacin. Additionally, the
pathology report described an atypical T-cell infiltrate in the dermis
and positive TCR gene rearrangement raising concern for cutaneous
T-cell lymphoma (CTCL). At this time, the patient was referred to us
for work-up of his potentially malignant skin condition.
At
presentation to our institution, lesions consisted of 3 to 4 inches in
diameter, diffuse, raised, well-circumscribed erythematous red scaly
plaques with a lack of active ulceration (Figure 1).
Because of the concern for CTCL, further skin biopsies were performed.
Results showed a histiocytic infiltrate admixed with very few small
atypical lymphocytes (Figure 2) and long beaded bacilli consistent with mycobacterium, located in the superficial and deep dermis (Figure 3). Isolates demonstrated not only MAI, but also the presence of Mycobacterium simiae.
Bone marrow core biopsy revealed a normocellular bone marrow
constituents being populated by foamy histiocytes that are disposed in
loosely formed granulomas (Figure 4).
The admixed T-cells were clonal and appeared to lack CD7 by flow
cytometry, but were positive for CD3, CD4, and CD26. However, the
absence of epidermotropism, lymphohistiocytic infiltrate with acid-fast
mycobacteria, and ultimately the marked paucity of T-cells and
lymphocytes, rendered a clonal lymphoproliferative disorder involving
the skin exceedingly unlikely. With CTCL effectively ruled out and
compelling evidence of disseminated MAI infection, investigation into
an underlying immune defect was performed. Evaluation of acquired
immunodeficiencies associated with MAI infections was done, with
negative antibodies to HIV and HTLV 1 and 2, and so a search for a
primary immunodeficiency with susceptibility to non-tuberculous
mycobacteria ensued. Based on family history implying X-linked
inheritance, NEMO deficiency seemed most likely. A single base change
of the last base of exon 1b (c.1-16G>C) of IKKg resulting in
abnormally spliced transcripts and reduced expression of NEMO was
found. This datum confirmed the diagnosis of NEMO deficiency.
The
patient was followed by the National Institutes of Health (NIH) clinic
in Bethesda, Maryland. She was given gamma interferon and all the
listed antibiotics and was improving along with tapering his
corticosteroid use.
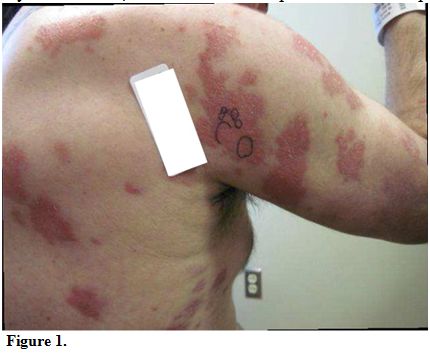 |
Figure 1. |
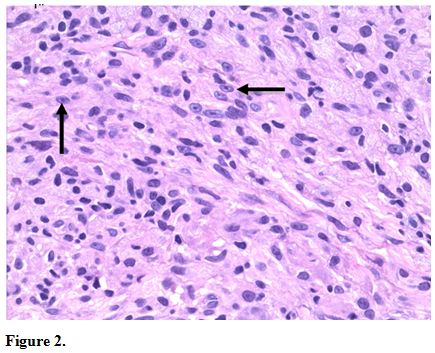 |
Figure 2. |
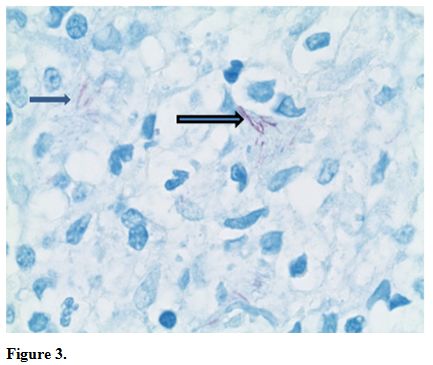 |
Figure 3. |
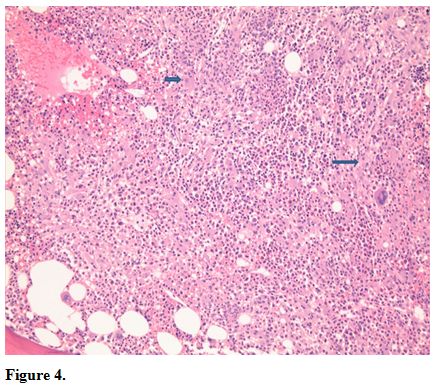 |
Figure 4. |
Discussion
The transcription factor nuclear factor-kB (NF-kB) is a key
transcription factor involved in regulating innate and adaptive immune
responses as well as ectodermal development. NF-kB essential
modulator (NEMO or inhibitor of NF-kB kinase gamma (IKKg)) is a 419
amino acid regulatory protein encoded by IKBKG on the X chromosome that
is critical for NF-kB activation. In the resting state,
NF-kB
proteins are held by inhibitors of NF-kB (IkB) proteins. Activation of
numerous cell receptors causes activation of the IkB kinase complex
(IKK) including IKKa, IKKb, and IKKg or NEMO. The IKK complex then
leads to phosphorylation of IKb proteins followed by ubiquitination
thus freeing NF-kB to undergo nuclear translocation and activation of
gene transcription.[10]
Amorphic NEMO mutations are lethal in
boys, but hypomorphic mutations result in a combined immunodeficiency
characterized by infectious susceptibility to bacterial, viral,
pneumocystis, and mycobacterial infections starting in the first year
of life. They can present with pneumonia, bacteremia, skin and soft
tissue abscesses, enteritis or colitis, encephalitis or meningitis,
sinusitis, or osteomyelitis.[9,11]
The immune phenotype of NEMO
deficiency is variable but can include hypogammaglobulinemia with
elevated IgM and IgA and reduced specific antibody responses due to
impaired CD40-mediated B cell class switch recombination. TLR signaling
and NK cell cytotoxicity are also diminished; T cell numbers and
proliferation are variable.[9,11]
Due to perturbations in
other pathways relying on NF-kB signaling, patients with NEMO
deficiency may present with other manifestations including X-linked
hypohydrotic ectodermal dysplasia, invasive pneumococcal disease and
incontinentia pigmenti. While the majority of these diseases tend to
present in childhood; exceptions are possible, and other primary
immunodeficiency syndromes with defective innate and adaptive immunity
may present later on in adulthood.
Hypomorphic mutations of the
NEMO gene are responsible in the majority of patients who present with
the rare NEMO deficiency syndrome.[12,13] Ectodermal dysplasia (ED),
one of the better characterized disorders resulting from deficiency of
NEMO protein, shows the typical phenotype including thickened
skin, conical teeth, absence of sweat glands, and thin, sparse
hair.[14] Similar to all NEMO syndrome variants, ED patients have a
crippled innate immune system leading to a poor response to bacterial
and fungal invasion, as well as difficulties in antibody
production.[15] Hypohydrotic ectodermal dysplasia with immune
deficiency (HED-ID) results from an immunological aberration in the
gene encoding the NF- κB essential modulator (NEMO; also known as IκB
kinase γ subunit [IKKγ]).[16] Classically, a hypomorphic coding
mutation on the X-chromosome is responsible for the decreased function
in NEMO, which leads to HED-ID.[4] Yet, in 2010, Mooster et al.
described a patient that had a normal coding sequence, but a
securely deficient NEMO stemming from mutations in the noncoding region
of the gene.[17] Female carriers of this mutation develop anomalies of
teeth, hair, skin, nails, and the CNS; however, the severity in female
carriers depends mainly on X-chromosome lyonization, and thus is
extremely variable.[16] Male patients who have HED-ID suffer from
reduced or absent sweat glands and hair follicles,
dysgammaglobulinemia, and recurrent pyogenic infections of the
integumentary, skeletal, and gastrointestinal systems.[16] Also, due to
lack of development of mucous glands and other anatomic abnormalities
such as cleft palate, upper and lower respiratory tract infections are
particularly prominent in these patients.[18] In addition, atopic
disease appears to be highly prevalent in HED-ID. In one series there
was a 71% prevalence of eczema, 65% prevalence of asthma or recurrent
wheezing, and 26% prevalence of food and drug allergies.[15,16] This
marked propensity of atopic disease is thought to be a result of
impaired barrier function seen in the various organ systems affected by
the ectodermal dysplasia syndromes.[18]
The vast majority of
invasive pneumococcal disease (IPD) cases are unexplained, but in 2007
Ku et al described a child that had a hemizygous mutation in NEMO
leading to a narrow clinical phenotype of susceptibility to Streptococcus pneumoniae.[19]
This patient had very mild signs consistent with anhydrotic
ectodermal dysplasia, and from the age of 15 months was plagued several
times with pneumococcal diseases.[19] He developed buccal cellulitis
and periorbital cellulitis, due to S. pneumoniae
serotype 33.[19] Despite being vaccinated with the heptavalent and
23-valent pneumococcal vaccinations, by the age of 2 years and seven
months, he again developed blood and hip infections by S. pneumoniae
serotype 23.[19] The immunological profile of this patient showed that
the antibody response to the immunizations was significantly
blighted.[19] In addition overwhelming infections with Staphylococcus aureus, Pseudomonas species and Hemophilus influenza
have also been described. Susceptibility is also greatly variable among
patients with some manifesting no infections to mild to very severe
septic forms of disease.
Incontinentia pigmenti (IP), also
known as Bloch-Sulzberger syndrome, is an X-linked dominant
genodermatosis affecting primarily female newborns and is typically
lethal in males.[20] The estimated prevalence of IP is about
0.2/100,000 and results from a mutation in the NEMO gene leading to an
inaccurate gene product and defective NF-κB activation.[21] Even in
patients with the same mutation, the phenotypic expression of this
disease is highly variable. Due to the difficulty of diagnosis in mild
cases, in 2013 Minic et al. proposed an updated version of the IP
diagnostic criteria. There are four clinical stages IP patients
progress through usually starting in the early neonatal period. These
stages may occur concomitantly or sequentially and include the
vesicular, verruciform, hyperpigmented, and hypopigmented stages.[21]
Linear vesicles, appearing within the first two months following birth,
characterize the vesicular stage, to which follow verrucous
hyperkeratotic plaques, thus indicating the verrucous stage. Brown to
bluish-gray hyperpigmentation following Blaschko lines designates the
third stage, which is followed by linear hypopigmented macules in the
final stage.[21] The cutaneous findings are treated nonspecifically
with topical steroids and emollients. However, in up to 80% of IP cases
other extracutaneous clinical manifestations are present and comprise
abnormalities of the teeth, eyes, hair, CNS, musculoskeletal systems,
and the immune system.[21] In June of 2014, Marques et al. published a
case report highlighting the importance of early detection of IP.
Seizures, ischemic strokes, strabismus, and cranial anomalies may
result, and a multidisciplinary approach must be taken to address these
extracutaneous signs.[20]
Santos et al., in 2006, uncovered an
MSMD-causing NEMO mutation that curiously did not affect NF-κB
activation. Rather, the mutations selectively impair CD40-triggered,
and NF-κB/c-Rel mediated generation of IL-12 by monocytes, and thus
explaining the specific susceptibility to mycobacteria.[4] In their
case reports, they describe a family with four maternally related males
in successive generations with severe Mycobacterium avium intracellulare
(MAI) infections. One of their patients was rather similar to our
previous report and did not manifest disease until 13 years of age with
extensive granulomatous cutaneous lesions initially thought to be
sarcoidosis. These lesions turned out to be MAI of the skin.[4] Other
mutations in the IL-12/23-INF-γ circuit leading to MSMD, have been
reported to result in infections with several other mycobacterial
species, including M. chelonae, M. fortuitum, M. smegmatis, M. kansasii, and M. szulgai.
However, to our knowledge, these species have not been found in cases
related to mutations in NEMO, and we have not seen a report suggesting
infection with M. simiae,
like in our patient described previously. Its role as a pathogen in
this case is very questionable. We cannot differentiate between
infection and colonization, particularly if the lung is not involved.
The presence of M. Simiae in
the skin biopsies might also be related to the immune defect and was
not reported previously in the NEMO deficiency patients.
Conclusion
The occurrence of disseminated low-virulence atypical mycobacteria is rare in immunocompetent adults. When challenged with a patient such as the one we described, it is important to consider a range of genetic defects that may be causing this susceptibility. The primary considerations include IL-12/IFN-γ receptor defects resulting in MSMD, GATA2 deficiency, and acquired autoantibodies to IFN-γ, and highlights others that should be considered in the differential diagnosis (Table 1). Although NEMO deficiency typically manifests in childhood, our case demonstrates the phenotypic heterogeneity resulting from a genetic mutation in NEMO. Thus, clinical suspicion for defects in the NEMO gene should be high, and prompt genetic and immunological testing performed, when immunocompetent adults present with atypical mycobacterial infections.
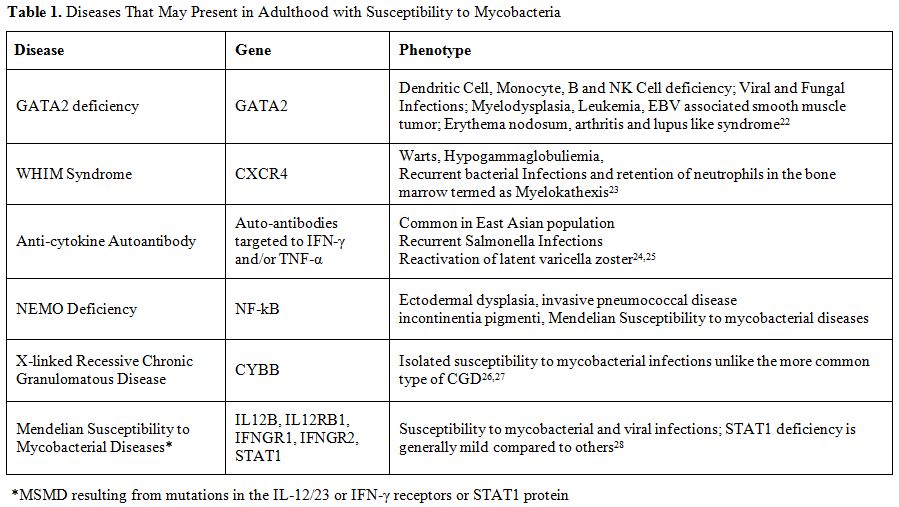 |
Table 1. Diseases That May Present in Adulthood with Susceptibility to Mycobacteria |
References
 .
. [TOP]