Refractory Thrombocytopenia and Neutropenia: a Diagnostic Challenge
Emmanuel Gyan1,2,3, François Dreyfus3,4 and Pierre Fenaux3,5
1 Service d’hématologie et thérapie cellulaire, Centre hospitalier universitaire, Tours, France
2 Team 2 "Leukemic Niche and Redox metabolism", UMR CNRS 7292 GICC, Université François Rabelais, Tours, France
3 Groupe Francophone des Myélodysplasies, Hôpital Saint Louis, AP-HP, Paris, France
4 Service d’hématologie, Hôtel-Dieu, AP-HP, Paris, France
5 Service d’hématologie séniors, Hôpital Saint Louis, AP-HP and Paris 7 University, Paris, France
Corresponding author: Prof. Emmanuel Gyan, MD,
PhD. Service d’hématologie et thérapie cellulaire, Centre hospitalier
universitaire de Tours, 2 boulevard Tonnellé, 37044 TOURS Cedex 9. Tel:
+ 33 2 47 47 37 12, Fax: +33 2 47 47 38 11. E-mail:
emmanuel.gyan@univ-tours.fr
Published: February 18, 2015
Received: December 20, 2014
Accepted: January 28, 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015018, DOI
10.4084/MJHID.2015.018
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The 2008 WHO classification
identified refractory cytopenia with unilineage dysplasia (RCUD) as a
composite entity encompassing refractory anemia, refractory
thrombocytopenia (RT), and refractory neutropenia (RN), characterized
by 10% or more dysplastic cells in the bone marrow respective lineage.
The diagnosis of RT and RN is complicated by several factors.
Diagnosing RT first requires exclusion of familial thrombocytopenia,
chronic auto-immune thrombocytopenia, concomitant medications, viral
infections, or hypersplenism. Diagnosis of RN should also be made after
ruling out differential diagnoses such as ethnic or familial
neutropenia, as well as acquired, drug-induced, infection-related or
malignancy-related neutropenia. An accurate quantification of dysplasia
should be performed in order to distinguish RT or RN from the
provisional entity named idiopathic cytopenia of unknown significance
(ICUS). Cytogenetic analysis, and possibly in the future somatic
mutation analysis (of genes most frequently mutated in MDS), and flow
cytometry analysis aberrant antigen expression on myeloid cells may
help in this differential diagnosis. Importantly, we and others found
that, while isolated neutropenia and thrombocytopenia are not rare in
MDS, those patients can generally be classified (according to WHO 2008
classification) as refractory cytopenia with multilineage dysplasia or
refractory anemia with excess blasts, while RT and RN (according to WHO
2008) are quite rare. These results suggest in particular that
identification of RT and RN as distinct entities could be reconsidered
in future WHO classification updates. |
Background: WHO Classification of MDS
Myelodysplastic syndromes (MDS) are marrow stem cell disorders
characterized by ineffective hematopoiesis leading to blood cytopenias,
a variable proportion of blasts, and a propensity to evolve to acute
myeloblastic leukemia (AML). The first classification of MDS was
published by the French-American-British group in 1982, individualizing
five entities named refractory anemia (RA), refractory anemia with
ringed sideroblasts, RA with excess blasts (RAEB), RA with excess
blasts in transformation (RAEB-T), and chronic myelomonocytic leukemia
(CMML).[1] This FAB MDS classification, mainly based on the morphologic
features of the blood and the bone marrow was refined in 2002[2] and
finally in 2008 by the World Health Organization,[3] that shifted the
RAEB-T category to AML by lowering the threshold of bone marrow blasts
for AML diagnosis from 30% to 20%, also excluded CMML from MDS,
individualized MDS with isolated deletion of the long arm of chromosome
5 (del 5q), and took into account the number of morphologically
dysplastic myeloid lineages. This led to separate, in patients without
excess of marrow blasts, those with multilineage dysplasia (refractory
cytopenia with multilineage dysplasia or RCMD, with or without ringed
sideroblasts) from patients with unilineage dysplasia (refractory
cytopenia with unilineage dysplasia or RCUD) (Table 1).
RCUD as a Distinct Diagnostic Group in the 2008 WHO Classification
RCUD was thus identified as a new MDS group, containing three
arbitrarily defined subgroups: refractory anemia (RA), refractory
neutropenia (RN) and refractory thrombocytopenia (RT). It is important
to consider that these diagnoses are mainly based on the bone marrow
finding of a unique dysplastic lineage, contrarily to what their name
would intuitively suggest. The characteristics of WHO-defined RCUD are
detailed below.
Common characteristics of RCUD.
Marrow findings should be unilineage dysplasia defined as the presence
of ≥ 10% dysplastic cells in one myeloid lineage. Less than 5% blasts
are observed. The blood should contain < 1% blasts. Cases of
unilineage dysplasia with 1% circulating blasts should be classified as
MDS-U. If 2-4% circulating blasts are observed, the diagnostic
classification is RAEB-1. Even though RARS has unilineage dysplasia, it
is recognized as a distinct entity and not included in RCUD. Therefore,
RA diagnosis is considered when only erythroid dysplasia is present and
if < 15% ringed sideroblasts.
For the diagnosis of MDS, cytopenias are defined as hemoglobin < 10 g/dL, absolute neutrophil count (ANC) < 1.8x109/L, and platelet count < 100x109/L.
Importantly, two cytopenias are accepted for the diagnosis of RCUD,
provided there is only one dysplastic lineage in the bone marrow. In
case of pancytopenia associated with only one dysplasia in the bone
marrow, the classification should be MDS-U (Table 1).
Also, the cytopenia does not always correspond to the bone marrow
dysplastic lineage. In a series of 44 patients with a single cytopenia
with unilineage dysplasia described by Verburgh et al, 18 (41%)
presented with a cytopenia in a lineage not affected by dysplasia.[4]
This discrepancy creates an ambiguity in the understanding of the RCUD
subgroups, theoretically characterized by one ‘refractory cytopenia’
(RA, RN, or RT), since a unique cytopenia in a patient with MDS may be
associated in some cases with ≥ 10% bone marrow dysplasia in another or
several lineages. There is thus an ‘unilineage paradox’, where the
WHO-defined RCUD can be associated with one or two cytopenias not
corresponding with the affected lineage in the bone marrow, whereas MDS
with only one cytopenia – which could be identified as ‘isolated
thrombocytopenia’ (IT) or ‘isolated neutropenia’ (IN) – are common.
This issue will be discussed below.
In refractory anemia (RA),
signs of dyserythropoiesis may be observed on blood smears, such as
macrocytosis, anisochromasia or dimorphism, with or without
anisocytosis and poikilocytosis, which are markers of clonal
heterogeneity in a chimeric bone marrow. Neutrophils and platelets are
usually normal in number and morphology. However, the presence of
moderate neutropenia or thrombocytopenia remains consistent with the
diagnosis of RA. Bone marrow cellularity is generally increased, but
can be normal or decreased. Dyserythropoiesis is defined as 10% or more
dysplastic erythroid precursors. Dysery-thropoiesis is not specific for
RCUD compared to other types of MDS. If a dysplasia is present in a
second lineage, it should always be < 10%.
In refractory
neutropenia (RN), dysgranulopoiesis can be identified in the blood by
the presence of nuclear hypolobation and hypogranulation of
neutrophils. In the bone marrow, dysplasia in the granulocytic lineage
is ≥10%, with no significant dysplasia (<10%) in the erythroid or
megakaryocytic lineage.
Refractory Thrombocytopenia (RT) is mainly
characterized in the blood by isolated thrombo-cytopenia. A second
cytopenia may be associated. In the bone marrow, RT is characterized by
≥10% dysplasia evaluated on at least 30 megakaryocytes.
Dysmegakaryopoiesis may include hypolobated megakaryocytes,
multinucleated megakaryocytes and micromegakaryocytes. The other cell
lineages are not affected, or may display non-significant dysplasia
(<10%).
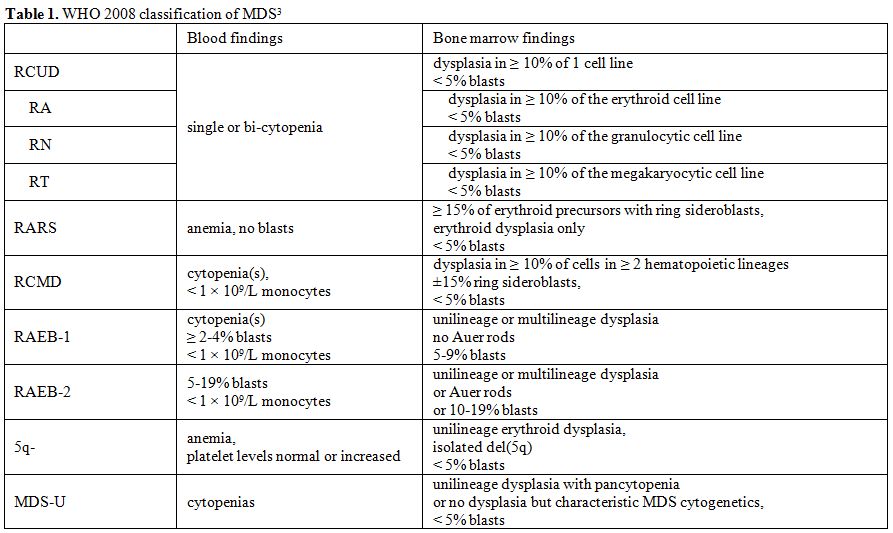 |
Table 1.WHO 2008 classification of MDS[3] |
Differential Diagnosis of RT
Following the exclusion of pseudothrombocytopenia, isolated
thrombo-cytopenia of RT should mainly be distinguished from chronic
immunologic thrombocytopenic purpura (ITP) and familial
thrombocytopenia (Table 2). RT
may be overlooked if bone marrow evaluation is not performed. For this
reason, the bone marrow examination should be performed in any patient
with an isolated confirmed thrombocytopenia above the age of 60
years.[5] A complete workup for thrombocytopenia should be performed
with viral serology, careful medical history with an inquiry about all
possible concomitant medications is needed. Cytogenetic studies are of
clear interest in this distinction, since 20q deletion has frequently
been reported in RT,[6–8] or more rarely other cytogenetic
abnormalities such as del(5q).[9] Furthermore, even in MDS, an
autoimmune destruction of platelets can contribute to thrombocytopenia.
Platelet lifespan studies (and of their sequestration) by radioisotopic
methods can be of interest to analyze the various mechanisms of
thrombocytopenia,[10] and help in therapeutic decision-making.[11]
Anti-platelet autoantibodies have a low sensitivity for the diagnosis
of ITP,[12] and, although they are frequently positive in MDS[13] but
they do not help very much to identify a mixed pathophysiology of
thrombocytopenia.[10] Platelet morphology on blood smears can be
helpful for diagnostic orientation. Giant platelets or
microthrombocytes can be secondary to hereditary thrombocytopenias of
childhood,[14] or associated infections. Associated morphological
abnormalities such as Pelger-Huët bilobed nuclei, or evidence of
dysgranulopoiesis may be suggestive of MDS, whereas abnormal
hematopoietic cells may orient the diagnosis towards a hematologic
malignancy.
|
|
Table
2. Differential diagnosis of RT |
Differential Diagnosis of RN
During
workup for neutropenia, sepsis-associated, drug induced,
hemodialysis-associated, auto-immune, familial or “ethnic” neutropenia,
should be ruled out (Table 3).[15]
Acute or cyclic neutropenias are not consistent with the diagnosis of
RN. Post-infectious neutropenia is mostly seen after viral infections
such as varicella, rubella, influenza, measles, hepatitis, Epstein-Barr
virus or HIV infections, and may sometimes be prolonged. Chronic
moderate isolated neutropenia can be secondary to concomitant
medications (such as clozapine, chlorpromazine, ticlopidine, or
sulfasalazine), auto-immune disorders, and ethnic/familial
neutropenias, characterized by an excessive margination of
granulocytes.[16] Autoimmune neutropenia is mainly associated with
autoimmune diseases such as lupus erythematosus (LE) or rheumatoid
arthritis (Felty’s syndrome),[17] or large granular lymphocyte
leukemia.[18] Neutropenia associated with other cytopenias may be
suggestive of splenomegaly, dietary deficiencies or hematologic
malignancies, and should be explored appropriately.
|
|
Table 3. Differential diagnosis of RN |
Getting Appropriate Material for Morphological Diagnosis
The
diagnosis of MDS, and particularly RCUD, relies on the availability of
high quality bone marrow samples, and on the exclusion of other
diseases. Morphological bone marrow examination, with an iron stain and
cytogenetic study still represents the cornerstone of MDS diagnosis. In
a study comparing bone marrow smears, bone marrow imprints, and bone
marrow biopsies, the best accuracy in 86 MDS was achieved with BM
smears. Interestingly, for patients with a diagnosis of RCUD,
inter-observer accuracy was 100% with BM smears, compared with only 60%
with BM sections.[19]
Distinguishing between RCUD and Borderline Entities
The WHO 2008
classification proposed an entity named idiopathic cytopenia of unknown
significance (ICUS), defined as a condition with less than 10%
dysplastic cells, fewer than 5% blasts in the bone marrow and no
cytogenetic abnormalities.[3,20] These patients most often present with
mild cytopenias, and if the morphologist is unaware of the complete
medical history, the diagnosis might be reported as “abnormalities not
sufficient for the diagnosis of MDS”, when the cytogenetic study is
normal. Differential diagnosis of ICUS, like for RCUD, includes
autoimmune disorders, drug intake, chronic infections, paroxysmal
nocturnal hemoglobinuria, and appropriate explorations need to be
carried out.[21,22] ICUS patients should be followed to document or
exclude hematological evolution to an authentic MDS, most importantly
by repetition of the BM examination with cytogenetic studies if the
cytopenia worsens or if a second cytopenia develops. One should also
bear in mind that dysplastic changes can be seen in up to 9,5% of the
erythroid or granulocytic bone marrow cells in elderly persons and in
smokers.[23]
Another borderline entity is idiopathic dysplasia of
unknown significance (IDUS). This is a rare condition characterized by
no or only mild cytopenias (hemoglobin ≥ 11 g/dL, neutrophils ≥ 1500/mm3, and platelets ≥ 100000/mm3,
associated with > 10% dysplasia in one lineage.[24] Most patients
are asymptomatic young patients referred to the hematology departments
because of macrocytosis or detection of Pseudo-Pelger-Huët
abnormalities. As for ICUS, these patients should have regular
follow-up and repeated diagnostic investigations in case of hematologic
evolution, likely to detect overt MDS. To harmonize the identification
of the minimal changes sufficient for MDS diagnosis, a recent
collaborative work has set up a list of morphological findings with a
high sensitivity/specificity, a high reproducibility and a high
prognostic value of a morphology-based score.[25]
The role of
cytogenetic analysis is important in the identification of RCUD, since
cytogenetic abnormalities will support the diagnosis of MDS as opposed
to ICUS.[21] The most common cytogenetic abnormality in RCUD is
del(20q). In a cytogenetic and mutational study of 305 MDS with
del(20q) whose samples were referred to the MLL Munich Leukemia
laboratory, the most represented diagnostic category was RCUD (133
patients, 43.6%), among which 80.5% had del(20q) as sole
abnormality.[26] High-throughput sequencing can also help in the
diagnosis of MDS in difficult cases by detecting mutations frequently
associated with MDS, including TET-2, ASXL1, SF3B1, SRSF2, RUNX1 and
DNMT3A.[27,28] On the other hand isolated mutations of TET2, ASXL1 or
DNMT3a can be found in elderly apparently healthy persons.[29]
RT and RN are Rare
Apart from RA,
the other RCUD (RT and RN) appear to be rare. In a cytomorphologic
study of 3156 MDS patients from the Düsseldorf MDS registry, the
diagnosis of RCUD was made in 218 (7%). When the Düsseldorf group
revaluated, by WHO 2008 diagnostic criteria, 193 RA according to WHO
2001, the following diagnoses were found: 37 RCUD (19%), 6 MDS-U (3%),
111 RCMD (58%), and 39 5q- syndromes (20%), but a higher proportion of
RCUD (45%) was found in the Japanese registry.[30] To assess the RCUD
and MDS-U categories in 196 patients with less than 5% marrow blasts,
Maassen et al. found 28% RA, 6% RT, 13% RN, 20% patients with no
cytopenia, and 34% patients with bicytopenia.[31] Another retrospective
study on 293 MDS in a single institution identified 5 RN (1.7%) and 6
RT (2.0%) only.[32] Furthermore, in a study combining 228 MDS patients
from the Italian, Düsseldorf and GFM registries presenting with
isolated neutropenia (IT) (< 1.5 x 109/L) or isolated thrombocytopenia (IT) (< 100 x 109/L)
and no anemia, we found only 3 (1%) RT and no RN (Gyan et al.,
submitted). The most frequent diagnosis found in patients with IT or IN
was RCMD (32 %) and RAEB 1 (18 %), which occurred at similar frequency
in both types. Furthermore, during evolution, RT or RN patients often
develop additional cytopenias,[33] which is consistent with the
hypothesis that RT and RN are early presentations of refractory
cytopenias with multilineage dysplasia. This observation further
suggests that real WHO-defined RT and RN are very rare – if they even
exist – whereas MDS patients with only one cytopenia most often show
dysplasia in multiple lineages.
Another important issue adds to
the difficulty of identifying RT and RN. Following publication of the
WHO 2008 classification, a study evaluating the inter-observer
variability in MDS diagnosis found a discrepancy rate of 27%, mostly in
the categories with unilineage dysplasia.[34] This was recently
confirmed by a study of 50 cases of unilineage dysplasia where an
agreement of only 21% was present between observers. Additionally, the
threshold of 2% blasts for the revised IPSS calculation was subject to
a 30% discordance rate.[35] The diagnosis of RT or RN thus remains
difficult and does not to date reflect an international and reliable
consensus on diagnostic criteria. The fact that these extremely rare
entities are at the frontiers of RCMD and ICUS/IDUS may be a likely
explanation.
Prognosis of RT and RN
RCUD is
associated with a more favorable outcome than RCMD.[4,36] In a
comparative study between the Düsseldorf and the Japanese MDS
registries, median overall survival of RCUD and RCMD was 202 months vs.
109 months in the Japanese cohort, respectively, and 142 months vs. 36
months in the German cohort, respectively, with statistical
significance.[30] It is important to try to distinguish RCUD patients
with a high and low risk of evolution to RAEB or AML. In a series of
126 patients with RCUD, RT diagnosis was associated with shorter OS
(median 15.9 months) then RA (median 48.2 months) and RN (median 35.9
months, p<0.001).[33] In another study, the number of RT and RN was
too low to identify a statistically different outcome, but median
survival was 32.5 months and 72 months for RT and RN, respectively.[32]
In a bone marrow flow cytometry analysis of patients with RCUD, Oka et
al. described a lower content of CD19+ or CD10+ lymphoid cells in the
marrow blast region (CD45int/side scatterlow) of patients in whom
circulating blasts appeared during follow-up, compared to patients who
did not experience disease evolution to higher risk MDS or AML.[37]
In
a study evaluating the prognostic value of multilineage dysplasia,
Verburgh et al. found a favorable impact of unilineage dysplasia and of
a single dysplasia.[4] ANC < 500/mm3
has been described as an adverse prognostic factor in Low/Int-1 risk
MDS by two independent teams, with a shorter leukemia-free survival but
surprisingly, no increase in infection-related deaths.[38,39] Beyond
the number or cytopenias, the depth of neutropenia and thrombocytopenia
have been incorporated as prognostic factors into the revised IPSS
prognostic score.[40]
Diagnostic Tools for the Diagnosis of RT and RN
Flow cytometry
(FC) is able to identify aberrant expression patterns of lineage
antigens in the erythroid, granulo-monocytic and lymphoid lineages, and
a collaborative effort has proposed guidelines for the FC recognition
of dysplasia.[41] Since RCUD displays a variable level of dysplastic
cells in one lineage only, FC may be a valuable tool for the
identification of MDS FC signatures. Moreover, a FC score may help to
distinguish MDS from other nonmalignant reactive or secondary
cytopenias,[42,43] and support the diagnosis of IDUS,[24] which may
represent a pre-phase of MDS. The Ogata score, based on a 4-color
analysis of 13 antigens, has shown a sensitivity of 70% and a
specificity of 92% in the whole MDS group.[43] For RCUD, the
sensitivity was 62%, and a specificity reaching 97% in distinguishing
MDS from immune cytopenias.[43] Additionally, a FC score is likely to
bring prognostic information in MDS even when the blast count is below
5%, with a high correlation with transfusion dependency, cytogenetics,
and the IPSS score.[44] In addition, a higher number of aberrantly
expressed antigens detected by FC has been associated with worse
survival.[45] Altogether, the available data support the use of FC as a
diagnostic tool to increase the accuracy of RCUD diagnosis, as well as
for the diagnosis of differential conditions, such as PNH.
Identification
of recurrent mutations with deep sequencing, such as TET-2, ASXL1,
TP53, RAS, SF3B1, SRSF2, RUNX1 and others[46] may help to delineate RN
and RT from other non-MDS conditions. However, as said above,
mutational analysis as a tool for RT or RN diagnosis may be hampered by
the fact that mutations of TET2, DNMT3a and ASXL1 can be seen
individually in elderly healthy persons.[29]
Conclusions
The
challenge of RT and RN resides in the paucity of diagnostic criteria,
the possible overlap with non-MDS disorders, and in the rarity of true
cases of these subgroups of RCUD. Furthermore, isolated refractory
cytopenias are frequent in other MDS categories. The workup of such
patients should include a complete screening for differential
diagnosis, cytogenetic analysis, an expert review of the bone marrow
smears, and the help of emerging diagnostic tools such as flow
cytometry and molecular biology. The clinical relevance of their
distinction from RA or RCMD could be reconsidered in a future revision
of the WHO classification of MDS.
Acknowledgements
Funding. This work was not supported by any academic, associative, or industrial funding.
Author contributions. E.G., F.D., and P.F. analyzed literature data and wrote the paper.
Conflict-of-interest. E.G.
received research grants from Celgene, Janssen, Fresenius Kabi, and
Novartis. P.F. and F. D. declare no conflict-of-interest.
References
- Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the
classification of the myelodysplastic syndromes. Br J Haematol
1982;51:189–199. http://dx.doi.org/10.1111/j.1365-2141.1982.tb08475.x
PMid:6952920
- Jaffe
ES Pathology and Genetics: Tumours of Haematopoietic and Lymphoid
Tissues (World Health Organization Classification of Tumours). 2002
- Swerdlow SH, Campo E, Harris NL, et al. Who Classification of Tumors of Haematopoietic and Lymphoid Tissues. 2008
- Verburgh
E, Achten R, Louw VJ, et al. A new disease categorization of low-grade
myelodysplastic syndromes based on the expression of cytopenia and
dysplasia in one versus more than one lineage improves on the WHO
classification. Leukemia 2007;21:668–677. PMid:17301818

- Provan
D, Stasi R, Newland AC, et al. International consensus report on the
investigation and management of primary immune thrombocytopenia. Blood
2009;115:168–186.
http://dx.doi.org/10.1182/blood-2009-06-225565 PMid:19846889 - Padhi
S, Varghese R, Phansalkar M, Sarangi R Isolated deletion of the long
arm of chromosome 20 [del(20q12)] in myelodysplastic syndrome: a case
report and literature review. Singapore Med J 2013;54:e185–e189.
http://dx.doi.org/10.11622/smedj.2013119 PMid:24068064
- Sashida
G, Takaku T-I, Shoji N, et al. Clinico-hematologic features of
myelodysplastic syndrome presenting as isolated thrombocytopenia: an
entity with a relatively favorable prognosis. Leuk Lymphoma
2003;44:653–658. http://dx.doi.org/10.1080/1042819031000063507
PMid:12769343
- Haase D, Fonatsch C, Freund
M, et al. Cytogenetic findings in 179 patients with myelodysplastic
syndromes. Ann Hematol 1995;70:171–187.
http://dx.doi.org/10.1007/BF01700373 PMid:7748962
- Chang
J, Park C-J, Seo E-J, et al. A case of refractory thrombocytopenia with
5q deletion: myelodysplastic syndrome mimicking idiopathic
thrombocytopenic purpura. Ann Lab Med 2014;34:466–8.
http://dx.doi.org/10.3343/alm.2014.34.6.466 PMid:25368823
PMCid:PMC4215410
- Hebbar M, Kaplan C,
Caulier MT, et al. Low incidence of specific anti-platelet antibodies
detected by the MAIPA assay in the serum of thrombocytopenic MDS
patients and lack of correlation between platelet autoantibodies,
platelet lifespan and response to danazol therapy. Br J Haematol
1996;94:112–115.http://dx.doi.org/10.1046/j.1365-2141.1996.6322262.x PMid:8757517
- Sarpatwari
A, Provan D, Erqou S, et al. Autologous 111 In-labelled platelet
sequestration studies in patients with primary immune thrombocytopenia
(ITP) prior to splenectomy: a report from the United Kingdom ITP
Registry. Br J Haematol 2010;151:477–487.
http://dx.doi.org/10.1111/j.1365-2141.2010.08377.x PMid:20950403
- Chan
H, Moore JC, Finch CN, et al. The IgG subclasses of platelet-associated
autoantibodies directed against platelet glycoproteins IIb/IIIa in
patients with idiopathic thrombocytopenic purpura. Br J Haematol
2003;122:818–824. http://dx.doi.org/10.1046/j.1365-2141.2003.04509.x
PMid:12930395
- Chabannon C, Molina L,
Pégourié-Bandelier B, et al. A Review of 76 Patients with
Myelodysplastic Syndromes Treated with Danazol. Cancer 1994;94:3073–80.
http://dx.doi.org/10.1002/1097-0142(19940615)73:12<3073::AID-CNCR2820731228>3.0.CO;2-# - Patel
PD, Samanich JM, Mitchell WB, Manwani D A unique presentation of
Wiskott-Aldrich syndrome in relation to platelet size. Pediatr Blood
Cancer 2011;56:1127–1129. http://dx.doi.org/10.1002/pbc.22920
PMid:21488158
- Gibson C, Berliner N How we evaluate and treat neutropenia in adults. Blood 2014;124:1251–8; quiz 1378. http://dx.doi.org/10.1182/blood-2014-02-482612 PMid:24869938
- Bishop
CR, Rothstein G, Ashenbrucker HE, Athens JW Leukokinetic studies. XIV.
Blood neutrophil kinetics in chronic, steady-state neutropenia. J Clin
Invest 1971;50:1678–1689. http://dx.doi.org/10.1172/JCI106657
PMid:5097574 PMCid:PMC442068
- Campion G,
Maddison PJ, Goulding N, et al. The Felty syndrome: a case-matched
study of clinical manifestations and outcome, serologic features, and
immunogenetic associations. Medicine (Baltimore) 1990;69:69–80.
http://dx.doi.org/10.1097/00005792-199069020-00001
- Saway
PA, Prasthofer EF, Barton JC Prevalence of granular lymphocyte
proliferation in patients with rheumatoid arthritis and neutropenia. Am
J Med 1989;86:303–307.
http://dx.doi.org/10.1016/0002-9343(89)90300-8 - Gong
X, Lu X, Wu X, et al. Role of bone marrow imprints in haematological
diagnosis: a detailed study of 3781 cases. Cytopathology 2012;23:86–95.
http://dx.doi.org/10.1111/j.1365-2303.2010.00825.x PMid:21129051
- Wimazal
F, Fonatsch C, Thalhammer R, et al. Idiopathic cytopenia of
undetermined significance (ICUS) versus low risk MDS: the diagnostic
interface. Leuk Res 2007;31:1461–8.
http://dx.doi.org/10.1016/j.leukres.2007.03.015 PMid:17507091
- Giagounidis
A, Haase D Morphology, cytogenetics and classification of MDS. Best
Pract Res Clin Haematol 2013;26:337–53.
http://dx.doi.org/10.1016/j.beha.2013.09.004 PMid:24507811
- Valent
P, Horny H, Bennett JM, et al. Definitions and standards in the
diagnosis and treatment of the myelodysplastic syndromes : Consensus
statements and report from a working conference. Leuk Res. 2007 Jun; 31(6); 727-36 PMid: 17257673
- Fernández-Ferrero
S, Ramos F Dyshaemopoietic bone marrow features in healthy subjects are
related to age. Leuk Res 2001;25:187–189.
http://dx.doi.org/10.1016/S0145-2126(00)00109-0
- Valent
P, Horny H-P Minimal diagnostic criteria for myelodysplastic syndromes
and separation from ICUS and IDUS: update and open questions. Eur J
Clin Invest 2009;39:548–553.
http://dx.doi.org/10.1111/j.1365-2362.2009.02151.x PMid:19453651
- Della
Porta MG, Travaglino E, Boveri E, et al. Minimal morphological criteria
for defining bone marrow dysplasia: a basis for clinical implementation
of WHO classification of myelodysplastic syndromes. Leukemia
2015;29:66–75. http://dx.doi.org/10.1038/leu.2014.161 PMid:24935723
- Bacher
U, Haferlach T, Schnittger S, et al. Investigation of 305 patients with
myelodysplastic syndromes and 20q deletion for associated cytogenetic
and molecular genetic lesions and their prognostic impact. Br J
Haematol 2014;164:822–33. http://dx.doi.org/10.1111/bjh.12710
PMid:24372512
- Bejar R, Stevenson K,
Abdel-Wahab O, et al. Clinical effect of point mutations in
myelodysplastic syndromes. N Engl J Med 2011;364:2496–506.
http://dx.doi.org/10.1056/NEJMoa1013343 PMid:21714648 PMCid:PMC3159042
- Bejar
R, Stevenson KE, Caughey B a, et al. Validation of a prognostic model
and the impact of mutations in patients with lower-risk myelodysplastic
syndromes. J Clin Oncol 2012;30:3376–82.
http://dx.doi.org/10.1200/JCO.2011.40.7379 PMid:22869879
PMCid:PMC3438234
- Jaiswal
S, Fontanillas P, Flannick J, et al. Age-Related Clonal Hematopoiesis
Associated with Adverse Outcomes. N Engl J Med 2014 Dec
25;371(26):2488-98. PMid: 25426837
- Matsuda
A, Germing U, Jinnai I, et al. Differences in the distribution of
subtypes according to the WHO classification 2008 between Japanese and
German patients with refractory anemia according to the FAB
classification in myelodysplastic syndromes. Leuk Res
2010;34:974–80. http://dx.doi.org/10.1016/j.leukres.2009.11.015
PMid:20022110
- Maassen
A, Strupp C, Giagounidis A, et al. Validation and proposals for a
refinement of the WHO 2008 classification of myelodysplastic syndromes
without excess of blasts. Leuk Res 2013;37:64–70.
http://dx.doi.org/10.1016/j.leukres.2012.09.021 PMid:23122806
- Marinier
DE, Mesa H, Rawal A, Gupta P Refractory cytopenias with unilineage
dysplasia: a retrospective analysis of refractory neutropenia and
refractory thrombocytopenia. Leuk Lymphoma 2010;51:1923–6.
http://dx.doi.org/10.3109/10428194.2010.506568 PMid:20919862
- Breccia
M, Latagliata R, Cannella L, et al. Refractory cytopenia with
unilineage dysplasia: analysis of prognostic factors and survival in
126 patients. Leuk Lymphoma 2010;51:783–8.
http://dx.doi.org/10.3109/10428191003682759 PMid:20302387
- Font
P, Loscertales J, Benavente C, et al. Inter-observer variance with the
diagnosis of myelodysplastic syndromes (MDS) following the 2008 WHO
classification. Ann Hematol 2013;92:19–24.
http://dx.doi.org/10.1007/s00277-012-1565-4 PMid:22948274
- Font
P, Loscertales J, Soto C, et al. Interobserver variance in
myelodysplastic syndromes with less than 5 % bone marrow blasts:
unilineage vs. multilineage dysplasia and reproducibility of the
threshold of 2 % blasts. Ann Hematol 2014 Nov 13 PMid:25387664
- Cazzola
M Risk assessment in myelodysplastic syndromes and
myelodysplastic/myeloproliferative neoplasms. Haematologica
2011;96:349–352. http://dx.doi.org/10.3324/haematol.2010.030023
PMid:21357714 PMCid:PMC3046263
- Oka S,
Muroi K, Fujiwara S, et al. Prediction of Progression from Refractory
Cytopenia with Unilineage Dysplasia by Analysis of Bone Marrow Blast
Cell Composition. J Clin Exp Hematop 2012;52:63–66.
http://dx.doi.org/10.3960/jslrt.52.63 PMid:22706533
- Cordoba
I, Gonzalez-Porras JR, Such E, et al. The degree of neutropenia has a
prognostic impact in low risk myelodysplastic syndrome. Leuk Res
2012;36:287–292.
http://dx.doi.org/10.1016/j.leukres.2011.10.025 PMid:22133642 - Breccia
M, Loglisci G, Salaroli A, et al. Neutropenia at baseline could
indicate poor prognosis in low/intermediate risk myelodysplastic
syndrome patients. Leuk Res 2012;36:546–547.
http://dx.doi.org/10.1016/j.leukres.2012.01.003 PMid:22309889
- Greenberg
PL, Tuechler H, Schanz J, et al. Revised international prognostic
scoring system for myelodysplastic syndromes. Blood 2012;120:2454–65.
http://dx.doi.org/10.1182/blood-2012-03-420489 PMid:22740453
- Westers
TM, Ireland R, Kern W, et al. Standardization of flow cytometry in
myelodysplastic syndromes: a report from an international consortium
and the European LeukemiaNet Working Group. Leukemia 2012;26:1730–41.http://dx.doi.org/10.1038/leu.2012.30 PMid:22307178
- Ogata
K, Kishikawa Y, Satoh C, et al. Diagnostic application of flow
cytometric characteristics of CD34+ cells in low-grade myelodysplastic
syndromes. Blood 2006;108:1037–44.
http://dx.doi.org/10.1182/blood-2005-12-4916 PMid:16574954
- Della
Porta MG, Picone C, Pascutto C, et al. Multicenter validation of a
reproducible flow cytometric score for the diagnosis of low-grade
myelodysplastic syndromes: results of a European LeukemiaNET study.
Haematologica 2012;97:1209–17.
http://dx.doi.org/10.3324/haematol.2011.048421 PMid:22315489
PMCid:PMC3409819
- Van de Loosdrecht AA,
Westers TM, Westra AH, et al. Identification of distinct prognostic
subgroups in low- and intermediate-1-risk myelodysplastic syndromes by
flow cytometry. Blood 2008;111:1067–77.
http://dx.doi.org/10.1182/blood-2007-07-098764 PMid:17971483
- Kern
W, Haferlach C, Schnittger S, Haferlach T Clinical utility of
multiparameter flow cytometry in the diagnosis of 1013 patients with
suspected myelodysplastic syndrome: correlation to cytomorphology,
cytogenetics, and clinical data. Cancer 2010;116:4549–63.
http://dx.doi.org/10.1002/cncr.25353 PMid:24144312
- Kohlmann
A, Bacher U, Schnittger S, Haferlach T Perspective on how to approach
molecular diagnostics in acute myeloid leukemia and myelodysplastic
syndromes in the era of next-generation sequencing. Leuk Lymphoma
2014;55:1725–1734. http://dx.doi.org/10.3109/10428194.2013.856427
PMid:24144312
[TOP]

