Importance of Classical Morphology in the Diagnosis of Myelodysplastic Syndrome
Rosangela Invernizzi, Federica Quaglia and Matteo Giovanni Della Porta
Department of Internal Medicine, University of Pavia, IRCCS Policlinico San Matteo Foundation, Pavia, Italy
Corresponding author:Rosangela Invernizzi, Clinica
Medica 3, Fondazione IRCCS Policlinico San Matteo, Viale Golgi 19,
27100 Pavia, Italy. Tel.: +39 0382 502160. Fax: +39 0382 526223.
E-mail:
r.invernizzi@smatteo.pv.it
Published: May 1, 2015
Received: March 25, 2015
Accepted: April 23, 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015035, DOI
10.4084/MJHID.2015.035
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(http://creativecommons.org/licenses/by/2.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Myelodysplastic syndromes (MDS) are
hematopoietic stem cell disorders characterized by dysplastic,
ineffective, clonal and neoplastic hematopoiesis. MDS represent a
complex hematological problem: differences in disease presentation,
progression and outcome have necessitated the use of classification
systems to improve diagnosis, prognostication, and treatment selection.
However, since a single biological or genetic reliable diagnostic
marker has not yet been discovered for MDS, quantitative and
qualitative dysplastic morphological alterations of bone marrow
precursors and peripheral blood cells are still fundamental for
diagnostic classification. In this paper, World Health Organization
(WHO) classification refinements and current minimal diagnostic
criteria proposed by expert panels are highlighted, and related
problematic issues are discussed. The recommendations should facilitate
diagnostic and prognostic evaluations in MDS and selection of patients
for new effective targeted therapies. Although, in the future,
morphology should be supplemented with new molecular techniques, the
morphological approach, at least for the moment, is still the
cornerstone for the diagnosis and classification of these disorders. |
Introduction
Myelodysplastic syndromes (MDS) are clonal hematopoietic stem cell
disorders characterized by dysplastic, ineffective and neoplastic
hematopoiesis. The risk of evolution to acute myeloid leukemia (AML) is
variable, and the clinical outcome is greatly heterogeneous. Therefore,
MDS constitute a complex hematological problem that gives rise to
difficulties in diagnosis and therapeutic decision-making.[1]
Since a single biological or genetic reliable diagnostic marker has not
yet been discovered for MDS, quantitative and qualitative dysplastic
alterations of bone marrow precursors and of peripheral blood cells are
still fundamental for diagnostic classifications.[2]
While the detection of increased blast cells may facilitate the
diagnosis in advanced forms, in the early forms, especially with modest
morphological abnormalities, a correct diagnosis is based mainly on the
exclusion of other diseases. Some bone marrow failure syndromes can
indeed mimic the MDS,[3,4] and the formulation of a correct diagnosis is fundamental for both prognostic evaluation and therapeutic approach.
In
this review the meaning of morphology in MDS is examined; World Health
Organization (WHO) classification refinements and current minimal
morphological criteria for defining dysplastic involvement are
highlighted, and several problematic issues are discussed.
Diagnosis and Classification
Currently, the reference classification of MDS is still the WHO classification, published in 2001 and updated in 2008.[5-7]
This classification system is based on an integrated multidisciplinary
approach that uses all available information (morphology,
cytochemistry, immunophenotype, genetics, clinical aspects) to define
biologically homogeneous and clinically relevant entities, that can be
usefully applied in clinical practice. The WHO classification improved
the prognostic value of the former FAB classification,[8]
by recognizing more specific categories on the basis of cytogenetic
findings as well as cellular morphology and allowed to evaluate more
accurately emerging therapies that target specific genetic
abnormalities.[9,10]
The suspicion of MDS arises
on the basis of an abnormal blood count with evidence of different
combinations of anemia, neutropenia, and thrombocytopenia in an
appropriate clinical setting. Anemia is often macrocytic, associated
with a significantly reduced reticulocyte count. Obviously, all causes
of reactive cytopenia/dysplasia should be excluded as well as other
clonal stem cell disorders and congenital abnormalities (Table 1).
The minimal diagnostic criteria for MDS include the presence of bone
marrow specific alterations, i.e. one or more of the following
characteristics: dysplasia in at least 10% of at least one of the major
hematopoietic lineages, at least 15% ring sideroblasts or 5-19%
myeloblasts in bone marrow smears. Certain chromosomal abnormalities
detected by conventional karyotyping or FISH in the presence of a
refractory cytopenia, but no morphological evidence of dysplasia, are
considered presumptive evidence for MDS (Table 2).[6,11,12]
Since morphology alone is often insufficient to reach a final
diagnosis, it should be integrated, but not replaced, by other
investigations such as flow cytometry, molecular studies, in vitro
culture of hematopoietic progenitors.[2,13,14] However, if multilineage dysplasia, chromosomal aberrations and proof of clonality are absent, the diagnosis may be difficult.
On
the basis of the proportion of peripheral blood and bone marrow blasts,
defined by a morphological examination, two broad categories of MDS are
recognized: forms with <2% peripheral blood blasts and <5% bone
marrow blasts (lower risk subtypes), including refractory cytopenias
with unilineage dysplasia (RCUD), refractory anemia with ring
sideroblasts (RARS), refractory cytopenia with multilineage dysplasia
(RCMD), myelodysplastic syndrome-unclassified (MDS-U) and MDS
associated with isolated del(5q), and forms characterized by at least
2% peripheral blood blasts and/or at least 5% bone marrow blasts
(higher risk subtypes), including refractory anemia with excess
blasts-1 (RAEB-1) and RAEB-2 (Table 3).
Chronic myelomonocytic leukemia (CMML), characterized by persistent
monocytosis, is placed into the category of
myelodysplastic/myeloproliferative neoplasms together with atypical
chronic myeloid leukemia (ACML), BCR-ABL1 negative, juvenile
myelomonocytic leukemia (JMML) and refractory anemia with ring
sideroblasts associated with marked thrombocytosis (RARS-T), which is
still a provisional entity.[15,16]
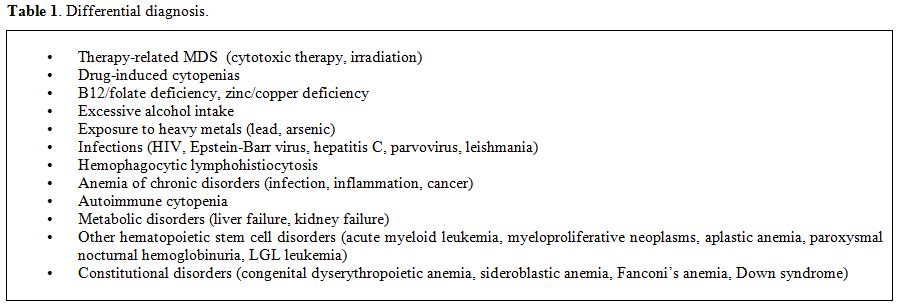 |
Table 1. Differential diagnosis. |
 |
Table 2. Recurrent chromosomal abnormalities and their frequency in MDS.[6] |
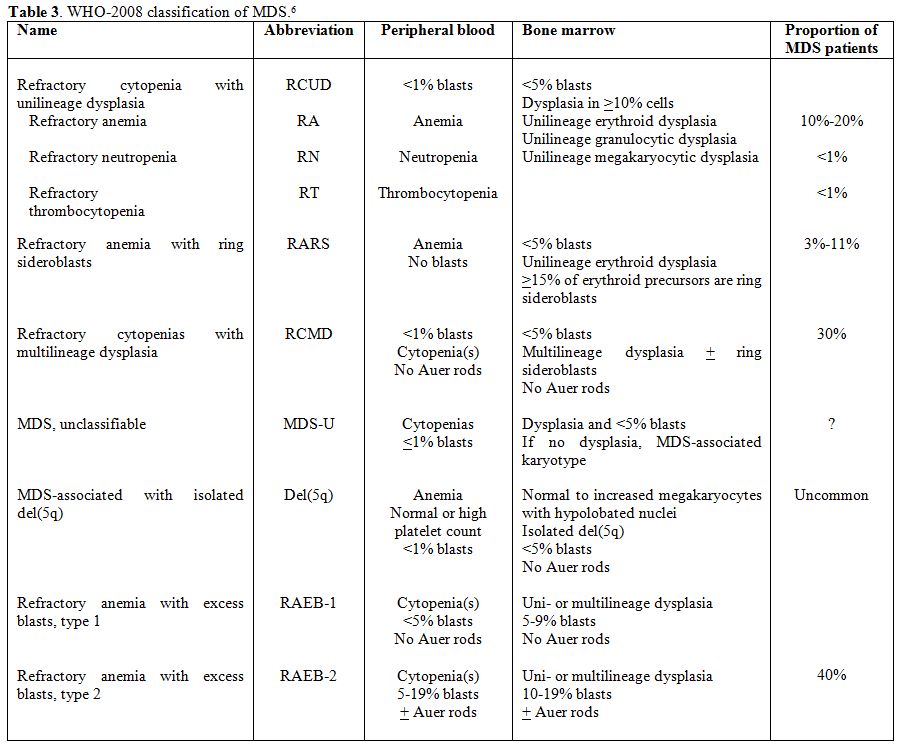 |
Table 3. WHO-2008 classification of MDS.[6] |
Morphological Features
The diagnosis of MDS is mainly based on morphological findings of peripheral blood and bone marrow.[17-20]
Morphological examination has several advantages: it is a simple,
technically easy, not expensive method, which gives quick results;
moreover, it has prognostic importance, and should be supplemented, but
not replaced, by other tests. The morphological examination requires
peripheral blood smear, bone marrow aspirate, and bone marrow trephine
biopsy.
Peripheral blood and bone marrow specimens should be
collected before any definitive therapy. No case of MDS should be
reclassified while the patient is on growth factor therapy. Since
prolonged exposure to anticoagulants can cause artifacts, the slides
for the assessment of dysplasia should be made from freshly obtained
specimens. On bone marrow aspirate smears and/or biopsy touch
preparations, MGG or similar staining and iron staining could possibly,
but not necessarily, be supplemented by cytochemical dyes to identify
bone marrow cells and maturation stages: myeloperoxidase and Sudan
black detect myeloid cells by staining cytoplasmic granular contents
and better identify Auer rods, periodic acid-Schiff detects lymphocytic
cells and certain abnormal erythroid cells by staining cytoplasmic
glycogen, esterases distinguish myelocytic from monocytic maturation
stages. On bone marrow aspirates, the cellularity should be enough to
perform a 500 cells differential count, whereas, on peripheral blood
smears, a differential count of 200-cell leukocyte is recommended. The
blood and marrow smears should be examined for the percentages of
blasts, dysplastic cells and ring sideroblasts. At least 100
erythroblasts, 100 granulocytic cells, and 30 megakaryocytes should be
evaluated.[6]
Assessment of Blasts
An
increase of blast cells has to be considered as a sign of
myelodysplasia. An International Working Group on Morphology of MDS
(IWGM-MDS) of hematopathologists and hematologists, in order to improve
diagnostic accuracy, agreed on some recommendations for the definition
and enumeration of blasts.[21] First, blast
percentage should be determined by visual inspection. Flow cytometric
assessment of CD34+ cells is not recommended, as not all blasts express
CD34 antigen and flow cytometry analysis can be affected by peripheral
blood dilution of the sample.[6] Myeloblasts,
monoblasts, promonocytes, and megakaryoblasts should be counted as
blasts; dysplastic megakaryocytes and proerytrhoblasts must not be
counted as blasts except in the rare cases of “pure” acute
erythroleukemia. Blast lineage could be assessed by flow cytometry,
cytochemistry or immunocytochemistry. In severely cytopenic patients,
buffy coat smears of peripheral blood may facilitate performing the
differential count. The diagnostic and prognostic importance of an
accurate count of the blasts should be emphasized.[22]
According to WHO, 20% bone marrow or peripheral blood blasts is the
threshold for the diagnosis of AML, whereas, according to the revised
International Prognostic Scoring System, the forms with <2% bone
marrow blasts are to be distinguished from those with >2% blasts, as
they have a better prognosis.[23] Moreover, they were
included in the MDS-U subtype patients with 1% blasts in the blood and
fewer than 5% blasts in the bone marrow.[24]Blasts
have variable size, ovoid or irregularly outlined nuclei with loose
chromatin pattern and variable number of nucleoli, basophilic
cytoplasm, with the absence of an evident Golgi zone. They are defined
as granular or agranular and may contain Auer rods, whose presence
allows the automatic diagnosis of RAEB-2. Myeloblasts showing strongly
basophilic cytoplasm could be misinterpreted as immature erythroid
precursors. Erythroid precursors, however, have relatively mature
clumped chromatin and are often larger than myeloblasts at early
stages. Granular blasts should be distinguished from normal or
dysplastic promyelocytes. Promyelocytes are usually characterized by a
well recognizable Golgi zone; dysplastic promyelocytes, however, are
often hyper- or hypogranulated and may present a less evident Golgi
area than normal promyelocytes (Figure 1).It
is worth noting that in the forms with recurrent cytogenetic
abnormalities, such as t(8;21)(q22;q22), inv(16)(p13.1q22) or
t(16;16)(p13.1;q22) and t(15;17)(q22;q12) the diagnosis of AML should
be made even with fewer than 20% bone marrow blasts. These forms are
considered clinical-pathological-genetic entities with peculiar
features.
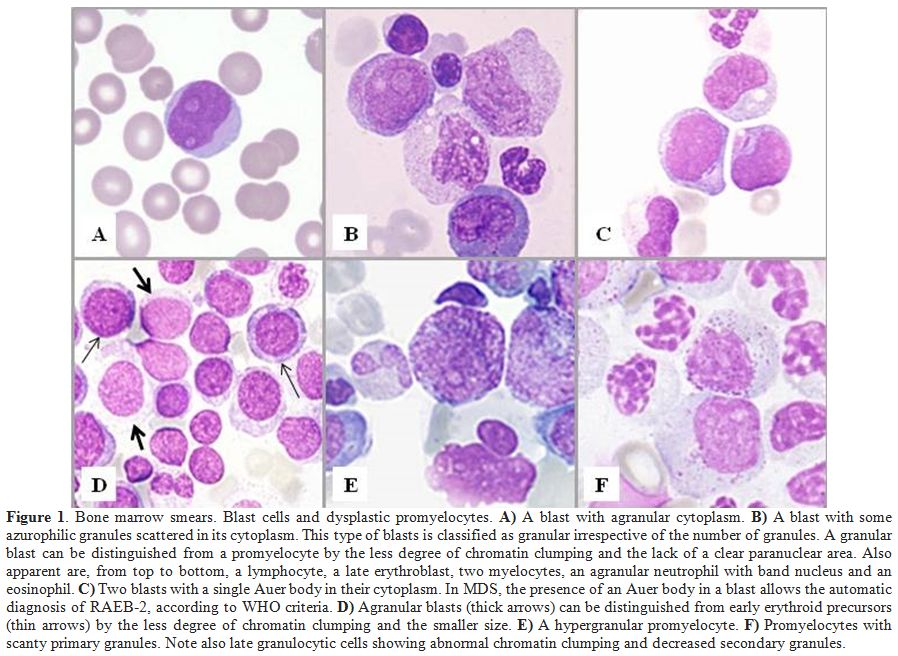 |
Figure 1. Bone marrow smears. Blast cells and dysplastic promyelocytes. A) A blast with agranular cytoplasm. B)
A blast with some azurophilic granules scattered in its cytoplasm. This
type of blasts is classified as granular irrespective of the number of
granules. A granular blast can be distinguished from a promyelocyte by
the less degree of chromatin clumping and the lack of a clear
paranuclear area. Also apparent are, from top to bottom, a lymphocyte,
a late erythroblast, two myelocytes, an agranular neutrophil with band
nucleus and an eosinophil. C)
Two blasts with a single Auer body in their cytoplasm. In MDS, the
presence of an Auer body in a blast allows the automatic diagnosis of
RAEB-2, according to WHO criteria. D)
Agranular blasts (thick arrows) can be distinguished from early
erythroid precursors (thin arrows) by the less degree of chromatin
clumping and the smaller size. E) A hypergranular promyelocyte. F)
Promyelocytes with scanty primary granules. Note also late granulocytic
cells showing abnormal chromatin clumping and decreased secondary
granules. |
Assessment of Monocytic Cells
The IWGM-MDS also defined the different maturation stages of monocytic cells.[25]
A promonocyte differs from a monoblast for the irregular nuclear
outline but has similar immature chromatin pattern; it is a blast
equivalent and should be counted as such. Thus, the distinction between
a monoblast and a promonocyte has no practical importance as they are
regarded as having the same significance. An atypical/immature monocyte
is characterized by a more condensed chromatin pattern and less evident
nucleoli, but its distinction from a promonocyte can be very difficult.
Monocytic cells can be better identified with the nonspecific esterase
reaction. Monoblasts and promonocytes, however, are rare in MDS, and
their presence is rather indicative of CMML or AML with monocytic
differentiation.
Assessment of Dysplasia
The
precise recognition and quantification of dysplasia is critical for a
correct application of the WHO classification for the following main
reasons: WHO proposal introduced uni- versus multilineage dysplasia as
a diagnostic criterion in MDS with fewer than 5% bone marrow blasts,
increasing the prognostic value of the classification;[26,27]
the finding, in an appropriate clinical setting, of dysplastic
morphological alterations in at least 10% of the cells of at least one
myeloid lineage is the most important criterion for the diagnosis of
RCUD. This subtype is rather difficult to recognize because of the
minimal percentage of blasts in the bone marrow and the low incidence
of chromosome abnormalities.[28,29]
The dysplastic abnormalities of the cell nucleus and/or cytoplasm to be taken into account are listed in Table 4 and illustrated in Figure 2.
Whereas variable degrees of dyserythropoiesis are commonly observed in
various hematological, as well as non-hematological disorders, the
morphological abnormalities of the granulocytic and megakaryocytic
series are more specific and significant for the diagnosis. However, no
single morphological finding is diagnostic for MDS, that sometimes
remains a diagnosis of exclusion.
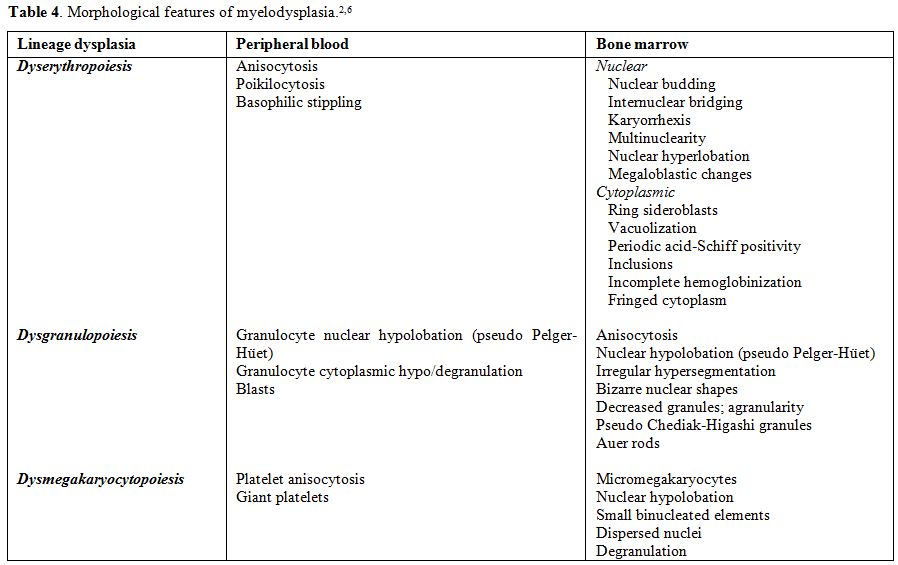 |
Table 4. Morphological features of myelodysplasia.[2,6] |
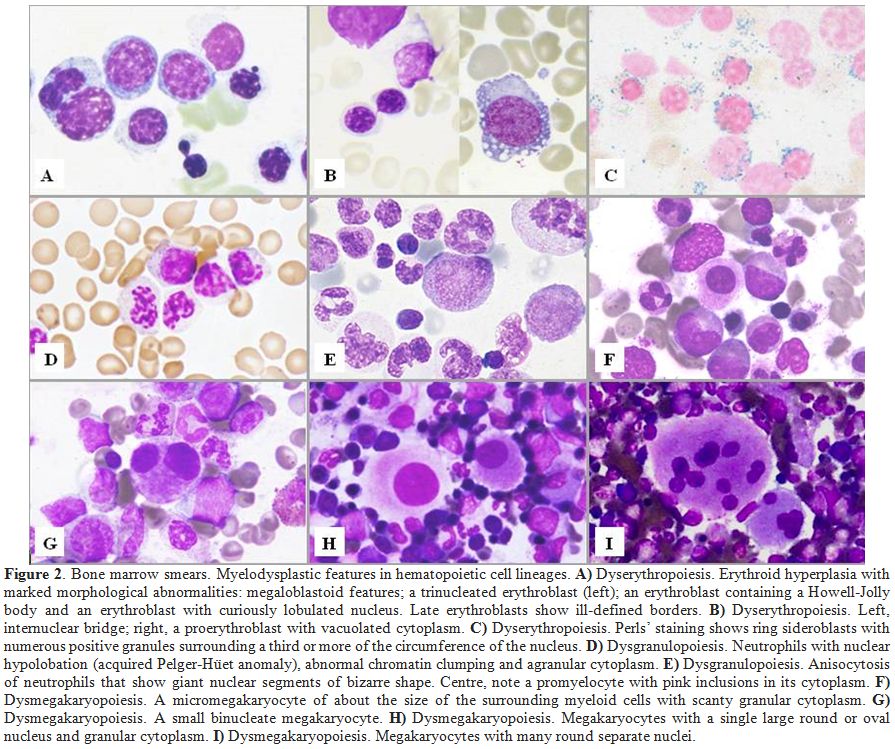 |
Figure 2. Bone marrow smears. Myelodysplastic features in hematopoietic cell lineages. A)
Dyserythropoiesis. Erythroid hyperplasia with marked morphological
abnormalities: megaloblastoid features; a trinucleated erythroblast
(left); an erythroblast containing a Howell-Jolly body and an
erythroblast with curiously lobulated nucleus. Late erythroblasts show
ill-defined borders. B) Dyserythropoiesis. Left, internuclear bridge; right, a proerythroblast with vacuolated cytoplasm. C) Dyserythropoiesis.
Perls’ staining shows ring sideroblasts with numerous positive granules
surrounding a third or more of the circumference of the nucleus. D)
Dysgranulopoiesis. Neutrophils with nuclear hypolobation (acquired
Pelger-Hüet anomaly), abnormal chromatin clumping and agranular
cytoplasm. E)
Dysgranulopoiesis. Anisocytosis of neutrophils that show giant nuclear
segments of bizarre shape. Centre, note a promyelocyte with pink
inclusions in its cytoplasm. F) Dysmegakaryopoiesis. A micromegakaryocyte of about the size of the surrounding myeloid cells with scanty granular cytoplasm. G) Dysmegakaryopoiesis. A small binucleate megakaryocyte. H) Dysmegakaryopoiesis. Megakaryocytes with a single large round or oval nucleus and granular cytoplasm. I) Dysmegakaryopoiesis. Megakaryocytes with many round separate nuclei. |
Dysgranulopoiesis
Hypo-agranularity
of neutrophils is considered a highly specific dysplastic feature;
usually, it is more evident in peripheral blood smears and better
assessable with Sudan black or peroxidase reaction.[30]
According to the recently published IWGM-MDS proposal for refining the
definition of dysgranulopoiesis, neutrophils could be recognized as
dysplastic in the presence of one of the following morphological
features: at least 2/3 reduction of the content of granules, pseudo
Pelger anomaly of the nucleus, not-Pelger abnormalities of nuclear
segmentation, macropolycytes, abnormal clumping of the chromatin and
the presence of more than four nuclear projections.[31]
Dysmegakaryopoiesis
Micromegakaryocytes
are highly specific for dysmegakaryopoiesis, but there is still no
consensus on their definition. It is recommended to consider as
micromegakaryocyte a megakaryocyte of about the size of the surrounding
myeloid cells, with scanty granular cytoplasm. Other categories of
dysplastic megakaryocytes are illustrated in Figure 2:
medium sized megakaryocytes with a single, ovoid, eccentric nucleus,
pathognomonic of the 5q- syndrome; or with 2 nuclei of similar or
different size, close one to another; mature megakaryocytes with
numerous small round separated nuclei.
Dyserythropoiesis and Ring Sideroblasts
As
already mentioned, morphological abnormalities of erythroid cells, as
megaloblastic features and non-round nuclei, are commonly observed in
many hematological as well as non-hematological disorders, and have a
low diagnostic power. Only ring sideroblasts are considered highly
specific dysplastic changes. Recommendations for the definition of ring
sideroblasts have been provided by the IWGM-MDS.[21]
They are defined as erythroblasts characterized by at least 5 siderotic
granules surrounding at least a third of the nuclear circumference, as
a result of the iron accumulation within mitochondria, including some
deposited as mitochondrial ferritin.[32] A high
microscopic magnification is necessary to distinguish these granules.
In some cases, ring sideroblasts constitute <15% of erythroid
precursors: in such cases the diagnosis of MDS with RS would not be
possible. However, ring sideroblasts would be considered as unequivocal
expression of dyserythropoiesis. On the contrary, type 1 sideroblasts,
characterized by <5 siderotic granules, are also present in the
normal bone marrow, whereas type 2 sideroblasts show at least five
non-perinuclear siderotic granules. In type 1 and type 2 sideroblasts,
siderotic granules represent aggregates of ferritin molecules that are
stored in lysosomes.
Erythroid Predominant MDS (MDS-E)
Recently,
the term of MDS-E or MDS Ery has been proposed to indicate forms of MDS
with marked erythroid hyperplasia. Marked erythroid hyperplasia (50% or
greater) with or without left-shifted erythroid maturation can be seen
in approximately 15% of patients with MDS and is often associated with
the presence of ring sideroblasts.[33] In this
condition, the count of blasts should be performed on non-erythroid
cells, excluding lymphocytes and plasma cells, and for the diagnosis of
MDS, it should be lower than 20%. There is an ongoing discussion
regarding the subclassification of MDS-E since low-risk MDS such as RA
may be upgraded to a higher risk category if blasts were calculated as
a percentage of non-erythroid cells.[34,35] Thus,
once the diagnosis of MDS is established, blast enumeration should be
derived from all nucleated marrow cells. On the other hand, similar
demographic and laboratory characteristics were reported in MDS-E in
comparison with MDS cases with less than 50% erythroid precursors.
Problematic Issues
The
problems in the morphological diagnosis of MDS are mainly due to the
non-specificity of dysplastic changes. Morphological alterations may be
observed even in healthy bone marrow and in patients with non-clonal
disorders; moreover, poor quality of marrow specimens and various
artifacts may cause misinterpretation. On the other hand, recent
studies have demonstrated discrepancy in morphological diagnosis in
rather high proportions of cases as well as low reproducibility of the
WHO 2008 criteria. Unfortunately, unanimous agreement on the type of
morphological alterations that characterize MDS and on the threshold to
be considered is still missing.[36]Several
studies have addressed the impact of the single morphological
abnormalities and the degree of dysplasia on prognosis, and grading
systems have been proposed to increase the diagnostic accuracy of MDS.[26,29,37-39]A
Japanese-German study concerning patients with MDS without excess
blasts, 5q-syndrome excluded, showed the adverse prognostic
significance of three parameters: the presence of at least 10% of
micromegakaryocytes, dysmegakaryocytopoiesis > 40% and
dysgranulopoiesis >10%. The authors suggested using these threshold
values for the identification of multilineage dysplasia.[26]
In a very detailed cytomorphological study on 3156 patients of the
Düsseldorf register, no differences were observed in the frequency of
dysplastic changes in relation to the WHO subtype of MDS and no single
morphological abnormality had prognostic significance. Also, these
authors recommended using 40% as a threshold value for
dysmegakaryopoiesis.[40]On
the other hand, dysplastic features may also be observed in the normal
bone marrow, as reported by some authors in the late '90s.[41,42]
A more recent work has shown dysgranulopoiesis >10% in 46% of the
bone marrow aspirates from 120 healthy donors, with multilineage
dysplasia in 26% of the subjects; however, the counting of cells with
pseudo Pelger anomaly and micromegakaryocytes did not exceed 10% and
total dysmegakaryopoiesis 40%. The concordance rate between the four
investigators was modest in dysgranulopoiesis but poor in
dyserythropoiesis and dysmegakaryopoiesis; raising the threshold from
10% to an arbitrary 20% for all lineages led to a higher concordance
rate. In conclusion, the 10% cut-off for dyshematopoietic cells is
questionable in patients without cytopenia and should be revised for
future consensus recommendations.[43] Interestingly,
another study showed discordance in the morphological diagnosis between
the reference and peripheral centers in 12% of 915 MDS cases referred
to MD Anderson Cancer Center, with a majority reclassified as having
higher-risk disease with implications for therapy selection and
prognosis calculation.[44] Finally, a Spanish group
showed a poor reproducibility of the WHO criteria for cases with 5-9%
marrow blasts or up to 1% circulating blasts as well as for the
percentage of dysplastic erythroid cells.[45]It
should be emphasized the possible role of the barriers that can hinder
a correct diagnostic definition: poor quality of marrow specimen, lack
of clinical information, lack of available cytogenetic results,
inter-observer variability in the assessment of dysplasia.[46]
The application of well codified reproducible criteria could allow a
more objective morphological evaluation, and thus a correct
implementation of the WHO classification.
Morphological Score
In
a retrospective study of 318 patients with MDS, a group of patients
with other types of non-clonal cytopenias used as pathological
controls, and a group of normal subjects, bone marrow hematopoietic
cells were carefully examined and classified according to their nuclear
and cytoplasmic morphological alterations to identify minimal
reproducible morphological criteria to define marrow dysplasia and to
evaluate the prognostic relevance of the degree of dysplasia.[47]
The most discriminant morphological features for dyserythropoiesis,
dysgranulopoiesis and dysmegakaryopoiesis were identified. For each
parameter, the optimal cut-off value to discriminate between MDS and
controls and the weight in the recognition of BM dysplasia were
determined to develop a score for defining minimal morphological
criteria for MDS (Table 5).
This score showed high sensitivity and specificity (>90%). The
diagnostic value and reproducibility of the proposed criteria were
independently validated (Table 6).
There was a high inter-operator agreement, especially for patients with
excess blasts. Very interestingly, erythroid score value did not
significantly affect survival while granulocytic or megakaryocytic
score levels had a significant effect on overall survival. Also,
multilineage dysplasia showed an independent unfavorable prognostic
value. Moreover, a close association was found between ring
sideroblasts and SF3B1 mutations and between severe granulocytic dysplasia and mutations of ASXL1, RUNX1, TP53 and SRSF2 genes.In
conclusion, this morphological score improving the objectivity and
reproducibility of microscopic analysis might be very useful in the
work-up of patients with suspected MDS. On the other hand, prognostic
systems including the evaluation of the degree of bone marrow dysplasia
should be adopted for clinical decision-making.
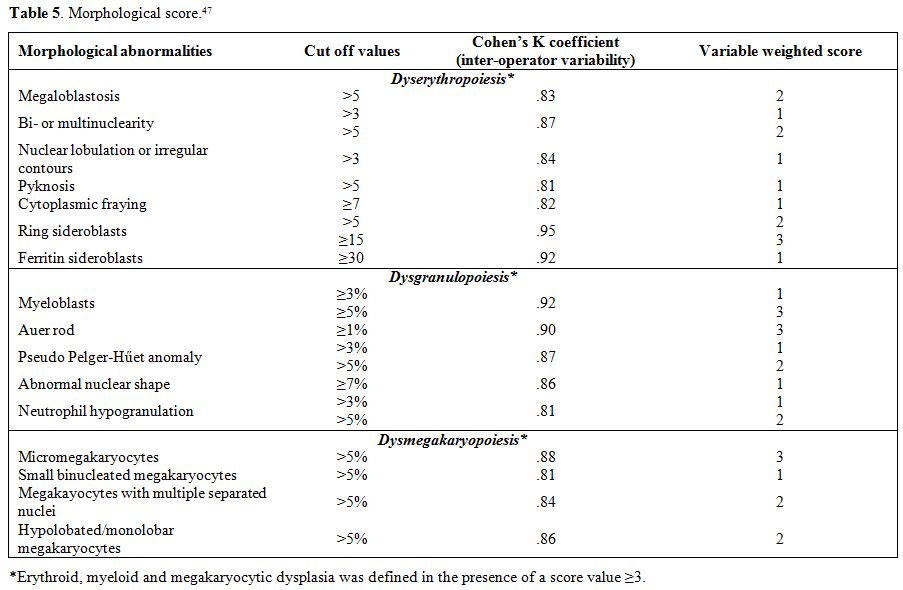 |
Table 5. Morphological score.[47] |
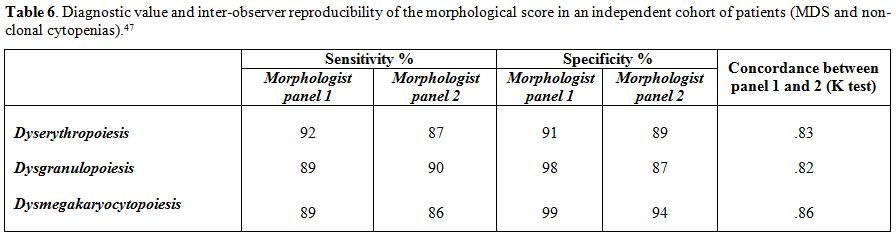 |
Table 6. Diagnostic value and
inter-observer reproducibility of the morphological score in an
independent cohort of patients (MDS and non-clonal cytopenias).[47] |
Histopathology
A
bone marrow trephine biopsy may increase the diagnostic accuracy and
help in refining the prognostic scoring system for MDS. It provides
information on cellularity and stroma and is essential for the
identification of MDS with fibrosis and hypoplastic MDS.[48-52] In these peculiar entities (10-15% of patients) that have a particular prognostic significance,[52,53]
diagnosis may be very difficult using bone marrow aspirates. In this
regard, a scoring system for the differential diagnosis between MDS and
other myeloid neoplasms with fibrosis, and between MDS and other
cytopenias with reduced bone marrow cellularity was developed.[47]Bone
marrow biopsy also allows a better evaluation of megakaryocytes and may
show the presence of aggregates or clusters of blasts, a typical
finding in aggressive subtypes.[35,54]
Moreover, it can provide material for additional diagnostic procedures,
such as immunohistochemistry, in situ hybridization or molecular
analysis.
Recommendations for Diagnosis
The
combination of manifest bone marrow dysplasia and clonal cytogenetic
abnormality allows a conclusive diagnosis, but this is possible for
only a part of patients. Diagnosis may be particularly difficult in
patients with <5% bone marrow blasts and only one cytopenia. If a
patient with a clinical and laboratory suspect of MDS has inconclusive
morphological features, a presumptive diagnosis of MDS can be made in
the presence of a specific chromosomal abnormality demonstrating
clonality. If there is only unilineage dysplasia, in the absence of
recurrent cytogenetic abnormalities, without increase of peripheral or
bone marrow blasts, with less than 15% ring sideroblasts, an
observation period of 6 months and repeating bone marrow examination is
recommended prior to making the diagnosis of MDS. For patients with
persistent cytopenia(s) (at least 6 months), in the absence of
morphological or cytogenetic evidence sufficient for a definitive
diagnosis of MDS, the term "idiopathic cytopenia of undetermined
significance" (ICUS) should be used (Figure 3).
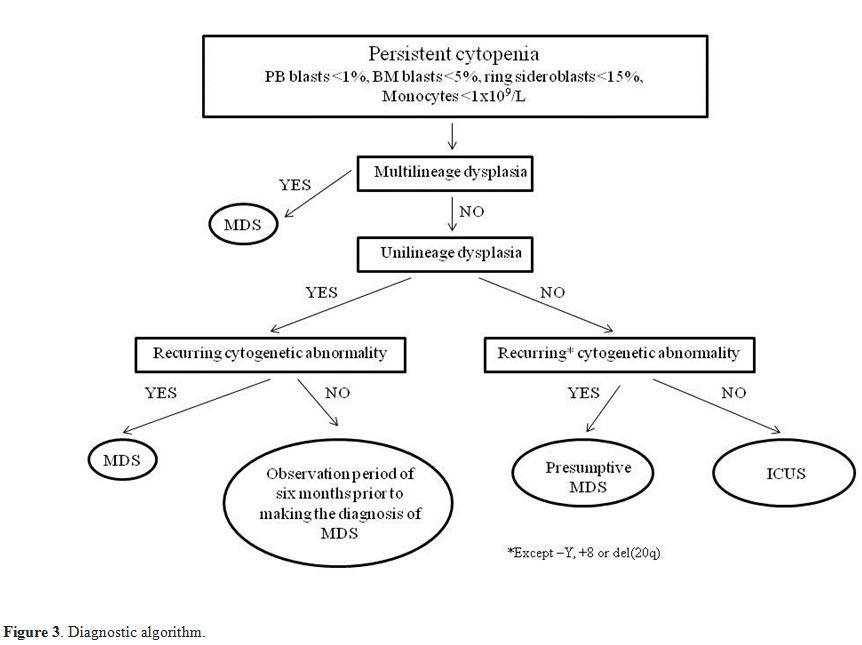 |
Figure 3. Diagnostic algorithm. |
Newly Defined Entities
The term ICUS was first proposed by the IWGM-MDS at a meeting in
Lisbon in 2005, and subsequently used in the 2008 WHO classification
and by others. ICUS and idiopathic dysplasia of undetermined
significance (IDUS) are conditions in which the criteria for the
diagnosis of MDS are not satisfied, even if cytopenia or dysplasia is
present.[6,55-58] ICUS is
characterized by persistent primary cytopenia, in the absence of
morphological or cytogenetic abnormalities specific of MDS, whereas in
IDUS there are morphological and/or karyotypic dysplastic alterations,
casually observed, in the absence of cytopenia. In ICUS, cytopenia may
concern one or more hematopoietic lineages; therefore, the terms of
idiopathic anemia, neutropenia, thrombocytopenia, or bi/pancytopenia of
uncertain significance have been proposed. The groups of cases so far
described are numerically small, except the one obtained from the MDS
registry of Düsseldorf.[59] In both ICUS and IDUS, a
neoplastic clone can be found already at diagnosis, and progression to
an overt MDS or another myeloid malignancy is possible after a variable
period. Thus, these conditions should be considered as a potential
pre-phase of myeloid neoplasms, and have to be closely monitored for
the unpredictable course.
Conclusions
Despite the WHO diagnostic and classification criteria, the
morphological diagnosis of MDS is still often critical and requires
considerable expertise.[60] On the other hand, as
more specific treatments are becoming available, an accurate diagnosis
is increasingly important. Recently, the use of new molecular
techniques, including gene expression profiling and analysis of point
mutations, has allowed to detect, even in patients with normal
karyotype, clonal abnormalities of considerable diagnostic and
prognostic meaning.[61-64] However, although in the
future morphology and cytogenetics should be integrated with the new
molecular techniques to classify MDS,[65] for the moment the morphological approach continues to be fundamental at least at the beginning of the diagnostic algorithm.
Acknowledgments
This work was supported by a grant from Fondazione IRCCS Policlinico San Matteo, Pavia, Italy.
References
- Cazzola M, Della Porta MG, Travaglino E, Malcovati
L. Classification and prognostic evaluation of myelodysplastic
syndromes. Sem Oncol 2011; 38:627-634.
http://dx.doi.org/10.1053/j.seminoncol.2011.04.007 PMid:21943669.
 .
. - Malcovati
L, Hellström-Lindberg E, Bowen D, et al. diagnosis and treatment of
primary myelodysplastic syndromes in adults. Recommendations from the
European LeukemiaNet. Blood 2013; 122:2943-2964.
http://dx.doi.org/10.1182/blood-2013-03-492884 PMid:23980065 PMCid:PMC3811170. . - Yamazaki H, Nakao S. Border between aplastic anemia and myelodysplastic syndrome. Int J Hematol 2013; 97:558-563. http://dx.doi.org/10.1007/s12185-013-1324-x PMid:23613266. .
- Gondek
LP, DeZern AE. I walk the line: how to tell MDS from other bone marrow
failure conditions. Curr Hematol Malig Rep 2014; 9:389-399. http://dx.doi.org/10.1007/s11899-014-0224-3 PMid:25079655. .
- Vardiman
JW, Harris NL, Brunning RD. The World Health Organization (WHO)
classification of the myeloid neoplasms. Blood 2002; 100:2292-2302. http://dx.doi.org/10.1182/blood-2002-04-1199 PMid:12239137. .
- Swerdlow
SH, Campo E, Harris NL, et al. WHO classification of tumours of
haematopoietic and lymphoid tissues. IARC Press, Lyon, 2008.
- Vardiman
JW, Thiele J, Arber DA et al. The 2008 revision of the World Health
Organization (WHO) classification of myeloid neoplasms and acute
leukemia: rationale and important changes. Blood 2009; 114:937-951. http://dx.doi.org/10.1182/blood-2009-03-209262 PMid:19357394. .
- Bennett
JM, Catovky D, Daniel MT, et al. Proposals for the classification of
the myelodysplastic syndromes. Br J Haematol 1982; 51:89-99. http://dx.doi.org/10.1111/j.1365-2141.1982.tb08475.x .
- Malcovati
L, Porta MG, Pascutto C, et al. Prognostic factors and life expectancy
in myelodysplastic syndromes classified according to WHO criteria: a
basis for clinical decision making. J Clin Oncol 2005; 23:7594-7603. http://dx.doi.org/10.1200/JCO.2005.01.7038 PMid:16186598. .
- Malcovati
L, Germing U, Kuendgen A, et al. Time dependent prognostic scoring
system for predicting survival and leukemic evolution in the
myelodysplastic syndromes. J Clin Oncol 2007; 25:3503-3510.
http://dx.doi.org/10.1200/JCO.2006.08.5696 PMid:17687155. . - Valent
P, Horny HP, Bennett JM, et al. Definitions and standards in the
diagnosis and treatment of the myelodysplastic syndromes: Consensus
statements and report from a working conference. Leuk Res 2007;
727-736. http://dx.doi.org/10.1016/j.leukres.2006.11.009 PMid:17257673. .
- Platzbecker
U, Santini V, Mufti GJ, et al. Update on developments in the diagnosis
and prognostic evaluation of patients with myelodysplastic syndromes
(MDS): Consensus statements and report from an expert workshop. Leuk
Res 2012; 36:264-270. http://dx.doi.org/10.1016/j.leukres.2011.11.005 PMid:22137318. .
- Malcovati
L, Della Porta MG, Lunghi M, et al. Flow cytometry evaluation of
erythroid and myeloid dysplasia in patients with myelodysplastic
syndrome. Leukemia 2005; 19:776-783. http://dx.doi.org/10.1038/sj.leu.2403680 PMid:15789068. .
- Bacher
U, Haferlach T, Kem W, Weiss T, Scnittger S, Haferlach C. The impact of
cytomorphology, cytogenetics, molecular genetics, and immunophenotyping
in a comprehensive diagnostic workup of myelodysplastic syndromes.
Cancer 2009; 115:4524-4532. http://dx.doi.org/10.1002/cncr.24501 PMid:19569249. .
- Orazi
A, Germing U. The myelodysplastic/myeloproliferative neoplasms:
myeloproliferative diseases with dysplastic features. Leukemia 2008;
22:1308-1319. http://dx.doi.org/10.1038/leu.2008.119 PMid:18480833. .
- Cazzola
M, Malcovati L, Invernizzi R. Myelodysplastic/myeloproliferative
neoplasms. Hematology Am Soc Hematol Educ Program. 2011; 2011:264-272. http://dx.doi.org/10.1182/asheducation-2011.1.264 PMid:22160044. .
- Goasguen
JE, Bennett JM. Classification and morphologic features of the
myelodysplastic syndromes. Semin Oncol 1992; 19:4-13. PMid:1736369. .
- Komrokji
RS, Zhang L, Bennett JM. Myelodysplastic syndromes classification and
risk stratification. Hematol Oncol Clin N Am 2010; 24:443-457. http://dx.doi.org/10.1016/j.hoc.2010.02.004 PMid:20359636. .
- Wang SA. Diagnosis of myelodysplastic syndromes in cytopenic patients. Hematol Oncol Clin N Am 2011; 25:1085-1110. http://dx.doi.org/10.1016/j.hoc.2011.09.009 PMid:22054736. .
- Giagounidis A, Haase D. Morphology, cytogenetics and classification of MDS. Best Practice Res Clin Haematol 2013; 26:337-353. http://dx.doi.org/10.1016/j.beha.2013.09.004 PMid:24507811. .
- Mufti
GJ, Bennett JM, Goasguen J, et al. Diagnosis and classification of MDS:
International Working Group on Morphology of MDS (IWGM-MDS) consensus
proposals for the definition and enumeration of myeloblasts and ring
sideroblasts. Haematologica 2008; 93:110-115. http://dx.doi.org/10.3324/haematol.13405 PMid:18838480. .
- Wang
H, Wang XQ, Xu XP, Lin GW. Bone marrow blast level predicts prognosis
in patients with refractory cytopenia with multilineage dysplasia. Eur
J Haematol 2009; 83:550-558. http://dx.doi.org/10.1111/j.1600-0609.2009.01343.x PMid:19737310. .
- Greenberg
PL, Tuechler H, Schanz J, et al. Revised international prognostic
scoring system for myelodysplastic syndromes. Blood 2012;
120:2454-2465. http://dx.doi.org/10.1182/blood-2012-03-420489 PMid:22740453. .
- Knipp
S, Strupp C, Gattermann N, et al. Presence of peripheral blasts in
refractory anemia and refractory cytopenia with multilineage dysplasia
predicts an unfavourable outcome. Leuk Res 2008; 32:33-37.
http://dx.doi.org/10.1016/j.leukres.2007.02.021 PMid:17412418. . - Goasguen
JE, Bennett JM, Bain BJ, Vallespi T, Brunning R, Mufti GJ for the
International Working Group on Morphology of Myelodysplastic Syndrome
(IWGM-MDS). Morphologic evaluation of monocytes and their precursors.
Haematologica 2009; 94:994-997.http://dx.doi.org/10.3324/haematol.2008.005421 PMid:19535346 PMCid:PMC2704310. .
- Matsuda
A, Germing U, Jinnai I, et al. Improvement of criteria for refractory
cytopenia with multilineage dysplasia according to the WHO
classification based on prognostic significance of morphological
features in patients with refractory anemia according to the FAB
classification. Leukemia 2007; 21:678-686. http://dx.doi.org/10.1038/sj.leu.2404571 . .
- Verburgh
E, Achten R, Louw VJ, et al. A new disease categorization of low-grade
myelodysplastic syndromes based on the expression of cytopenia and
dysplasia in one versus more than one lineage improves on the WHO
classification. Leukemia 2007; 21:668-677. http://dx.doi.org/10.1038/sj.leu.2404564 . .
- Buckstein
R, Jang K, Friedlich J, et al. Estimating the prevalence of
myelodysplastic syndromes in patients with unexplained cytopenias: a
retrospective study of 322 bone marrows. Leuk Res 2009; 33:1313-1318. http://dx.doi.org/10.1016/j.leukres.2009.02.010 PMid:19282029. .
- Liu
D, Chen Z, Xue Y, et al. The significance of bone marrow cell
morphology and its correlation with cytogenetic features in the
diagnosis of MDS-RA patients. Leuk Res 2009; 33:1029-1038.
http://dx.doi.org/10.1016/j.leukres.2009.02.011 PMid:19411106. . - Hast
R, Nilsson I, Widell S, Ost A. Diagnostic significance of dysplastic
features of peripheral blood polymorphs in myelodysplastic syndromes.
Leuk Res 1989; 13:173-178
http://dx.doi.org/10.1016/0145-2126(89)90142-2. . - Goasguen
JE, Bennett JM, Bain BJ, et al. Proposal for refining the definition of
dysgranulopoiesis in acute myeloid leukemia and myelodysplastic
syndromes. Leuk Res 2014; 38:447-453. http://dx.doi.org/10.1016/j.leukres.2013.12.020 PMid:24439566. .
- Cazzola
M, Invernizzi R, Bergamaschi G, et al. Mitochondrial ferritin
expression in erythroid cells from patients with sideroblastic anemia.
Blood 2003;101:1996-2000. http://dx.doi.org/10.1182/blood-2002-07-2006 PMid:12406866. .
- Zhou J, Orazi A, Czader MB. Myelodysplastic syndromes. Semin Diagn Pathol 2011; 28:258-272. http://dx.doi.org/10.1053/j.semdp.2011.08.005 PMid:22195404. .
- Wang
SA, Tang G, Fadare O, et al: Erythroid-predominant myelodysplastic
syndromes: enumeration of blasts from nonerythroid rather than total
marrow cells provides superior risk stratification. Mod Pathol 2008;
21:1394-1402. http://dx.doi.org/10.1038/modpathol.2008.142. .
- Valent
P, Orazi A, Büsche G, et al. Standards and impact of hemathopathology
in myelodysplastic syndromes (MDS). Oncotarget 2010; 1:483-496.
PMid:21317447 PMCid:PMC3248141. .
- Cantù
Rajnoldi A, Fenu S, Kerndrup G, van Wering ER, Niemeyer CM, Baumann I
for the European Working Group on Myelodysplastic Syndromes in
Childhood (EWOG-MDS). Evaluation of dysplastic features in
myelodysplastic syndromes: experience from the morphology group of the
European Working Group of MDS in Childhood (EWOG-MDS). Ann Hematol
2005; 84:429-433. http://dx.doi.org/10.1007/s00277-005-1034-4 PMid:15838669. .
- Matsuda
A, Jinnai I, Yagasaki F, et al. Refractory anemia with severe
dysplasia: clinical significante of morphological features in
refractory anemia. Leukemia 1998; 12:482-485. http://dx.doi.org/10.1038/sj.leu.2400966 PMid:9557604. .
- Tassin
F, Dewé W, Schaaf N, et al. A four-parameter index of marrow dysplasia
has predictive value for survival in myelodysplastic syndromes. Leuk
Lymph 2000; 36:485-496. http://dx.doi.org/10.3109/10428190009148396 PMid:10784393. .
- Matsuda
A, Jinnai I, Miyazaki Y, Tomonaga M. Proposals for a grading system for
diagnostic accuracy of myelodysplastic syndromes. Clin Leuk 2008;
2:102-106. http://dx.doi.org/10.3816/CLK.2008.n.012 . .
- Germing
U, Strupp C, Giagounidis A, et al. Evaluation of dysplasia through
detailed cytomorphology in 3156 patients from the Düsseldorf Registry
on myelodysplastic syndromes. Leuk Res 2012; 36:727-734.
http://dx.doi.org/10.1016/j.leukres.2012.02.014 PMid:22421409. . - Ramos
F, Fernandez-Ferrero S, Suarez D, et al. Myelodysplastic syndrome: a
search for minimal diagnostic criteria. Leuk Res 1999; 23:283-290. http://dx.doi.org/10.1016/S0145-2126(98)00166-0 . .
- Bain BJ. The bone marrow aspirate of healthy subjects. Br J Haematol 1996; 94:206-209. http://dx.doi.org/10.1046/j.1365-2141.1996.d01-1786.x PMid:8757536. .
- Parmentier
S, Schetelig J, Lorenz K, et al. Assessment of dysplastic
hematopoiesis: lessons from healthy bone marrow donors. Haematologica
2012; 97:723-730. http://dx.doi.org/10.3324/haematol.2011.056879 PMid:22180437 PMCid:PMC3342975. .
- Naqvi
K, Jabbour E, Bueso-Ramos C, et al. Implications of discrepancy in
morphologic diagnosis of myelodysplastic syndrome between referral and
tertiary care centers. Blood 2011; 118:4690-4693.
http://dx.doi.org/10.1182/blood-2011-03-342642 PMid:21868570 PMCid:PMC4081364. . - Senent
L, Arenillas L, Lu-o E, Ruiz JC, Sanz J, Florensa L. Reproducibility of
the World Health Organization 2008 criteria for myelodysplastic
syndromes. Haematologica 2013; 98:568-575. http://dx.doi.org/10.3324/haematol.2012.071449 PMid:23065505 PMCid:PMC3659988. .
- Glauser
TA, Sagatys EM, Williamson JC, et al. Current pathology practices in
and barriers to MDS diagnosis. Leuk Res 2013; 37:1656-1661. http://dx.doi.org/10.1016/j.leukres.2013.10.007 PMid:24220584. .
- Della
Porta MG, Travaglino E, Boveri E, et al. Minimal morphological criteria
for defining bone marrow dysplasia: a basis for clinical implementation
of WHO classification of myelodysplastic syndromes. Leukemia 2015;
29:66-75. http://dx.doi.org/10.1038/leu.2014.161 PMid:24935723. .
- Lambertenghi-Deliliers
G, Annaloro C, Oriani A, Soligo D. Myelodysplastic syndrome associated
with bone marrow fibrosis. Leuk Lymphoma 1992; 8:51-55. http://dx.doi.org/10.3109/10428199209049817 PMid:1493471. .
- Tuzuner
N, Cox C, Rowe JM, Watrous D, Bennett JM. Hypocellular myelodysplastic
syndromes (MDS): new proposals. Br J Haematol 1995; 91:612-617. http://dx.doi.org/10.1111/j.1365-2141.1995.tb05356.x PMid:8555063. .
- Thiele
J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A.
European consensus on grading bone marrow fibrosis and assessment of
cellularity. Haematologica 2005; 90:1128-1132.
PMid:16079113. . - Bennett
JM, Orazi A. Diagnostic criteria to distinguish hypocellular acute
myeloid leukemia from hypocellular myelodysplastic syndromes and
aplastic anemia: recommendations for a standardized approach.
Haematologica 2009; 94:264-268. http://dx.doi.org/10.3324/haematol.13755 PMid:19144661 PMCid:PMC2635414. .
- Della
Porta MG, Malcovati L, Boveri E, et al. Clinical relevance of bone
marrow fibrosis and CD34-positive cell clusters in primary
myelodysplastic syndromes. J Clin Oncol 2009; 27:754-762.
http://dx.doi.org/10.1200/JCO.2008.18.2246 PMid:19103730. . - Buesche
G, Teoman H, Wilczak W, et al. Marrow fibrosis predicts early fatal
marrow failure in patients with myelodysplastic syndromes. Leukemia
2008; 22:313-322. http://dx.doi.org/10.1038/sj.leu.2405030 PMid:18033321. .
- Orazi A, Czader MB. Myelodysplastic syndromes. Am J Clin Pathol 2009; 132:290-305. http://dx.doi.org/10.1309/AJCPRCXX4R0YHKWV PMid:19605823. .
- Wimazal
F, Fonatsch C, Thalhammer R, et al. Idiopathic cytopenia of
undetermined significance (ICUS) versus low risk MDS: the diagnostic
interface. Leuk Res 2007; 31:1461-1468. http://dx.doi.org/10.1016/j.leukres.2007.03.015 PMid:17507091. .
- Valent
P, Horny HP. Minimal diagnostic criteria for myelodysplastic syndromes
and separation from ICUS and IDUS: update and open questions. Eur J
Clin Invest 2009; 39:548-553.
http://dx.doi.org/10.1111/j.1365-2362.2009.02151.x PMid:19453651. . - Valent
P, Jäger E, Mitterbauer-Hohendanner G, et al. Idiopathic bone marrow
dysplasia of unknown significance (IDUS): definition, pathogenesis,
follow up, and prognosis. Am J Cancer Res 2011; 1:531-541.
PMid:21984971 PMCid:PMC3186051. . - Valent
P, Bain BJ, Bennett JM, et al. Idiopathic cytopenia of undetermined
significance (ICUS) and idiopathic dysplasia of uncertain significance
(IDUS), and their distinction from low risk MDS. Leuk Res 2012; 36:1-5.
PMid:21920601. .
- Schroeder
T, Ruf L, Bernhardt A, et al. Distinguishing myelodysplastic syndromes
(MDS) from idiopathic cytopenia of undetermined significance (ICUS):
HUMARA unravels clonality in a subgroup of patients. Ann Oncol 2010;
21:2267-2271. http://dx.doi.org/10.1093/annonc/mdq233 PMid:20439346. .
- Bennett
JM. Morphological classification of the myelodysplastic syndromes: how
much more education of diagnosticians is necessary? Haematologica 2013;
93:490-491. http://dx.doi.org/10.3324/haematol.2013.084418 PMid:23543153 PMCid:PMC3659978. .
- Bejar
R, Levine R, Ebert BL. Unraveling the molecular pathophysiology of
myelodysplastic syndromes. J Clin Oncol 2011; 29:504-515. http://dx.doi.org/10.1200/JCO.2010.31.1175 PMid:21220588 PMCid:PMC3969457. .
- Yoshida
K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing
machinery in myelodysplasia. Nature 2011; 478:64-69. http://dx.doi.org/10.1038/nature10496 PMid:21909114. .
- Cazzola
M, Della Porta MG, Malcovati L. The genetic basis of myelodysplasia and
its clinical relevance. Blood 2013; 122:4021.4034.. .
- Malcovati
L, Papaemmanuil E, Ambaglio I, et al. Driver somatic mutations identify
distinct disease entities within myeloid neoplasms with myelodysplasia.
Blood. 2014; 124:1513-1521. http://dx.doi.org/10.1182/blood-2014-03-560227 PMid:24970933 PMCid:PMC4148773. .
- Van't Veer M, Haferlach T. Should clinical hematologists put their microscopes on eBay? Haematologica 2014; 99:1533-1534. http://dx.doi.org/10.3324/haematol.2014.114710 PMid:25271310 PMCid:PMC4181246. .
TOP]