Received: March 27, 2015
Accepted:April 28, 2015
Mediterr J Hematol Infect Dis 2015, 7(1): e2015037, DOI 10.4084/MJHID.2015.037
This article is available on PDF format at:

Andrea Pellagatti and Jacqueline Boultwood
Leukaemia
& Lymphoma Research Molecular Haematology Unit, Nuffield Division
of Clinical Laboratory Sciences, Radcliffe Department of Medicine,
University of Oxford, and BRC Blood Theme, NIHR Oxford Biomedical
Centre, Oxford University Hospitals, Oxford, United Kingdom
| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract The 5q- syndrome is the most distinct
of the myelodysplastic syndromes (MDS) and patients with this disorder
have a deletion of chromosome 5q [del(5q)] as the sole karyotypic
abnormality. Several genes mapping to the commonly deleted region of
the 5q- syndrome have been implicated in disease pathogenesis in recent
years. Haploinsufficiency of the ribosomal gene RPS14 has been shown to
cause the erythroid defect in the 5q- syndrome. Loss of the microRNA
genes miR-145 and miR-146a has been associated with the thrombocytosis
observed in 5q- syndrome patients. Haploinsufficiency of CSNK1A1 leads
to hematopoietic stem cell expansion in mice and may play a role in the
initial clonal expansion in patients with 5q- syndrome. Moreover, a
subset of patients harbor mutation of the remaining CSNK1A1 allele.
Mouse models of the 5q- syndrome, which recapitulate the key features
of the human disease, indicate that a p53-dependent mechanism underlies
the pathophysiology of this disorder. Importantly, activation of p53
has been demonstrated in the human 5q- syndrome. Recurrent TP53
mutations have been associated with an increased risk of disease
evolution and with decreased response to the drug lenalidomide in
del(5q) MDS patients. Potential new therapeutic agents for del(5q) MDS
include the translation enhancer L-leucine. |
The 5q- syndrome: candidate genes and pathophysiology
The myelodysplastic syndromes (MDS) are heterogeneous clonal
hematopoietic stem cell (HSC) malignancies characterized by ineffective
hematopoiesis, peripheral blood cytopenias, and typically patients have
a hypercellular bone marrow. MDS patients frequently show disease
progression (approximately 40% of cases) to acute myeloid leukemia
(AML).[1]
Chromosomal monosomies and deletions
are commonly observed in MDS. Cytogenetic abnormalities are present in
approximately 50% of de novo MDS and 80% of therapy-related MDS cases.[2]
Interstitial deletion within the long arm of chromosome 5 [del(5q)] is
one of the most common karyotypic abnormalities reported in de novo
MDS, occurring in approximately 10-20% of patients with this disorder.[2]
Patients
are defined as 5q- syndrome when they have a del(5q) as the sole
karyotypic abnormality and a medullary blast count of less than 5%.[3,4] The 5q- syndrome was first described by Van den Berghe et al in 1974[5]
and is the most distinct of the MDS with a clear genotype-phenotype
relationship. Patients with the 5q- syndrome show macrocytic anemia,
hypolobulated megakaryocytes, a normal or high platelet count and a
good prognosis with approximately 10% of patients transforming to AML.[6,7]
The
del(5q) is considered to mark the location of one or more genes the
loss of which may affect important processes involved in normal
hematopoiesis.[8] The commonly deleted region (CDR) of
the 5q- syndrome was identified over 20 years ago using molecular
mapping and fluorescent in situ hybridization techniques by Boultwood
et al[9] and was progressively narrowed to a ~1.5Mb interval at 5q32-q33 flanked by the DNA marker DS5413 and the GLRA1 gene.[10,11]
Genomic annotation of the CDR of the 5q- syndrome highlighted several
promising candidate genes mapping to the CDR, including the tumor
suppressor gene SPARC, the ribosomal protein gene RPS14 and several microRNA genes.[10,12]
Mutation screening of all 40 genes within the CDR was performed in ten
5q- syndrome patients using Sanger sequencing several years ago and no
mutations were identified.[12] The absence of
mutations in genes in the CDR was suggestive of haploinsufficiency (a
dosage effect resulting from the loss of one allele of a gene)[13] being an important mechanism in the 5q- syndrome.
In a study published in 2007, the transcriptome of bone marrow CD34+ cells was investigated in a cohort of ten patients with the 5q- syndrome using microarray-based gene expression profiling.[12] Several candidate genes mapping to the CDR of the 5q- syndrome showed haploinsufficiency in 5q- syndrome patients, including RPS14, encoding a component of the 40S ribosomal subunit, and CSNK1A1, encoding a serine/threonine kinase.[12] Crucially, these two genes would be shown in subsequent studies[14,15] to have an important role in the molecular pathogenesis of the 5q- syndrome.
In a landmark study by Ebert et al in 2008, RPS14 was identified as a 5q- syndrome gene using a RNA-mediated interference (RNAi)-based screen of each gene within the CDR.[14] Knockdown of RPS14
to haploinsufficient levels in normal HSC resulted in a block in
erythroid differentiation with relative preservation of megakaryocytic
differentiation. Forced expression of RPS14 in primary bone marrow cells from 5q- syndrome patients rescued the phenotype, demonstrating the important role of RPS14 in the 5q- syndrome.[14] In addition, RPS14
haploinsufficiency resulted in a block in the processing of
pre-ribosomal RNA and in abrogation of 40S ribosomal subunit formation.[14]
Studies by Pellagatti et al have shown that CD34+ cells from patients
with the 5q- syndrome have defective expression of many ribosomal- and
translation-related genes.[16,17] The results of
these studies suggest that the 5q- syndrome is a disorder of aberrant
ribosome biogenesis, and the 5q- syndrome is now considered to be a
ribosomopathy.[18] There is a strong analogy between the 5q- syndrome and Diamond-Blackfan anemia (DBA),[12] a disorder which is similarly caused by haploinsufficiency of ribosomal protein genes, including RPS19.[19]
A mouse model of the 5q- syndrome has been generated by Barlow et al using large-scale chromosomal engineering.[20] Mice with haploinsufficiency of the Cd74-Nid67 interval (which is syntenic to the CDR of the human 5q- syndrome and includes Rps14)
recapitulated the key features of the human disease, including a
macrocytic anemia and monolobulated megakaryocytes in the bone marrow.[20]
This ‘5q- mouse’ showed defective bone marrow progenitor development
and an accumulation of p53 protein with increased apoptosis was
observed in the bone marrow cells, similar to that observed in animal
models of DBA.[21] The progenitor cell defect could
be rescued by intercrossing the ‘5q- mouse’ with p53-deficient mice,
providing the first evidence that a p53-dependent mechanism underlies
the pathophysiology of the 5q- syndrome.[20]
Recently, a murine model for conditional, heterozygous inactivation of Rps14 in the bone marrow has been generated.[22] Rps14
haploinsufficient mice showed significantly reduced hemoglobin and red
blood cell counts with a significantly higher MCV. Bone marrow analysis
confirmed an erythroid differentiation defect, with a significant
increase in hypolobulated megakaryocytes. Rps14 haploinsufficient
mice also showed reduced protein synthesis and p53 induction in
late-stage erythroblasts. Genetic inactivation of p53 rescued the
erythroid phenotype: the erythroid differentiation defect was restored
in Rps14-/+p53-/+ mice.[22] This murine model shows that haploinsufficiency of Rps14 is sufficient to recapitulate the erythroid and megakaryocytic phenotype observed in the 5q- syndrome.
Importantly, induction of p53 and up-regulation of the p53 pathway has been shown to occur in the human 5q- syndrome.[23]
Immunohistochemical analysis showed moderate to strong p53 expression
in erythroid cells in bone marrow trephine sections from patients with
the 5q- syndrome, and gene expression profiling demonstrated that the
p53 pathway is significantly deregulated in the CD34+ cell population of these patients.[23]
The accumulation of p53 protein in bone marrow erythroid precursors of
patients with the 5q- syndrome has been confirmed in other studies.[24,25]
Activation
of p53 has been shown to occur selectively in erythroid cells
differentiated from human HSC with shRNA-based knockdown of RPS14.[24]
Induction of p53 resulted in erythroid-specific accumulation of p21,
cell cycle arrest and apoptosis, consistent with the failure of
erythropoiesis observed in the 5q- syndrome.[24]
Inhibition of p53 using pifithrin-α in culture rescued the erythroid
defect, suggesting that p53 activation may represent a therapeutic
target in MDS with del(5q).[24]
Thus several converging lines of evidence[20,21,23,24]
demonstrate that ribosomal stress leads to activation of the p53
pathway, a key effector of erythroid hypoplasia in both del(5q) MDS and
congenital ribosomopathies.
CSNK1A1. A recent study by Schneider et al has shown that CSNK1A1 plays a central role in the pathogenesis of del(5q) MDS.[15] CSNK1A1 encodes CK1α, a central regulator of β-catenin[26] which is a major driver of stem cell self-renewal.[15] Heterozygous inactivation of Csnk1a1 in mice led to β-catenin activation and expansion of HSCs,[15] suggesting that CSNK1A1
haploinsufficiency may be the mechanism underlying the initial clonal
expansion in patients with the 5q- syndrome. Mutations of CSNK1A1 were identified in approximately 7% of MDS del(5q) cases analyzed.[15] A CSNK1A1 mutation was reported in a del(5q) MDS patient in a previous study.[27] Interestingly, Schneider et al showed that expression of mutant CSNK1A1 resulted in β-catenin activation and HSC cell cycle progression. Thus CSNK1A1
mutations are recurrent in a small proportion of del(5q) MDS patients,
and there is evidence that these mutations may drive clonal dominance.
Furthermore, Csnk1a1 haploinsufficiency was shown to sensitize cells to casein kinase inhibition, indicating that CSNK1A1 is a potential new therapeutic target for the treatment of del(5q) MDS.[15]
MicroRNA genes.
It has been suggested that haploinsufficiency of miR-145 and miR-146a,
two miRNA genes that map within and adjacent to the CDR respectively,[12]
may be the cause of other key features of the 5q- syndrome, namely
hypolobulated megakaryocytes and thrombocytosis. The study by
Starczynowski et al showed down-regulation of miR-145 and miR-146a in
the CD34+ cells of patients with the 5q- syndrome.[28] Knockdown of these miRNAs in mouse HSCs resulted in thrombocytosis, mild neutropenia and megakaryocytic dysplasia.[28] Kumar et al have identified the FLI1 gene,
encoding a transcription factor involved in megakaryopoiesis, as a
critical target of miR-145 and have shown that patients with del(5q)
MDS have increased expression of FLI1.[29] Inhibition of miR-145 or overexpression of Fli-1 resulted in an increase in the production of megakaryocytic cells relative to erythroid cells.[29]
These data suggest that deficiency of miR-145 and miR-146a may underlie
the thrombocytosis observed in some 5q- syndrome patients.
Cell of Origin
The 5q- syndrome is a disorder originating in the human HSC. Using
immunophenotyping and FISH, Jaju et al showed B-cell involvement in one
of three cases with the 5q- syndrome.[30] In the
study of Nilsson et al, no T-cell involvement was observed in nine
patients with del(5q), but one patient had B-cell involvement.[31] A minimum of 94% of cells in the minor CD34+CD38- HSC compartment carried a del(5q) in all patients analyzed and 5q aberrations were found in 25-90% of purified CD34+CD19+ pro-B cells in three of five patients.[31]
These data strongly suggest that 5q deletions occur in HSCs with a
combined lympho-myeloid potential and that 5q deletions represent an
early event in MDS pathogenesis.[31] A subsequent gene expression profiling study of highly purified 5q-deleted CD34+CD38−Thy1+ cells in 5q- MDS identified a molecular signature supporting a HSC origin for this disorder.[32]
In
a recent study, Woll et al elegantly demonstrated that the MDS are
propagated by rare and distinct human cancer stem cells in vivo.[27]
A total of 34 somatic lesions, including del(5q) and driver mutations,
were identified in bulk bone marrow cells of 15 patient with lower-risk
MDS and all these lesions could be tracked back to the stem cell
compartment. In MDS cases with del(5q) and additional driver mutations,
acquisition of del(5q) preceded recurrent gene mutations, with the
exception of four MDS cases with sideroblastic anemia in which the
del(5q) was preceded by SF3B1
gene mutations. In all cases with isolated del(5q) or RAEB1/RCMD, the
del(5q) was predicted to be the first (or only) genetic lesion. These
data are compatible with the del(5q) being the initiating and
potentially the only genetic lesion required for the development of MDS
with isolated del(5q).
Treatment
The immunomodulatory drug lenalidomide has been shown to have
dramatic therapeutic efficacy in patients with the 5q- syndrome and
other MDS patients with del(5q).[33,34] A large
multicentre phase II trial by List et al evaluated lenalidomide
treatment response in 148 MDS patients with del(5q): transfusion
independency and a complete cytogenetic remission was achieved in 67%
and 45% of patients respectively.[33] Lenalidomide is
now considered the standard of care for the treatment of transfusion
dependent anemia in lower risk del(5q) MDS patients.[35]
Lenalidomide has been shown to inhibit the growth of MDS del(5q) erythroblasts but did not affect normal cells in culture.[36]
The mode of action of lenalidomide has been investigated in several
studies. Lenalidomide has been shown to upregulate several genes,
including the tumor suppressor gene SPARC
and the TGF-β family member activin A, in hematopoietic progenitor
cells from patients with del(5q) MDS and normal individuals.[36,37]
SPARC, located at 5q32-q33 within the CDR of the 5q- syndrome, has
anti-proliferative, anti-adhesive, and anti-angiogenic properties, all
known effects of immunomodulatory drugs.[38] Wei et al[39]
demonstrated that lenalidomide inhibits two phosphatases, Cdc25C and
PP2Acα. The genes for these phosphatases are located on 5q and are
deleted in most patients with del(5q) MDS. Cdc25C and PP2Acα are
co-regulators of the G2-M checkpoint in the cell cycle and thus their
inhibition by lenalidomide leads to G2 arrest and apoptosis. These data
suggest that haploinsufficiency of Cdc25C and PP2Acα in del(5q) MDS
causes an enhanced sensitivity to lenalidomide.[39]
Lenalidomide has been shown to promote degradation of p53 by inhibiting auto-ubiquitination of MDM2 in del(5q) MDS.[25]
It has been suggested that lenalidomide restores MDM2 functionality
in the 5q- syndrome to overcome p53 activation in response to
ribosomal stress.[25] A recent study reported that lenalidomide induces the ubiquitination and consequent degradation of CSNK1A1 by the CRBN-CRL4 E3 ubiquitin ligase.[40] Knockdown of CSNK1A1 sensitized primary CD34+ cells to lenalidomide, suggesting that haploinsufficiency of CSNK1A1 might increase lenalidomide sensitivity in del(5q) hematopoietic cells.[40]
These data implicate several genes mapping to del(5q) in the mode of action of lenalidomide in del(5q) MDS.
It
would be valuable to identify predictive factors for response to
lenalidomide, and an erythroid differentiation signature that predicts
response to lenalidomide in MDS has been identified.[41] A correlation of clinical response and response duration with induction of the microRNA miR-145 by lenalidomide in CD34+ cells from patients with MDS and the del(5q) has been recently proposed.[37] Importantly, the presence of TP53
mutation has been shown to influence negatively the response to
lenalidomide in del(5q) MDS in several studies. In the study by
Jädersten et al the probability of a complete cytogenetic response to
lenalidomide was significantly lower in TP53 mutated patients.[42] Another study confirmed the importance of TP53 mutational status for response to lenalidomide treatment: wild-type TP53 status showed a tendency for hematological response, while none of the cases with mutated TP53 achieved a complete cytogenetic response.[43]
In a recent study by Saft et al, immunohistochemical analysis of p53
was performed in bone marrow biopsies from 85 lower-risk del(5q) MDS
patients treated with lenalidomide.[44] Strong p53
expression in ≥1% of bone marrow progenitor cells was observed in 35%
of patients and was significantly associated with shorter survival,
higher risk of evolution to AML and a lower cytogenetic response rate
to lenalidomide.
Lenalidomide is clearly an effective treatment
for lower-risk, transfusion-dependent MDS patients with del(5q),
however not all patients respond to lenalidomide and approximately half
of MDS patients with del(5q) acquire resistance to the drug within two
to three years.[33] There is thus a clinical need for
novel treatments for MDS patients with del(5q). Potential new
therapeutic agents for this group of patients include the translation
enhancer L-leucine[45,46] and the p53 inhibitor cenersen[47] (Figure 1).
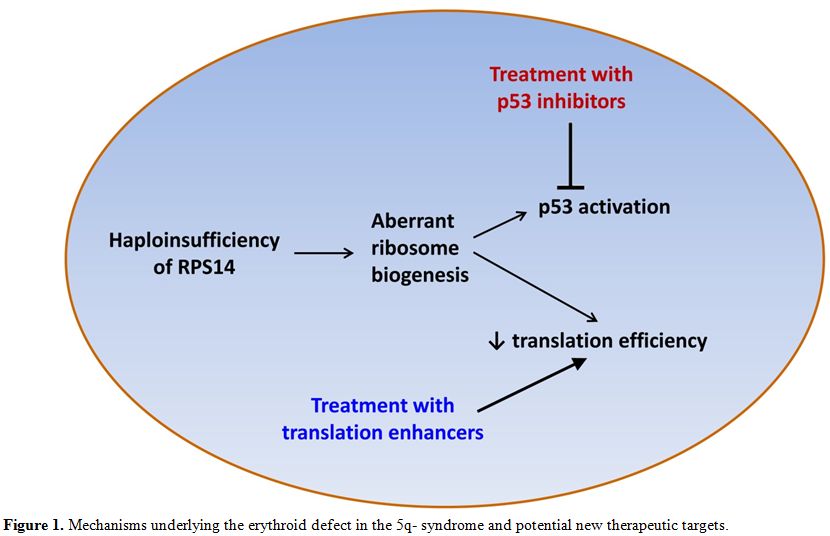 |
Figure 1. Mechanisms underlying the erythroid defect in the 5q- syndrome and potential new therapeutic targets. |
L-leucine. The HSCs of patients with the 5q- syndrome show defective ribosome biogenesis[14] and deregulation of many ribosomal- and translation-related genes.[17]
Defective ribosome biogenesis may result in a reduction in the
efficiency of mRNA translation and total protein production has been
shown to be significantly decreased in erythroid cells with knockdown
of RPS14.[46]
This defect in mRNA translation represents a potential therapeutic
target and there is evidence suggesting that the translation enhancer
L-leucine may have some efficacy in ribosomopathies. Pospisilova et al
described a DBA patient who showed a marked improvement in anemia and
became transfusion-independent after treatment with L-leucine.[48] Yip et al reported that L-leucine treatment of cultured erythroblasts derived from CD34+ cells of healthy controls with RPS14 knockdown and from CD34+ cells of del(5q) patients resulted in an increase in proliferation, erythroid differentiation and mRNA translation.[46]
Zebrafish models of del(5q) MDS and DBA treated with L-leucine showed
increased hemoglobinization and red cell numbers and reduced
developmental defects.[45] Similarly, L-leucine
treatment of a mouse model of DBA resulted in improved hemoglobin
concentration and in an increase in the number of erythrocytes.[49]
There is evidence suggesting that the enhanced erythroid progenitor
cell growth and differentiation observed in animal and cellular models
of the 5q- syndrome and DBA treated with L-leucine occurs through
activation of the mTOR pathway.[45,50] These data support the rationale for clinical trials of L-leucine as a therapeutic agent for the 5q- syndrome and DBA.
A
recent report described three MDS patients with isolated del(5q) who
were treated with L-leucine for up to three months. No adverse effects
were observed during L-leucine treatment, however none of the patients
showed an improvement in the cytopenia or transfusion requirements.[51] Leucine absorption tests may be useful in determining the optimal dose[48]
and in vitro measurement of basal and post-L-leucine translation levels
could help identifying patients that are more likely to respond to
L-leucine therapy.[48] It will be important to
determine the efficacy of L-leucine treatment in larger patient cohorts
within the context of clinical trials. Indeed, clinical trials
evaluating the therapeutic use of L-leucine in DBA are underway in the
US and in Russia (NCT02386267 and NCT01362595, www.clinicaltrials.gov).
Cenersen. Recently, cenersen, a clinically active 20-mer antisense oligonucleotide complementary to exon 10 of TP53, has been shown to suppress p53 expression and restore erythropoiesis in del(5q) MDS patient cells in culture.[47]
Cenersen treatment of RPS14-deficient erythroblasts significantly
reduced cellular p53 and PUMA expression, decreased apoptosis and
increased cell proliferation. Cenersen significantly suppressed nuclear
p53 in CD34+ cells isolated from
del(5q) MDS patients. Erythroid burst recovery increased in proportion
to the magnitude of p53 suppression without del(5q) clonal suppression.
Dexamethasone, a p53 antagonist, was added to lenalidomide treatment in
eight lower-risk, transfusion-dependent, del(5q) MDS patients with
acquired drug resistance. Transfusion independence was restored in five
patients, with expansion of erythroid precursors and decreased p53
expression. This study shows that targeted suppression of p53 restores
effective erythropoiesis in lenalidomide-resistant del(5q) MDS.[47] A clinical trial testing the benefits of cenersen in lower-risk MDS patients is in progress (NCT02243124, www.clinicaltrials.gov).
Data from AML and CLL clinical trials have shown increased cytotoxicity
and enhanced sensitivity to conventional chemotherapy when given in
combination with cenersen, with no safety issues associated with the
use of cenersen.[52,53] Transient pharmacological
inhibition of p53 has been shown not to increase the incidence of
cancer in a murine carcinogenicity model.[54]
Suppression of p53 could possibly be a therapeutic option in humans if
the tumor suppressor function of p53 is only transiently abrogated and
no long-term adverse effects are observed.
Disease Progression
Conclusions
Several cooperating events seem to be necessary in the development
of the 5q- syndrome. The studies described above show that p53
activation secondary to RPS14 haploinsufficiency underlies the anemia
observed in patients with the 5q- syndrome. Loss of the miRNA genes
miR-145 and miR146a seems to play a role in the development of the
megakaryocytic abnormalities observed in this disorder. The molecular
abnormality that confers a clonal growth advantage in the 5q- syndrome
has remained elusive for a long time, but it has now been shown that
haploinsufficiency of CSNK1A1 may be the cause of the initial clonal
expansion in 5q- syndrome patients. Thus molecular abnormalities
associated with the major features of the 5q- syndrome have now been
identified in a number of genes that map to the CDR.
Mutations of TP53
have been associated with disease progression in del(5q) MDS. Other
gene mutations have been described in a small number of del(5q)
patients that evolved to AML and their potential role in disease
transformation will need to be confirmed in larger patient cohorts. It
is possible that there are other events still unidentified that may be
important in disease pathogenesis. Whole genome sequencing studies may
identify new changes in intronic or regulatory regions of genes mapping
within the CDR of the 5q- syndrome or elsewhere in the genome that are
associated with the development and/or progression of this disorder.
Haploinsufficiency of other genes may also be involved in disease
pathogenesis.
Great progress has been made over the past decade in the elucidation of the molecular basis of the 5q- syndrome (Figure 2), and new insights into disease mechanisms are leading to the development of novel treatments for the 5q- syndrome.
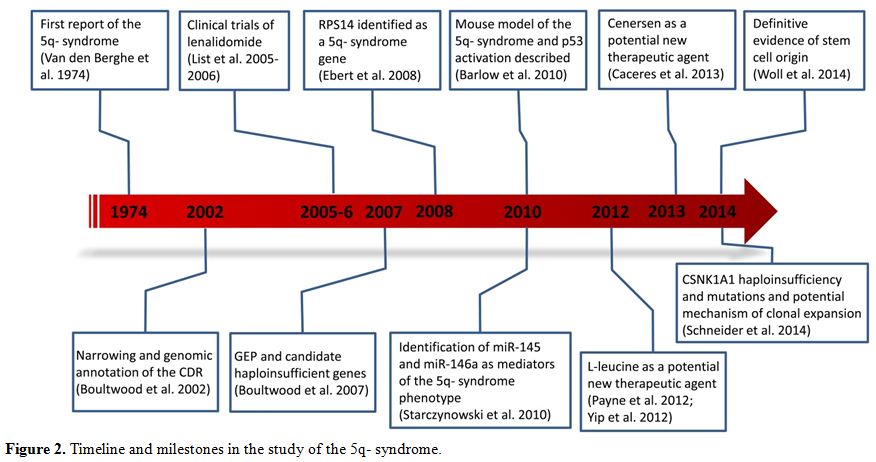 |
Figure 2. Timeline and milestones in the study of the 5q- syndrome. |
Acknowledgements
AP and JB acknowledge the support of Leukaemia & Lymphoma Research (UK).References
 . . . . . . . . . . .. ... . ... .... . .. . .. .. . . .. . . ..... . ... .. . . . . . . .. . . ..
. . . . . . . . . . .. ... . ... .... . .. . .. .. . . .. . . ..... . ... .. . . . . . . .. . . ..[TOP]