Received: October 9, 2015
Accepted: Jamuary 11, 2016
Mediterr J Hematol Infect Dis 2016, 8(1): e2016013, DOI 10.4084/MJHID.2016.013
This article is available on PDF format at:

| This is an Open Access article distributed
under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
|
Abstract Objectives: Our aim was to
study mannose-binding protein (MBP) polymorphisms in exonic and
promoter region and correlate it with associated infections and
vasoocculsive (VOC) episodes in sickle cell disease (SCD) patients
since MBP plays an important role in innate immunity by activating the
complement system. Methods: We studied the genetic polymorphisms in the Exon 1 (alleles A/O) and promoter region (alleles Y/X; H/L, P/Q) of the MBL2 gene, in SCD patients as an increased incidence of infections is seen in these patients. A PCR-based, targeted genomic DNA sequencing of MBL2 was used to study 68 SCD Omani patients and 44 controls (healthy voluntary blood donors). Results: In SCD patients, the frequency of the genotype related to the high production of MBL was 0.35 (YA/YA) and for intermediate/low production was 0.65 (YA/XA, XA/XA, YA/YO, XA/YO, YO/YO). The observed frequencies of MBL2 gene promoter polymorphism (-221, Y/X) were 44.4% and 20.5% for the heterozygous genotype Y/X and 3.2% and 2.2% for the homozygous (X/X) respectively between SCD patients and controls. MBL2 Exon1 gene mutations were 29.4% and 50% for the heterozygous genotype A/O and 5.9% and 6.8% respectively for the homozygous (O/O) genotype between SCD patients and controls. The distribution of variant MBL2 gene polymorphisms did not show any correlation in SCD patients with or without VOC attacks (p=0.16; OR-0.486; CI=0.177-1.33), however, it was correlated with infections (p=0.0162; OR-3.55; CI 1.25-10.04). Conclusions: Although the frequency of the genotypes and haplotypes of MBL2 in SCD patients did not differ from controls, overall in the SCD patient cohort the increased representation of variant alleles was significantly correlated with infections (p<0.05). However, these variant MBL2 polymorphisms did not seem to play a significant role in the VOC episodes in this SCD cohort. |
Introduction
Sickle cell disease (SCD) is characterized by a striking variability
in the clinical presentation ranging from an early-onset
life-threatening disease to a milder condition compatible with an
almost normal life course. Polymerization of deoxy Hb S with red blood
cell (RBC) deformation, desiccation and increased rigidity results in
painful vasoocclusive crises (VOC) and hemolytic anemia. Adherence of
sickle RBC stimulates endothelial cells to upregulate their adhesion
molecules, which accelerates the adhesion cascade.[1]
Activated endothelium also releases a broad range of cytokines,
including granulocyte-macrophage-colony-stimulating factor (GM-CSF),
interleukin (IL)-1, IL-3, IL-6, IL-8 and tumor necrosis factor (TNF-a),
and these have been detected in the plasma of patients with SCD.[2,3]
Neutrophils may also become activated during this cascade of
vasoocclusive events, and neutrophil adherence may contribute to
vasoocclusion,[3] as well as endothelial cell damage.[4]
Patients with SCD have an increased tendency to infection,[5] especially with encapsulated organisms, which is due in part to the poor splenic function,[6] but might also be a feature of altered neutrophil and monocyte function.[7]
There is now ample evidence indicative of an ongoing inflammatory state
between painful crises in SCD involving neutrophils, monocyte
activation and an abnormality of cytokine-regulated neutrophil
function, which may compromise the host defenses against certain
microorganisms.[8] In this context, polymorphism of
the mannose-binding lectin (MBL) has been documented as potential
immunogenetic modulating factors that could constitute an additional
risk of infection in SCD.[9] However, polymorphisms of the Fc receptor may, in fact, protect SCD patients from infections with H. influenzae.[10]
Mannose-binding lectin (MBL) is a serum protein of hepatic origin belonging to a family of Ca2+-dependent collagenous lectins, most of which are components of the innate immune system or natural immunity.[11,12] Mutations in the mannose binding protein gene have been associated with recurrent infections.[13-15] A single gene, MBL2, located on chromosome 10, codes for human MBL.[16,17]
Mannose-binding lectin may exert its action through binding to
mannose-rich, and N-acetyl-glucosamine oligosaccharides present on a
variety of microorganisms. Therefore, it activates the complement
system by MBL-associated serine proteases and by interacting with novel
receptors on phagocytes.[18-20]
The mannose
binding protein, being part of the innate immune system, is considered
particularly important in the vulnerable period of infancy before an
adequate specific immune protection was attained by the adaptive immune
system.[21]
Five single-nucleotide polymorphisms influencing serum MBL levels have been identified.[22] Three variant alleles have been described in exon 1 of the MBL2 gene.[23-25] These variants are due to 3 single-base pair substitutions at codon 54 (allele B), codon 57 (allele C), and codon 52 (allele D). They, independently, cause low serum MBL levels.[23] The normal wild type allele is commonly designated A, and the three mutant alleles O. All variant alleles reduce the amount of functional MBL subunits in heterozygous individuals 5-to 10-fold.[26] The serum MBL concentration is also dependent on some nucleotide substitutions in the promoter region of the MBL2 gene.[27,28] In particular, a polymorphism in codon -221 (X/Y type) has a significant effect on the MBL serum concentration with the Y promoter having high and the X having low MBL-expressing activity. [26-28]
Given the relatively high prevalence of SCD in the Omani population,[29]
we decided to study the genetic polymorphism of MBL in children and
adolescent patients with SCD. The aim of the study is to establish the
MBL genotypes in the SCD patients as well as in the Omani ethnic
population. The study also attempted clinical correlation with the
type, severity of infections and complication seen in SCD patients like
VOC’s.
Materials and Methods
Patients:
The study was conducted on 68 Omani SCD patients, aged between 3–18
years (mean age ± SD; 9.4± 3.9; M:F 37:31), who were enrolled into this
case-control study. They were treated at the Pediatric Hematology Unit,
Department of Child Health, Sultan Qaboos University Hospital (SQUH).
Forty-four ethnically matched healthy voluntary blood donors were
included in this study as a control group. The healthy controls were
ethnic Omani subjects who were volunteer blood donors aged between
21–44 years (mean age ± SD; 26.4± 2.5; M:F 31:13). They were screened
with a CBC and HPLC to confirm that they were indeed normal and then
their DNA was extracted for MBP polymorphism study. This study was
undertaken after approval by the institutional research and ethics
committee and written informed consents were obtained from the
patients, guardians in case of minor patients and controls before
enrollment. A thorough history and comprehensive examination were
conducted with particular emphasis on infections, history of
vasooclusive crises and SCD complications. The diagnosis of SCD was
confirmed by molecular studies in all the patients to characterize the
SCD subtypes and haplotypes. Amongst the 68 SCD patients, 56 patients
(82%) were HbSS, 11(17%) were Sickle Thal double heterozygotes, and
1(1%) was Sickle HbD [SD] double heterozygote. All the clinical and
laboratory details were obtained from the electronic medical file
records in all patients. Patients with infection symptoms were all
investigated to document a microbiologically blood culture/ urine
culture proven infection.
DNA studies:
A 5ml blood sample was collected in tubes containing EDTA. Genomic DNA
was isolated using the semi-automated ABI PRISM™ 6100 Nucleic Acid Prep
Station, [Applied Biosystems, Foster City, CA, USA] and samples were
stored at -20°C pending analysis. All the DNA polymorphisms were
studied by direct sequencing of the relevant PCR- amplified genome
segment on ABI PRISM™ 3100 Genetic Analyzer (Applied Biosystems, Foster
City, CA, USA).
Genotyping of Exon 1 of MBL2 gene: Genotyping of exon1 (codon 52, 54 and 57 for alleles D, B, C respectively) was done by multiplex PCR as previously described.[30]
Genotyping of the promoter region of MBL2 gene:
Genotyping of the promoter region was performed by direct sequencing of
the corresponding region of interest (-65 for P/Q alleles, -221 for X/Y
alleles and -618 for H/L alleles) by appropriate primers.[31]
Analysis of haplotypes and genotypes: Haplotypes of MBL2 gene were divided into three groups according to Garrett et al.[32]
Statistical Methods:
Data was analyzed using STATA ver. 11.1 (StataCorp, College Station,
TX, USA). Numerical data were expressed as mean, standard deviation,
range. Qualitative data were expressed as frequency and percentage.
Chi-square was used to study the statistical significance of
qualitative variables. Fisher’s exact test was used with Yates
correction wherever applicable. Odds ratio [OR] and 95% confidence
intervals [CI] were calculated for risk estimation. A two-sided p value
of less than 0.05 was considered as statistically significant. The
observed and expected genotype frequencies were analyzed by using
weighted least square estimates of allele frequencies and chi-square
goodness-of-fit test to see if Hardy-Weinberg’s proportions were
respected.
Results
The study initially had recruited 85 SCD patients and 50 voluntary
blood donor controls. However, as DNA results were available only in 68
SCD patients and 44 normal controls, this was the study population
analyzed in further details and reported herein. There were 37 males
and 31 females in the patient group. Their age ranged from 3 to 18
years with the mean age of 9.4 years.
The results of genotype and allele frequencies of the Exon-1 and promoter MBL2 gene polymorphisms are shown in Table 1.
MBP exon1 and promoter variants were grouped into haplotypes, and all
the possible groupings of the observed alleles A/O (B, C, D), H/L, Y/X
and P/Q, combined, homozygote and heterozygote, with SCD and no SCD
were analyzed and tabulated. In the patients with SCD the MBL2 exon-1
allele was significantly higher while the promoter alleles Q and Y were
lower than in controls, (p <0.05). Furthermore, the combined
genotype YA/YO was also down represented in the SCD affected, whereas,
the minor mutant alleles (O, X and Q) did not obey Hardy Weinberg’s
rule.
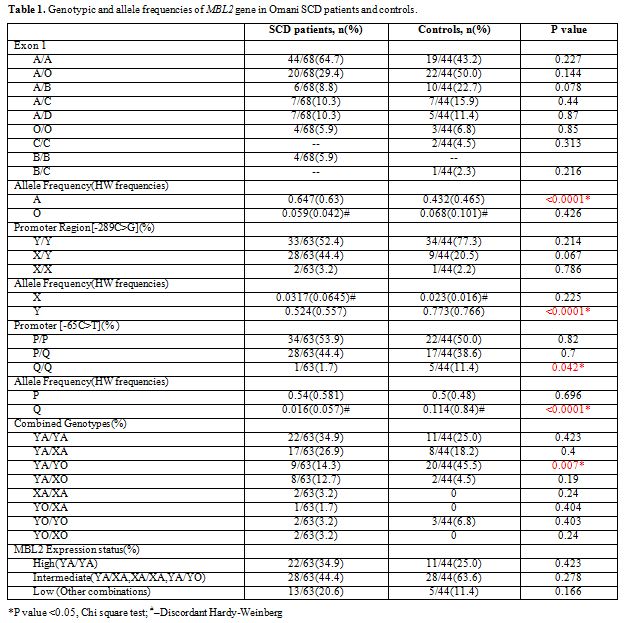 |
Table 1. Genotypic and allele frequencies of MBL2 gene in Omani SCD patients and controls. |
SCD patients were classified into three categories based on the clinical severity index according to our previous studies.[33,34]
Specifically, sickle cell disease patients were categorized as having a
mild, moderate, or severe systemic disease based on the history of
admissions/year for VOC events and associated clinical SCD
complications like Acute chest syndrome, Osteonecrosis, Splenic
sequestration, Stroke, Priapism and repeated infections. Patients with
more than three hospital admissions/year and/or SCD complications named
above were severe cases whereas those with less than one hospital
admission for VOC/year were mild, and those, needing between 1-3
admissions/year and SCD complications, were moderate cases.
Table 2 shows the distribution of MBL2
exon-1 A allele and the mutant allele O and significance of the
correlation between the MBL2 polymorphisms and presence or absence of
VOC or infections in this cohort of SCD patients. The homozygous or
heterozygous mutants [OO+AO] were less common in SCD patients with or
without VOC, but this difference was not statistically significant.
However, they were more frequent in SCD patients with infections, and
the difference was statistically significant (p<0.05).
Of 25
patients (37%), suspected to have an associated infection, 11(44%)
had a microbiologically had blood or urine documented culture. In this
group of 11 SCD patients, 6 had positive blood cultures (3-Gram
positive cocci, and one each with Bacillus spp., E.coli and Achromobacter spp.) The remaining 5 SCD patients had positive urine cultures (3-Klebsiella pneumoniae and 2-E. coli).
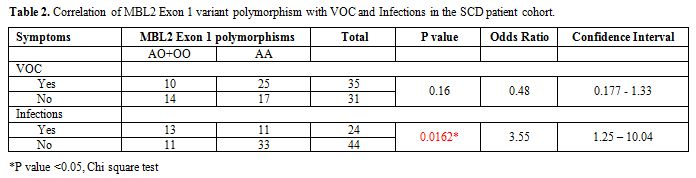 |
Table 2. Correlation of MBL2 Exon 1 variant polymorphism with VOC and Infections in the SCD patient cohort. |
Discussion
Acknowledgments
We wish to thank the Hospital Administration for allowing the use of hospital data. This work was supported by an internal grant (IG/MED/CHILD/06/01) from the College of Medicine and Health Sciences.
References

[TOP]