Eleftheria Lamprianidou1, Chryssoula Kordella1, Menelaos Papoutselis1, Zoi Bezyrgiannidou1, Evangelia Nakou1, Spyros Papamichos1, Emmanouil Spanoudakis1, Andreas Giannopoulos2, Katerina Zoi2 and Ioannis Kotsianidis1.
1 Department of Hematology, Democritus University of Thrace Medical School, Alexandroupolis, Greece.
2 Haematology Research Laboratory, Biomedical Research Foundation, Academy of Athens, Athens 11527, Greece.
Corresponding
author: Ioannis Kotsianidis, MD, PhD.
Department of Hematology, Democritus University of Thrace, Medical
School, Dragana, Alexandroupolis 68100, Greece. Tel: +302551030320;
Fax: +302551076154. E-mail:
ikotsian@med.duth.gr
Published: November 1, 2017
Received: August 31, 2017
Accepted: October 12, 2017
Mediterr J Hematol Infect Dis 2017, 9(1): e2017066 DOI
10.4084/MJHID.2017.066
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Myeloid
neoplasms with isolated isochromosome 17q [MN i(17q)] has been
described as a distinct entity with poor prognosis. However, literature
reports show a considerable clinical and molecular heterogeneity. We
describe a 58-year-old male patient who was diagnosed as refractory
anemia with multilineage dysplasia and ringed sideroblasts with
isolated i(17q). Though he initially responded well to erythropoietin,
he gradually progressed to an aggressive form of MDS/MPN refractory to
azacytidine and died 29 months after the first diagnosis. Notably, in
contrast to disease advancement, his karyotype reverted to normal,
whereas his mutational profile remained unchanged. To our knowledge,
this is the first report of karyotype normalization during disease
progression in patients with MN i(17q). It suggests that the i(17q)
anomaly is dispensable for the leukemic transformation and highlighting
the underlying clinical and molecular complexity which both has to be
resolved before the establishment of MN with isolated i(17q) as a
distinct entity.
|
Introduction
Isochromosome
17q is the most common isochromosome found in human cancer and
frequently occurs in different hematopoietic and non-hematopoietic
malignancies, including medulloblastoma, gastric and breast cancer,
chronic myelogenous leukemia (CML), acute myelogenous leukemia (AML),
myelodysplastic syndromes (MDS) and non-Hodgkin lymphomas.[1] In most hematological malignancies i(17q) is associated with aggressive disease and a complex karyotype,[2] but the presence of i(17q) as a sole abnormality is mostly restricted to the blast phase of CML, AML and MDS.[2,3]
Early reports suggested that MN patients with isolated i(17q) comprised
a distinct group with mainly myelodysplastic/ myeloproliferative
neoplasm (MDS/MPN) characteristics, resistant phenotype and dismal
prognosis.[4] These findings were further confirmed by largest series,[3,5,6]
which concluded that isolated i(17q) usually arise during disease
progression and reported a range of median overall survival (mOS) from
11 months in MDS/MPN[5] to only 4,5 months in a mixed AML and MDS patient cohort.[6] However, older series reported a considerably higher survival,[7]
while significant heterogeneity also appears to exist among the various
reports concerning MDS subtype at presentation. Similarly, the
mutational profile of such patients is complex and heterogeneous
encompassing mutations in various biologic functional categories.[3,6]
In
the present manuscript, we describe a 58-year old male patient with
isolated i(17q) who initially presented at our department as refractory
anemia with multilineage dysplasia with ringed sideroblasts (RCMD-RS).
Two years later he progressed to a fibrotic MDS/MPN with excess blasts,
while, at the same time, his karyotype converted to normal. Our case
supports the existence of recurrent characteristic features in MDS with
isolated i(17q) but argues on the effect on survival and the causality
of isolated i(17q) anomaly in leukemic progression.
Case Report
A
58-year old male patient was referred to our department due to
normocytic anemia and leucopenia in May 2008. His anamnesis included
mild diabetes type II and solitary congenital kidney with normal renal
function, and he was on no medication. A complete blood count performed
a year ago was showing hemoglobin (Hb) 121 g/L and average leukocyte
count (WBC) and platelet counts. The patient was in relatively good
condition complaining only of mild fatigue, and the physical
examination was unremarkable. Haemoglobin was 74 g/L, platelets 165x109/L, MCV 83 fl, WBC 2.98x109/L and the absolute neutrophil count (ANC) 1.4x109/L,
while biochemistry, including LDH, was normal. The rest laboratory
workup revealed erythropoietin serum level of 19.6 IU/L (normal range
3.3-24.4 IU/L), ferritin levels 418 ng/dl and B12 levels 1166 pg/ml.
The blood film showed dimorphic red cells, and hypogranular neutrophils
with ringed nuclei (Figure 1a)
and the bone marrow aspirate was consistent with refractory anaemia
with multilineage dysplasia, and 40% ringed sideroblasts (RCMD-RS).
Blast count was 1%. Bone marrow biopsy also revealed a hypercellular
marrow with trilineage dysplasia and grade I reticulin fibrosis by
Gomori's silver stain. Cytogenetic studies were performed in bone
marrow by using conventional G-banding analysis and showed 12/20
metaphases carrying the 46, XY, i(17q)(q10) and 8/20 residual normal
metaphases. By using PCR we found mutations in SETBP1, U2AF1 and KIT genes, whereas no mutations were found in SF3B1, JAK2, MPL, CALR, IDH1, IDH2, DNMT3A, SRSF2 and ASXL1
and no chimeric transcripts of BCR/ABL were detected. The patient was
transfused and started on recombinant erythropoietin (EPO) at
40.000IU/week. Two months after, Hb was 87 g/l, and we doubled the EPO
dose at 80.000IU/week. He remained untransfused for 7 months when his
Hb rose at 112 g/L, and although EPO was reduced to 40.000IU/week,
after 3 months the Hb levels reached 140 g/L, and EPO was discontinued.
He did not receive further EPO for 12 months, being asymptomatic with
normal Hb levels, whereas due to a synchronous rise in WBC and platelet
counts we performed JAK2V617F mutation analysis with negative results.
However, 24 months after diagnosis the patient developed splenomegaly 5
cm below left costal margin and low-grade fever peaking in the
afternoon. His Hb dropped at 84 g/L, his platelet count at 103x109/L and the WBC rose to 21.7x109/L. Peripheral blood smear showed 9% blasts, and immunophenotypic analysis by 4-color flow cytometry demonstrated a CD45low
population with typical myeloblast phenotype, positive for CD34, CD117,
CD33, HLA-DR and CD13, and negative for CD15, CD19, CD10, CD56, CD7 and
CD61 (Figure 1b). The aspirate
was unsuccessful (dry tap) and the bone marrow biopsy showed a
hypercellular marrow with myeloid and megakaryocytic hyperplasia,
decreased erythropoiesis, 9% blasts, and reticulin fibrosis grade 2-3,
whereas low-grade collagen fibrosis was also present. A new cytogenetic
study was pursued and, unexpectedly, revealed a regular 46, XY[30]
karyotype and normal FISH findings (Supplemental Figure S1), whereas his mutational profile remained unchanged. The patient received 5 courses of subcutaneous azacytidine at 75 mg/m2
for seven days on 28-day cycles, but his splenomegaly progressed, and
along with the ongoing fever he developed night sweats and weight loss.
We administered induction chemotherapy with cytarabine (Ara-C) 100 g/m2/d iv d1-7 and idarubicin 12 mg/m2/d iv d1-3, but the patient succumbed to sepsis after 23 days. The course of the patient’s CBC is shown in Figure 1c.
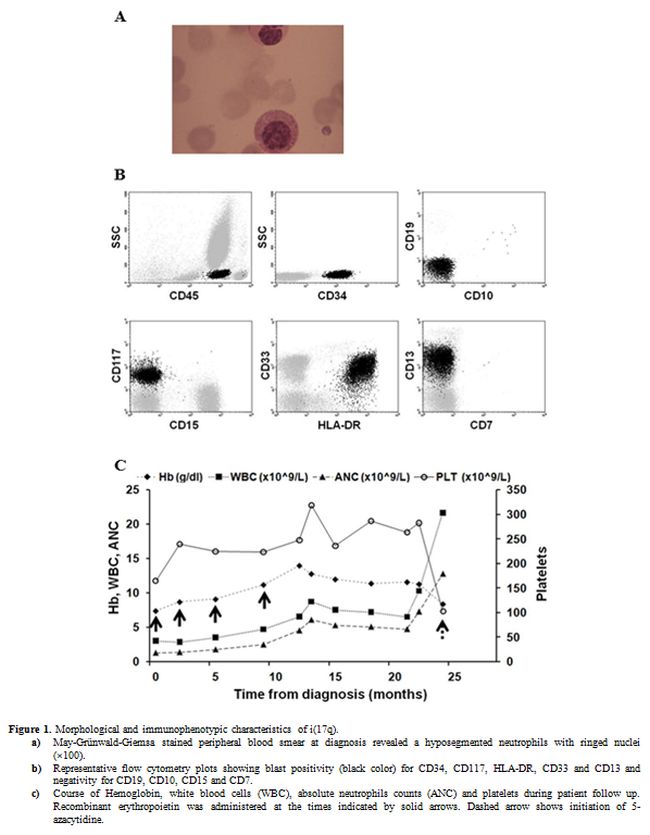 |
Figure 1. Morphological and immunophenotypic characteristics of i(17q).
a)
May-Grünwald-Giemsa stained peripheral blood smear at diagnosis
revealed a hyposegmented neutrophils with ringed nuclei (×100).
b)
Representative flow cytometry plots showing blast positivity
(black color) for CD34, CD117, HLA-DR, CD33 and CD13 and negativity for
CD19, CD10, CD15 and CD7.
c) Course of
Hemoglobin, white blood cells (WBC), absolute neutrophils counts (ANC)
and platelets during patient follow up. Recombinant erythropoietin was
administered at the times indicated by solid arrows. Dashed arrow shows
initiation of 5-azacytidine. |
Discussion
Patients
with myeloid neoplasms with isolated i(17q) share several common
characteristics regarding morphology, disease type, course, and
prognosis. However, there is still significant heterogeneity in all of
the above features among the reported patients (Table 1). By using 2008 WHO classification[8]
most patients with isolated i(17q) fall into the MDS/MPN category.
However, there are cases classified as various MDS, MPN and AML
subtypes including acute promyelocytic leukemia and even
hypereosinophilic syndrome.[2,3,5,6,9-11]
Another controversial issue is prognosis. Isolated i(17q) has
repeatedly been reported to confer a dismal outcome ranging from an mOS
of 4.5[6] to up to 14.5 months[3,5] in 3 large series.
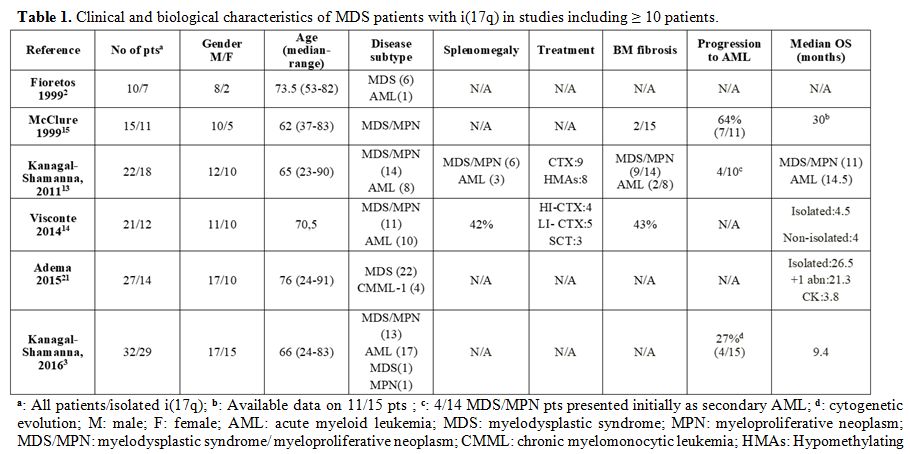 |
Table 1. Clinical and biological characteristics of MDS patients with i(17q) in studies including ≥ 10 patients. |
By contrast, in line with the 29-month survival of our patient, two other large series recorded an mOS of 30[7] and 26.5[11]
months. The exclusion of AML patients from the latter studies may
account for the discrepant findings; indeed, MDS patients with isolated
i(17q) are assigned to the intermediate-risk category by the revised
international prognostic scoring system (IPSS-R) and have a predicted
mOS of 32 months.[12] Of note, MDS/MPN patients fared
worse than the AML ones in one study, but 4 out of 14 MDS/MPN cases
were actually secondary AML.[5]
As concerns
treatment, myeloid neoplasms with isolated i(17q) are apparently
resistant to standard regimens, and such patients are candidates for
allogeneic transplantation early in the course of the disease.[5,10]
Data regarding the efficacy of hypomethylating agents in these patients
are very limited; Kanagal-Shamanna et al. reported on 5 patients, three
in azacytidine and two in decitabine, all of which failed to respond,
similarly to our patient.[5]
The more obvious
consequence of the formation of isochromosome 17q is the deletion of
one allele of TP53 gene located at 17p13. Loss of 17p might be
responsible for the Pelger-Huët dysgranulopoiesis,[13]
but coding mutations in the remaining allele are rare and usually
accompanied by additional cytogenetic abnormalities, rendering TP53 an
improbable player in the pathobiology of the syndrome.[2,3]
ASXL1, SRSF2, RAS, and SETBP1 are the most frequently mutated genes in isolated i(17q) (Table 2). The former three mutations may antedate the formation of i(17q), whereas SETBP1 mutations are associated with i(17q).[3] Our patient had mutations in SETBP1, U2AF1, and c-KIT, whereas ASXL1 was unmutated and no mutation analysis was performed in SRSF2 and RAS. Mutations in SETBP1,
mainly gain of function, are often observed in CMML, secondary AML and
atypical CML, while they are rare in childhood, de novo and
therapy-related AML.[14,15] Though linked with characteristics of poor prognosis, reports on the effect of SETBP1 mutations on survival are conflicting.[14,15] Our patient also had the U2AF1 (p.Q157P) mutation, rarely reported in MDS with isolated i(17q),[3] while it is more common in poor prognosis, advanced myelomonocytic leukemias.[16] U2AF1
mutations appear to lead to pathological splicing of several genes
involved in leukemogenesis and are strongly associated with leukemic
evolution and dismal outcome.[17] We also detected the KIT (p.D816V)
mutation, which is observed in over 90% of systemic mastocytosis cases
and at lower frequencies in patients with core binding factor AML
conferring a poor prognosis.[18] Activating mutations of KIT in AML are considered as a class I aberrations which provide a proliferative and survival advantage to the leukemic cells.[19] In MDS the above mutation is rarely found and is restricted to advanced stages,[20] whereas only one case of a different KIT
mutation has been reported so far as a sole molecular aberration in a
patient with AML with myelodysplasia-related changes and isolated
i(17q).[3] Interestingly, both U2AF1 and KIT mutations
antedated leukemic progression by two years in our patient, emphasizing
the often existing discordance between mutational profiles and clinical
course and the need for caution in the clinical translation of
molecular findings. In addition, the intriguing conversion of our
patient’s karyotype to normal during disease progression suggests that
genetic pathways unrelated to chromosome 17 are potentially involved in
the multifactorial pathobiology of this MDS entity. A hint of the
dispensability of i(17q) for the leukemic progression has been
previously reported in one patient with primary myelofibrosis who
developed a transient and progressively shrinking i(17q) clone without
changing his mutational profile. Another indication that the
development of i(17q) anomaly may represent an epiphenomenon and not
the initial oncogenic event is the fact that though the mutational
profile of our patient remained unchanged, the mutational load of each
mutated gene increased significantly during disease progression (data
not shown). Thus, the leukemogenic effect of one or more of SETBP1, U2AF1 and KIT mutants
appears to be independent of the i(17)q formation. Nevertheless, the
similarity in the clinical presentation of cases with isolated i(17)q
still suggests a role of this abnormality in the characteristic
features shared by these patients.
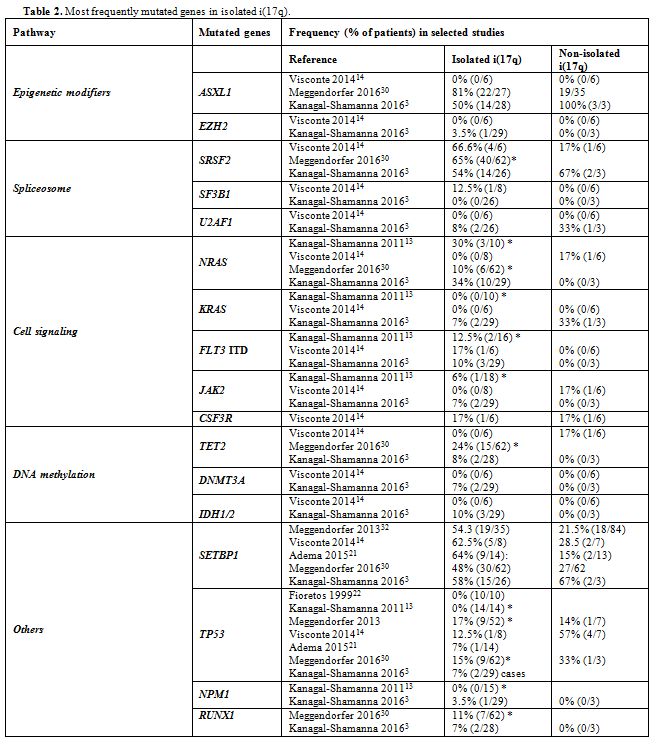 |
Table 2. Most frequently mutated genes in isolated i(17q). |
Conclusions
We
describe a patient with RCMD-RS who displayed most of the typical
characteristics of MDS/MPN with isolated i(17q) during his disease
course. Our patient is the second one with a diagnosis of acquired
idiopathic sideroblastic anemia with isolated i(17q) reported in the
literature[9] and his 29-month survival stresses the
fact that the prognosis of myeloid tumors with isolated i(17q)
potentially depends mainly on the MDS subtype at initial presentation.
More important, we report for the first time the paradoxical
disappearance of the i(17q) and karyotype normalization during disease
progression, a phenomenon that challenges the requirement and the
contribution of the i(17q) anomaly in the leukemogenic process. Our
case stresses the importance of the collection of an adequate amount of
clinical data and elucidation of the molecular basis of MDS to
accurately define and classify a new entity.
References
- Mertens F, Johansson B, Mitelman F. Isochromosomes in neoplasia. Genes Chromosomes Cancer. 1994;10(4):221-30. https://doi.org/10.1002/gcc.2870100402 PMid:7522535

- Fioretos
T, Strombeck B, Sandberg T, Johansson B, Billstrom R, Borg A, et al.
Isochromosome 17q in blast crisis of chronic myeloid leukemia and in
other hematologic malignancies is the result of clustered breakpoints
in 17p11 and is not associated with coding TP53 mutations. Blood.
1999;94(1):225-32. PMid:10381517
- Kanagal-Shamanna
R, Luthra R, Yin CC, Patel KP, Takahashi K, Lu X, et al. Myeloid
neoplasms with isolated isochromosome 17q demonstrate a high frequency
of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget.
2016;7(12):14251-8. https://doi.org/10.18632/oncotarget.7350 PMid:26883102 PMCid:PMC4924712
- Mitelman
F, Brandt L, Nilsson PG. Relation among occupational exposure to
potential mutagenic/carcinogenic agents, clinical findings, and bone
marrow chromosomes in acute nonlymphocytic leukemia. Blood.
1978;52(6):1229-37. PMid:719175
- Kanagal-Shamanna
R, Bueso-Ramos CE, Barkoh B, Lu G, Wang S, Garcia-Manero G, et al.
Myeloid neoplasms with isolated isochromosome 17q represent a
clinicopathologic entity associated with
myelodysplastic/myeloproliferative features, a high risk of leukemic
transformation, and wild-type TP53. Cancer. 2012;118(11):2879-88. https://doi.org/10.1002/cncr.26537 PMid:22038701
- Visconte
V, Tabarroki A, Zhang L, Hasrouni E, Gerace C, Frum R, et al.
Clinicopathologic and molecular characterization of myeloid neoplasms
harboring isochromosome 17(q10). American journal of hematology.
2014;89(8):862. https://doi.org/10.1002/ajh.23755 PMid:24796269
- McClure
RF, Dewald GW, Hoyer JD, Hanson CA. Isolated isochromosome 17q: a
distinct type of mixed myeloproliferative disorder/myelodysplastic
syndrome with an aggressive clinical course. British journal of
haematology. 1999;106(2):445-54. https://doi.org/10.1046/j.1365-2141.1999.01537.x PMid:10460605
- Vardiman
JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The
2008 revision of the World Health Organization (WHO) classification of
myeloid neoplasms and acute leukemia: rationale and important changes.
Blood. 2009;114(5):937-51. https://doi.org/10.1182/blood-2009-03-209262 PMid:19357394
- Lazarevic
V, Djordjevic V, Magic Z, Marisavljevic D, Colovic M. Refractory anemia
with ring sideroblasts associated with i(17q) and mutation of the TP53
gene. Cancer Genet Cytogenet. 2002;136(1):86-9. https://doi.org/10.1016/S0165-4608(02)00510-1
- Becher
R, Carbonell F, Bartram CR. Isochromosome 17q in Ph1-negative leukemia:
a clinical, cytogenetic, and molecular study. Blood.
1990;75(8):1679-83. PMid:2328318
- Adema
V, Larrayoz MJ, Calasanz MJ, Palomo L, Patino-Garcia A, Agirre X, et
al. Correlation of myelodysplastic syndromes with i(17)(q10) and TP53
and SETBP1 mutations. British journal of haematology.
2015;171(1):137-41. https://doi.org/10.1111/bjh.13355 PMid:25716545
- Greenberg
PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al.
Revised international prognostic scoring system for myelodysplastic
syndromes. Blood. 2012;120(12):2454-65. https://doi.org/10.1182/blood-2012-03-420489 PMid:22740453 PMCid:PMC4425443
- Lai
JL, Preudhomme C, Zandecki M, Flactif M, Vanrumbeke M, Lepelley P, et
al. Myelodysplastic syndromes and acute myeloid leukemia with 17p
deletion. An entity characterized by specific dysgranulopoiesis and a
high incidence of P53 mutations. Leukemia. 1995;9(3):370-81.
PMid:7885035
- Damm
F, Itzykson R, Kosmider O, Droin N, Renneville A, Chesnais V, et al.
SETBP1 mutations in 658 patients with myelodysplastic syndromes,
chronic myelomonocytic leukemia and secondary acute myeloid leukemias.
Leukemia. 2013;27(6):1401-3. https://doi.org/10.1038/leu.2013.35 PMid:23443343
- Meggendorfer
M, Haferlach C, Zenger M, Macijewski K, Kern W, Haferlach T. The
landscape of myeloid neoplasms with isochromosome 17q discloses a
specific mutation profile and is characterized by an accumulation of
prognostically adverse molecular markers. Leukemia. 2016;30(7):1624-7. https://doi.org/10.1038/leu.2016.21 PMid:26859077
- Makishima
H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, et al.
Mutations in the spliceosome machinery, a novel and ubiquitous pathway
in leukemogenesis. Blood. 2012;119(14):3203-10. https://doi.org/10.1182/blood-2011-12-399774 PMid:22323480 PMCid:PMC3321850
- Przychodzen
B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, et al.
Patterns of missplicing due to somatic U2AF1 mutations in myeloid
neoplasms. Blood. 2013;122(6):999-1006. https://doi.org/10.1182/blood-2013-01-480970 PMid:23775717 PMCid:PMC3739042
- Schnittger
S, Kohl TM, Haferlach T, Kern W, Hiddemann W, Spiekermann K, et al.
KIT-D816 mutations in AML1-ETO-positive AML are associated with
impaired event-free and overall survival. Blood. 2006;107(5):1791-9. https://doi.org/10.1182/blood-2005-04-1466 PMid:16254134
- Schlenk
RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L, et al.
Mutations and treatment outcome in cytogenetically normal acute myeloid
leukemia. The New England journal of medicine. 2008;358(18):1909-18. https://doi.org/10.1056/NEJMoa074306 PMid:18450602
- Bacher
U, Haferlach T, Kern W, Haferlach C, Schnittger S. A comparative study
of molecular mutations in 381 patients with myelodysplastic syndrome
and in 4130 patients with acute myeloid leukemia. Haematologica.
2007;92(6):744-52. https://doi.org/10.3324/haematol.10869 PMid:17550846
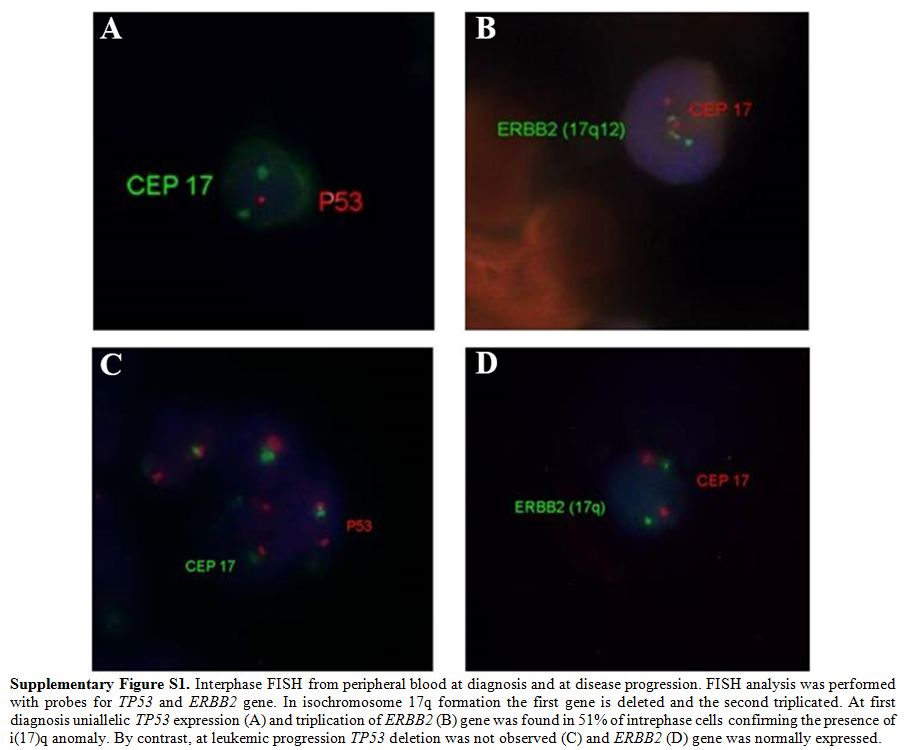 |
Supplementary Figure S1. Interphase FISH
from peripheral blood at diagnosis and at disease progression. FISH
analysis was performed with probes for TP53 and ERBB2 gene. In
isochromosome 17q formation the first gene is deleted and the second
triplicated. At first diagnosis uniallelic TP53 expression (A) and
triplication of ERBB2 (B) gene was found in 51% of intrephase cells
confirming the presence of i(17)q anomaly. By contrast, at leukemic
progression TP53 deletion was not observed (C) and ERBB2 (D) gene was
normally expressed. |
[TOP]