Roberto Ria, Franco Dammacco and Angelo Vacca.
Department of Biomedical
Sciences and Human Oncology, Section of Internal Medicine, University
of Bari “Aldo Moro” Medical School, Bari, Italy.
Corresponding
author: Roberto Ria, M.D.,
www.orcid.org/0000-0002-1515-0090.
Department of Biomedical Sciences, Section of Internal Medicine,
University of Bari “Aldo Moro” Medical School, Polyclinic, Piazza
Giulio Cesare 11, I-70124 Bari, Italy. Phone: +39-080-5593106, fax:
+39-080-5593106; e-mail:
roberto.ria@uniba.it
Published: January 1, 2018
Received: October 5, 2017
Accepted: December 23, 2017
Mediterr J Hematol Infect Dis 2018, 10(1): e2018011 DOI
10.4084/MJHID.2018.011
This article is available on PDF format at:

This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
The
heavy chain diseases (HCDs) are rare B-cell malignancies characterized
by the production of a monoclonal immunoglobulin heavy chain without an
associated light chain. There are three types of HCD, defined by the
class of immunoglobulin heavy chain produced: IgA (α-HCD), IgG (γ-HCD),
and IgM (μ-HCD). Alpha-HCD is the most common and usually occurs as
intestinal malabsorption in a young adult from a country of the
Mediterranean area. Gamma- and μ-HCDs are rarer and associated with a
B-cell non-Hodgkin lymphoma that produces an abnormal Ig heavy chain.
These patients may occasionally be diagnosed with a monoclonal
gammopathy of undetermined significance (MGUS). Fanconi syndrome, on
the other hand, can be primary (inherited) or secondary (acquired). The
only exception to this rule is the idiopathic form. Adult acquired
Fanconi syndrome can be a rare complication of a monoclonal gammopathy.
At diagnosis, most patients have an MGUS or smoldering multiple
myeloma, with renal failure and evidence of osteomalacia. During
follow-up, patients can develop an end-stage renal disease.
Chemotherapy provides little benefit on renal function.
|
Introduction
Heavy
chain diseases (HCDs) are a group of three rare B-cell neoplasms that
are clinically and morphologically distinct from one another, but have
in common the production of an abnormal immunoglobulin (Ig) heavy chain
incapable of binding light chains (LCs). The altered heavy chains
contain deletions, insertions, and point mutations that are acquired
during somatic hypermutation. These structural abnormalities typically
result in loss of a large portion of the constant-1 (CH1) domain of the
Ig heavy chain molecule responsible for LC binding, with different
effects on the variable (V), diversity (D), and joining (J) regions.[1-3]
In
the absence of an associated LC, the CH1 domain of the regular heavy
chain binds to heat-shock protein 78 (HSP78) and undergoes degradation
in the cell proteasome compartment. Regular heavy chains unassociated
with LCs are therefore never detected in serum or urine. In the HCDs,
the altered structure of the CH1 domain prevents the heavy chain from
binding both the LC and HSP78, thereby allowing it to bypass
degradation by the proteasome and be secreted into the serum or urine.[4]
In addition, recent work suggests that the altered heavy chain, which
forms part of the transmembrane B-cell receptor, may facilitate
antigen-independent aggregation and down-stream signaling by the
receptor, thereby conferring a growth advantage to neoplastic cells.[5]
This characteristic feature gives rise to three different HCDs,
depending on the heavy chain class that is produced—each with a unique
clinical presentation and characteristic findings on immunologic
evaluation and in biopsy specimens of involved tissues. Each HCD
appears to represent an unusual variant of a type of lymphoma, and none
of them can be defined a true plasma cell neoplasms.[6]
Guido
Fanconi, a Swiss pediatrician, described a child with a symptomatology
characterized by glycosuria, albuminuria, rickets, and dwarfism.[7] This syndrome bears his name.[8]
Environmental agents that cause Fanconi syndrome (FS) include exposure
to heavy metals (like cadmium, lead, mercury, platinum, uranium), other
substances (lysol, paraquat, toluene, the amino acid lysine taken as a
nutritional supplement). Moreover, it may be caused by various drugs,
including certain chemotherapeutic drugs (e.g., ifosfamide,
streptozotocin), antiretrovirals (e.g., didanosine, cidofovir,
tenofovir), and tetracycline. Acquired FS can also occur after renal
transplantation and in patients with multiple myeloma, amyloidosis,
intoxication with heavy metals or other chemicals, or vitamin D
deficiency.[9,10] FS may complicate plasma cell
dyscrasias when free LCs (FLCs) undergo homotypic polymerization within
the endo-lysosomal system of the proximal tubular epithelium to form
intracellular crystals. In this case, it is defined adult acquired FS,[11,12]
a rare complication of plasma cell dyscrasias, usually associated with
a monoclonal gammopathy of undetermined significance (MGUS). Overt
hematologic malignancies may occur, such as multiple myeloma,
Waldenström’s macroglobulinemia, or other lymphoproliferative
disorders.[11,12]
Heavy-Chains Diseases
Epidemiology and clinical features (Table 1).
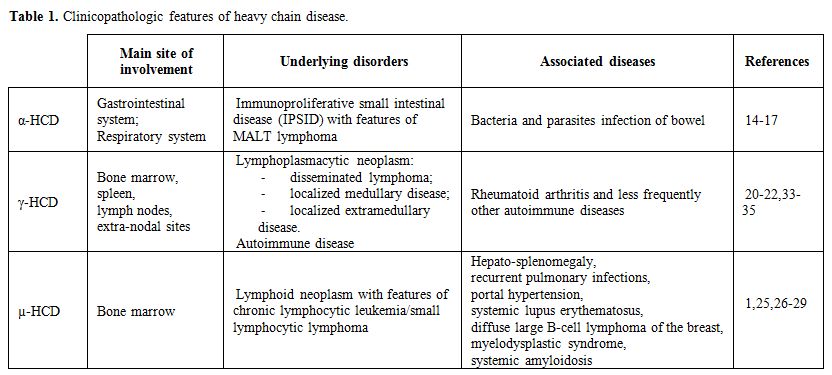 |
Table
1. Clinicopathologic features of heavy chain disease. |
α-HCD
– It is the most common of the three HCDs, with more than 400 cases
described in the literature since its initial description in 1968.[13]
It has a striking epidemiology, affecting primarily subjects of
Mediterranean, Northern African, and Middle Eastern descent,
particularly those of low socio-economic background, suggesting an
environmental, possibly infectious, pathogenetic mechanism.[14]
α-HCD
is most prevalent during the second and third decades of life, with a
slight male predominance, typically affects the gastrointestinal system
and rarely the respiratory tract. Lymphoma-like pictures have also been
described (Figure 1).
Malabsorption syndrome with weight loss that can cause growth
retardation, amenorrhea, alopecia, diarrhea, and abdominal discomfort
are the typical symptoms of gastrointestinal α-HCD. Nausea and emesis
can also be present. In advanced gastrointestinal α-HCD, ascites and
anasarca can be detected at the physical examination. Finger clubbing
and tetany can also be seen.[15] Generalized
lymphadenopathy and hepato-splenomegaly are hallmarks of the
lymphomatous forms that have been recognized as a distinct entity.[16]
Patients with respiratory tract involvement show a restrictive pattern
on pulmonary function tests. They present dyspnea, mild hypoxemia, and
diffuse pulmonary infiltrates and, in some cases, eosinophilia, skin
rash, hilar lymphadenopathy, and lymphomatous involvement of the
pharyngeal mucosa.[17]
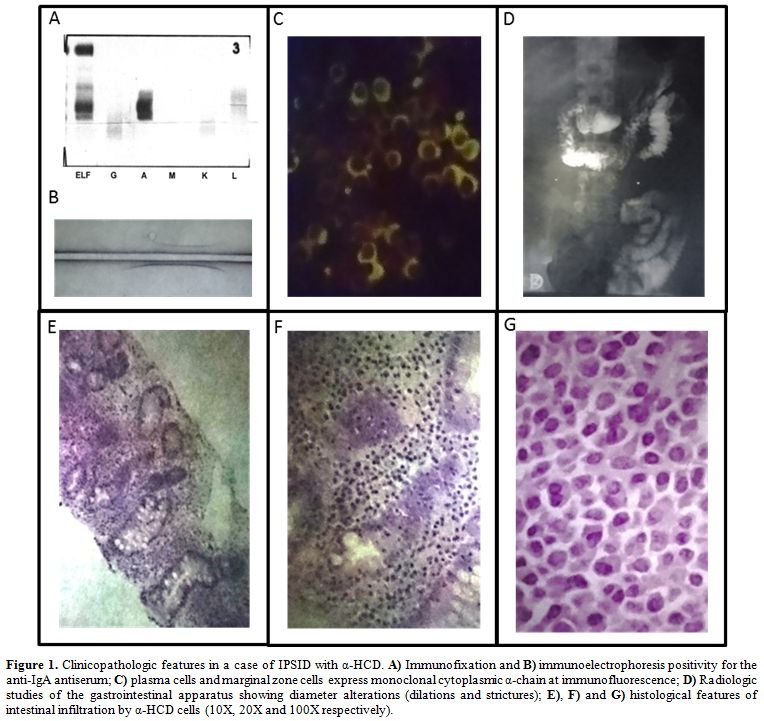 |
Figure 1. Clinicopathologic features in a
case of IPSID with α-HCD. A) Immunofixation and B)
immunoelectrophoresis positivity for the anti-IgA antiserum; C) plasma
cells and marginal zone cells express monoclonal cytoplasmic α-chain at
immunofluorescence; D) Radiologic studies of the gastrointestinal
apparatus showing diameter alterations (dilations and strictures); E),
F) and G) histological features of intestinal infiltration by α-HCD
cells (10X, 20X and 100X respectively). |
γ-HCD – It is also called Franklin’s disease, after the physician who first described it in 1964.[18]
It is uncommon, with approximately 130 cases reported in the
literature, being the age at diagnosis between 51 and 68 years,
with a female predominance.[19-21] An autoimmune
disease such as rheumatoid arthritis (the most common), Sjögren
syndrome, systemic lupus erythematosus, vasculitis, myasthenia gravis
and autoimmune cytopenias (particularly idiopathic thrombocytopenic
purpura) can be found in about 25% of patients.[22]
The manifestations of the associated autoimmune disease often herald
the diagnosis of γ-HCD by many years. A lymphoplasmacytic neoplasm is
present in 83% to 91% of these patients.
Three different
clinical patterns of γ-HCD have been described, based on the presence
or absence of an associated lymphoma. In 57% to 66% of patients, a
disseminated lymphoma associated with constitutional symptoms (i.e.,
fever, malaise, and weight loss) is present. Generalized
lymphadenopathy, splenomegaly, and hepatomegaly are present in 50% of
cases,[14,20] whereas roughly 25%
of patients present a lymphomatous bone marrow involvement (localized
medullary disease) or a localized extra-nodal disease (localized
extramedullary disease). This last form commonly involves the skin,
less frequently the thyroid or parotid gland, the oropharyngeal cavity,
and the gastrointestinal tract.[20,22]
Finally, 10-17% of patients have a pre-existing autoimmune disease with
a clinical presentation characterized by rheumatoid nodules, rashes,
synovitis, and joint deformities.
µ-HCD
– It is the rarest of the HCDs, with only 30 to 40 cases reported in
the literature. The first two patients were described in 1970 by Forte
et al.[23] and Ballard et al..[24] The disease occurs predominantly in Caucasian males, with a median age of 58 years at diagnosis.[14] Most patients with µ-HCD have a lymphoid neoplasm resembling chronic lymphocytic leukemia/small lymphocytic lymphoma.[1,25]
Palpable,
superficial lymphadenopathy can be identified in 40% of the patients.
Splenomegaly is frequent, and hepatomegaly can be found in about 25%.[14]
Rare associations of µ-HCD with recurrent pulmonary infections, portal
hypertension, and pancytopenia, systemic lupus erythematosus, diffuse
large B-cell lymphoma of the breast as well as myelodysplastic
syndrome, carpal tunnel syndrome, and systemic amyloidosis have been
reported.[26-29]
Diagnostic Approach.
The diagnosis of HCDs remains challenging due to their rarity, their
variable clinical presentation, and the skill required in interpreting
immunologic laboratory tests and tissue biopsies from affected
patients. A close collaboration between clinicians and pathologists is
usually needed. Two-dimensional immunoelectrophoresis has been shown to
be a useful diagnostic tool for all three types of HCD.[30]
α-HCD
- Common laboratory abnormalities include mild-to-moderate hypochromic
anemia, deficiency of vitamins and minerals, high levels of alkaline
phosphatase (the gastrointestinal isoform of the enzyme), electrolytic
disorders (i.e., hypoalbuminemia, hypocalcemia, hypokalemia, and
hypomagnesemia).[15] Serum protein electrophoresis
may appear normal or show hypogammaglobulinemia, but sometimes a broad
monoclonal band migrating to the α2 or β region of the electrophoretic
pattern can be seen. Positivity for the anti-IgA antiserum by
immunofixation is mandatory to confirm the diagnosis (Figure 1 panel A, B).[31]
The abnormal α-heavy chains can be detected in jejunal or gastric
fluids as well as in urine in only small amounts, but Bence Jones
proteinuria has never been detected.[31]
Radiologic studies of the gastrointestinal apparatus can show diameter alterations (dilations and strictures) (Figure 1 panel D), hypertrophic or pseudo-polypoid mucosa, or coarse mucosal folds.
Since
α-HCD typically affects the proximal small bowel at the level of the
duodenum or jejunum, endoscopy is mandatory and can reveal five
different patterns: i) infiltrative, ii) nodular, iii) ulcerations, iv)
mosaic, v) isolated mucosal fold thickening. The first two patterns are
most sensitive and characteristic for the diagnosis of α-HCD.[32]
The
histological features of α-HCD are those of an extranodal marginal zone
lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) (Figure 1 panel E, F, G),
also named immunoproliferative small intestinal disease (IPSID).
Bacterial or parasites infection (i.e., Campylobacter jejuni or
Helicobacter pylori) can be associated.[33-35] A
lymphoplasmacytic infiltrate rich in plasma cells can be detected in
the lamina propria of the bowel, and lymphoepithelial lesions may also
be present. The infiltrate can cause villous atrophy and is admixed
with small lymphocytes, resembling marginal zone B cells.[33,36-38]
All the α-heavy chain cells (plasma cells and marginal zone cells)
typically express monoclonal cytoplasmic α-chain without light chains (Figure 1 panel C). The immunophenotype of α-heavy chain cells is shown in Table 2. Finally, the presence of intestinal bacteria and parasites should be looked for on biopsy specimens.
γ-HCD
- Laboratory evidence of autoimmune diseases or bone marrow
infiltration can be detected during diagnostic procedures in patients
with γ-HCD. These comprise cytopenias, in particular, normochromic
normocytic anemia, Coombs-positive autoimmune hemolytic anemia, and
thrombocytopenia. In some cases, monoclonal plasmacytoid lymphocytes or
plasma cells are present in the circulation, as well as features of
chronic lymphocytic leukemia or plasma cell leukemia.[14]
Serum protein electrophoresis may appear normal or show a monoclonal
band migrating to the β-region of the electrophoretic pattern, where it
is often concealed by other proteins. Rarely, biclonal gammopathy with
an additional intact monoclonal Ig may be present.[20]
Positivity for the anti-IgG antiserum, without associated light chains,
by immunofixation is mandatory to confirm the diagnosis.
Due to their low molecular weight and existence as dimers, the abnormal γ-heavy chains often can be detected in the urine.[2] Small amounts of FLCs may be excreted in urine as Bence Jones protein.[39] Other laboratory findings include high serum levels of IgG, with normal serum FLCs.
The
pathologic heterogeneity of γ-HCD makes the histological diagnosis
rather difficult. Histological findings of γ-HCD are typically
associated with a lymphomatous infiltration of affected tissues such as
bone marrow, spleen, lymph nodes, as well as extra-nodal sites involved
in MALT lymphomas, such as skin, thyroid, salivary glands,
gastrointestinal tract, and conjunctiva.[21,22] The
infiltrate is formed by a mixed population of lymphocytes, plasmacytoid
lymphocytes, and plasma cells, similarly to a lymphoplasmacytic
lymphoma or, in some cases, it is more polymorphous showing
immunoblasts, eosinophils, and histiocytes in a variable number.
Atypical Reed-Sternberg–like cells have been described, thus inducing
Hodgkin lymphoma or certain types of peripheral T-cell lymphoma to be
considered in a morphologic differential diagnosis.[22]
Less frequently, γ-HCD can be similar to B-cell neoplasms, such as MALT
lymphoma, splenic marginal zone lymphoma, or other splenic small B-cell
lymphomas.[21] The association of γ-HCD with T-cell
large granular lymphocytic leukemia and extranodal marginal zone
lymphoma have also been reported.[40,41]
The immunophenotype of γ-heavy chain cells is shown in Table 2.
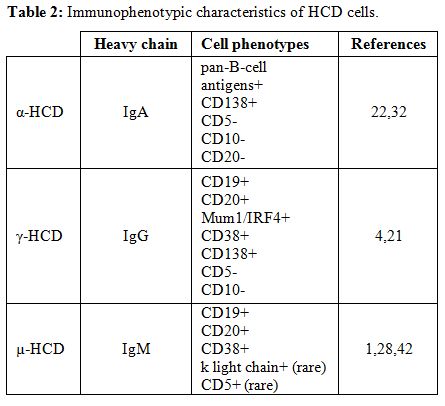 |
Table 2. Immunophenotypic characteristics of HCD cells. |
µ-HCD
– Hypoproliferative anemia related to bone marrow infiltration by
neoplastic cells is the most common laboratory abnormality of µ-HCD;
thrombocytopenia and lymphocytosis are less common.[27,28]
Serum protein electrophoresis is generally normal or shows a rather
broad monoclonal band. In a few cases, biclonal gammopathy with an
additional intact monoclonal Ig may be present.[1] Immunofixation that is positive with anti-µ but not with anti-kappa or anti-lambda LCs confirms the diagnosis.[42]
Bence Jones proteinuria is frequently detected because the neoplastic
cells also produce monoclonal LCs, usually of kappa type, that fail to
assemble with the truncated heavy chain;[1,24,42] however, Bence Jones proteinuria rarely causes renal complications.[43]
Bone
marrow smears and touch preparations show characteristic plasma cells
with prominent cytoplasmic vacuoles admixed with small, round
lymphocytes.[1,24,44,45] The immunophenotype of µ-heavy chain cells is shown in Table 2.
Treatment and Prognosis.
No standardized therapies are available for the HCDs, because of their
rarity and their clinicopathologic features characterized by
association with various conditions such as infectious, autoimmune and
lymphoproliferative diseases.
α-HCD
- Since it has a higher incidence in subjects of lower socio-economic
status, sanitation improvement would be expected to reduce its
occurrence. If untreated, α-HCD may initially progress locally, and
then spread systemically. Small bowel obstruction, perforation, and
intussusception that can be fatal are the dreadful local complications
of enlargement of the lymphomatous tissue. Other potential causes of
death are severe malnutrition and subsequent cachexia, as well as
infectious complications.[46]
If a bacterial or
parasitic gastro-intestinal infection is documented, it should be
eradicated with appropriate antimicrobial therapy. An empirically
administered antibiotic treatment is often recommended even in the
absence of a demonstrated infection. Metronidazole, ampicillin, or
tetracycline are the antibiotics of choice for this empiric therapy.
Antimicrobial treatment should be administered for a 6-months course,
although an early regression or a rapid improvement of symptoms is
typically observed in antibiotic-sensitive patients. A shorter
duration has been shown to cause a relapse of the disease.
Although
33-71% of patients with early-stage show clinical, laboratory, and
histological remission in response to antimicrobial treatment, disease
recurrences are frequent.[47] Refractory disease is
treated with either total abdominal radiation or, more commonly, with
doxorubicin-containing combination chemotherapy. Regimens such as CHOP
(cyclophosphamide, doxorubicin, vincristine, and prednisone), CHVP
(cyclophosphamide, doxorubicin, teniposide, and prednisone), or ABV
(doxorubicin, bleomycin, and vinblastine) have been shown to induce
better results than doxorubicin-free regimens such as COPP
(cyclophosphamide, vincristine, procarbazine, and prednisolone).[48-50] The complete remission rate after treatment with multi-drug chemotherapy is 64%, and 5-year overall survival is 67%.[51]
Surgical
debulking of the tumor mass can be pursued, followed by systemic
chemotherapy, but it should be limited to the management of
complications.[52] For patients with a refractory or
relapsing disease, high-dose therapy with autologous hematopoietic stem
cell transplantation should be considered.
γ-HCD
- Treatment of γ-HCD is typically tailored to the symptomatology of the
patient and to the presence of an accompanying autoimmune disease or
overt lymphoma. Chemotherapy is recommended in patients with
lymphomatous dissemination or with a localized medullary disease.
Chlorambucil, or melphalan and prednisone, or bortezomib and
prednisone, and rituximab in CD20-positive disease are the treatment of
choice for plasma cell–predominant disease. CHOP regimen (with
rituximab in CD20-positive cases) shows the best results in
aggressive/refractory patients. Inoue et al.[53] reported that the combination of fludarabine and rituximab was effective in patients with γ-HCD associated with pancytopenia.
While surgical resection or radiation therapy have been successfully employed in patients with localized extra-nodal disease,[22]
a ‘watch and wait’ strategy can be adopted in asymptomatic patients
without lymphoma. When co-existing autoimmune disorders are diagnosed,
they should be treated with immunosuppressive treatments according to
usual guidelines. Adequate prophylaxis and surveillance for infectious
complications are critical points. Prognosis is highly variable because
of the heterogeneity of γ-HCD and the lack of a standardized treatment.
Occasional
spontaneous remissions have been reported in patients with no overt
lymphoma, who are nonetheless expected to undergo a prolonged survival
without treatment.[22] A sustained, complete clinical
and immunologic remission is usually achieved in patients with treated,
localized lymphoma. The natural history of γ-HCD associated with
systemic lymphoma may be either aggressive and rapidly progressive, and
hence associated with poor prognosis, or exhibit an indolent course. In
the Mayo Clinic series, median survival was 7.4 years (range, one month
to more than two decades).[20]
µ-HCD
- Only a few data have been reported about the treatment and prognosis
of this disease because of its rarity. A ‘watch and wait’ approach is
recommended in patients with detectable monoclonal µ heavy chains who
are otherwise asymptomatic.[54] If and when an
underlying malignancy develops, useful treatment regimens are CHOP, CVP
(cyclophosphamide, vincristine, and prednisone), single-agent
fludarabine or cyclophosphamide.[14,45]
Reported median overall survival is approximately two years, ranging
from less than a month to over a decade. However, these data are likely
underestimated, since the presence of monoclonal µ heavy chains is
frequently missed on serum protein electrophoresis, especially in the
absence of an associated overt lymphoma. A spontaneous remission of
µ-HCD can rarely be observed.[14]
Myeloma-Associated Fanconi Syndrome (MAFS)
Pathophysiology.
The precise pathophysiology of MAFS is still unknown. The biological
and clinical features result from the impaired function of the proximal
renal tubule, consequent to reabsorption of some filtered substances.[55,56]
In particular, FLCs, generally of kappa isotype, secreted by plasma
cells are a major cause of MAFS or other plasma cell dyscrasias. In
virtually all cases, the diagnosis of FS precedes that of the
underlying hematological disorder, generally a ‘smoldering’ multiple
myeloma.[11,12]
FLCs slowly accumulate in the epithelial cells of the proximal tubule, forming crystals that can be demonstrated in all cases.[11,57,58]
Accumulation and crystallization take place in the lysosomes of tubular
cells and endoplasmic reticulum of plasma cells. These LCs have the
highest homology with a single germline variable segment sequence:
LCO2/O12 and non-polar amino acid residues exposed in the CDR1 region.[56-58]
This peculiar sequence is responsible for the crystallization because
of the resistance of the LC variable domain to proteolysis by several
enzymes such as cathepsin B[54,55] and of the
subsequent damage of the proximal tubule. The functional impairment of
the proximal kidney tubule can cause aminoaciduria, glycosuria with
normal glycemia, metabolic acidosis and increased clearances of uric
acid and phosphate. Phosphate loss is responsible for osteomalacia,
with bone pain and pseudo-fractures.[59,60]
Intracellular crystallization seems associated with the slowly
proliferative character of the tumor, in that the intracellular
accumulation of crystals impairs the proliferation of plasma cells, and
this may be a criterion for not treating the proliferative disease.[61-62]
In
contrast, when the LC variable domain derives from another germline
variable segment such as LCO8/O18, MAFS occurs without evidence of
intracellular crystallization, even when searched for by electron
microscopy.[63,64] In this case, defects of tubular
reabsorption and urine acidification are usually a consequence of the
direct toxicity of FLCs on epithelial cells of the proximal tubule, in
the absence of crystalline deposits.[65] Myeloma FLCs can interfere with the uptake of alanine, phosphate, and glucose.[66,67]
There has also been one report of LC-FS and nephrogenic diabetes
insipidus, suggesting that resistance to antidiuretic hormone can also
occur.[68] Interestingly, this patient had distal (type I), not proximal, renal tubular acidosis.
Clinical features and diagnostic approach.
Clinical manifestations of MAFS include defects in sodium-coupled
co-transport processes producing type II renal tubular acidosis,
aminoaciduria, phosphaturia, and glycosuria. The associated multiple
myeloma is often low-grade.[11] The offending
monoclonal FLCs are usually of the kappa type and possess uncommon
non-polar or hydrophobic residues in the complementarity-determining
region-1 (CDR1).[61] This unique proximal tubular
lesion may represent a subset of gammopathy-associated crystal-storing
histiocytosis, in which crystal-forming monoclonal Igs, composed of
heavy chains and typically kappa-FLC, accumulate in lysosomes of
histiocytes in soft tissues, kidney, bone marrow, spleen, liver,
stomach, and other organs.[69-70] Involvement of the
proximal tubule occurs specifically when the monoclonal FLCs are
overproduced, because intact Igs are not filtered through the
glomerulus.
The typical histological finding is intra-lysosomal
crystalline deposits of FLCs within epithelial cells of the proximal
tubule. There may be extra-renal crystal accumulation. FLCs of the VkI
subgroup are most frequently found,[61,62] although cases associated with lambda-FLCs have also been reported.[71]
The
diagnosis of FS can be made when a patient with a monoclonal plasma
cell disorder presents with hypophosphatemia, hypouricemia,
aminoaciduria, phosphaturia, and glycosuria. Bence Jones proteinuria is
usually present and is almost always of the kappa type. Crystal-storing
histiocytosis, an intra-lysosomal accumulation of monoclonal LCs that
aggregate in crystals, is observed in association with both plasma cell
and lymphoid disorders.[72,73] Although the type of
LCs involved is almost exclusively kappa, there is no consistent
association with a particular heavy chain. Crystals can form either in
histiocytes in soft tissues or parenchymal cells in bone marrow, lymph
nodes, spleen, liver, stomach, adrenal glands, proximal renal tubules,
and thyroid follicles. The initial clinical presentation depends on the
site of crystal formation and is, therefore, variable. Some patients
present with soft tissue masses in which predominantly histiocytes, but
also fibroblasts, contain crystals. The crystal formation in proximal
renal tubules is the fundamental diagnostic criterion of MAFS.
The
main laboratory abnormalities include aminoaciduria, renal glycosuria,
hypophosphatemia, hyperchloremic metabolic acidosis, hypokalemia,
proteinuria of tubular origin, and hypouricemia. The primary
manifestations include osteomalacia, polyuria, chronic acidosis, and
episodes of dehydration. FS frequently evolves into end-stage renal
failure.
Treatment and prognosis.
The prognosis in terms of survival is good in the absence of an overt
malignancy. Treatment includes symptomatic measures to prevent
osteomalacia by supplementation with phosphorus, calcium, and vitamin
D. Chemotherapy may benefit patients with rapidly progressive renal
failure or symptomatic malignancy.
Very few series of
LC-associated FS have been published, and the efficacy of the so-called
novel anti-myeloma agents, such as proteasome inhibitors and
immunomodulatory drugs (IMiDs), has not been evaluated. In most cases,
FS appears to progress toward end-stage renal disease slowly and rarely
results in symptomatic myeloma.[74] Accordingly,
therapeutic decisions should take into account treatment side effects,
particularly the potential risk of secondary myelodysplastic syndrome
from alkylating agents.[75]
All patients with an
associated overt lymphoid disorder should receive appropriate
chemotherapy, and treatment choices should be adapted to the degree of
renal failure. In patients with stages 1 to 3 chronic kidney disease,
bortezomib-based chemotherapy should be considered because of high
rates of both anti-myeloma response and recovery of renal function. In
addition, cyclophosphamide- or IMIDs-based regimens are good options to
treat bortezomib-refractory myeloma. Bendamustine may also be used.
High-dose melphalan/autologous stem cell transplantation (HDM/ASCT) may
be performed in selected non-responding patients, although the benefit
of this strategy remains to be proven. In the relapsed/refractory
setting, additional treatment options such as carfilzomib, pomalidomide
and monoclonal antibodies are now available. However, limited data have
been reported as regards their effects on patients with renal
impairment.[76]
In patients with stages 4 to 5
chronic kidney disease who are eligible for renal allograft,
chemotherapy, including HDM/ASCT, should be considered either before or
after transplantation. In patients who are not candidates for renal
transplantation, administration of chemotherapy does not result in
particular benefits.[76]
Acknowledgements
This
study was supported by the Italian Association for Cancer Research
(AIRC, Milan), Investigator grant 2013 (no. 14095), the five per
thousand Molecular Clinical Oncology Special Program (grant no. 9965;
to A. Vacca), and grants from MIUR PRIN 2009WCNS5C_004 (to R. Ria) and
2010NECHBX (to A. Vacca).
References
- Wahner-Roedler DL, Kyle RA. Mu-heavy
chain disease: presentation as a benign monoclonal gammopathy. Am J
Hematol. 1992;40:56-60. https://doi.org/10.1002/ajh.2830400112

- Fermand JP, Brouet JC. Heavy-chain diseases. Hematol Oncol Clin North Am. 1999;13:1281-94. https://doi.org/10.1016/S0889-8588(05)70127-1
- Goossens
T, Klein U, Kuppers R. Frequent occurrence of deletions and
duplications during somatic hypermutation: implications for oncogene
translocations and heavy chain disease. Proc Natl Acad Sci U S A.
1998;95:2463-8. https://doi.org/10.1073/pnas.95.5.2463
- Munshi
NC, Digumarthy S, Rahemtullah A. Case records of the Massachusetts
General Hospital. Case 13-2008. A 46-year-old man with rheumatoid
arthritis and lymphadenopathy. N Engl J Med. 2008;358:1838-48. https://doi.org/10.1056/NEJMcpc0800959
- Corcos D, Osborn MJ, Matheson LS. B-cell receptors and heavy chain diseases: guilty by association? Blood. 2011;117:6991-8. https://doi.org/10.1182/blood-2011-02-336164
- Harris
NL, Isaacson PG, Grogan TM, Jaffe ES. Heavy chain diseases. In:
Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of
tumours of the haematopoietic and lymphoid tissues. Lyon: IARC;
2008;196-9.
- Fanconi G. Der
fruhinfantile nephrotisch-glykosurische Zwergwuchs mit
hypophosphatamischer Rachitis. Jahrb Kinderheilkunde (Berlin).
1936;147:299-338.
- Lobitz S, Velleuer E. Guido Fanconi (1892-1979): a jack of all trades. Nat Rev Cancer. 2006;6:893-898. https://doi.org/10.1038/nrc2009
- Hall AM, Bass P, Unwin RJ. Drug-induced renal Fanconi syndrome. QJM. 2014;107:261-9. https://doi.org/10.1093/qjmed/hct258
- Foreman JW. Fanconi syndrome and other proximal tubule disorders. In: Comprehensive Clinical Nephrology, 5th Edition. Richard JJ, Feehally J, Floege J eds. 2014; pp 590-600.
- Maldonado
JE, Velosa JA, Kyle RA, Wagoner RD, Holley KE, Salassa RM. Fanconi
syndrome in adults. A manifestation of a latent form of myeloma. Am J
Med. 1975;58:354-364. https://doi.org/10.1016/0002-9343(75)90601-4
- Schillinger
F, Hopfner C, Montagnac R, Milcent T. IgG kappa myeloma with Fanconi's
syndrome and crystalline inclusions. Immunohistochemical and
ultrastructural study. Presse Méd . 1993;22:675-679.
- Seligmann
M, Danon F, Hurez D, Mihaesco E, Preud'homme JL. Alpha-chain disease: a
new immunoglobulin abnormality. Science. 1968;162:1396-7. https://doi.org/10.1126/science.162.3860.1396
- Wahner-Roedler DL, Kyle RA. Heavy chain diseases. Best Pract Res Clin Haematol.2005;18:729-46. https://doi.org/10.1016/j.beha.2005.01.029
- Doe WF, Henry K, Hobbs JR, Jones FA, Dent CE, Booth CC. Five cases of alpha chain disease. Gut. 1972;13:947-57. https://doi.org/10.1136/gut.13.12.947
- Takahashi
K, Naito M, Matsuoka Y, Takatsuki K. A new form of the alpha-chain
disease with generalized lymph node involvement. Pathol Res Pract.
1988;183:717-23. https://doi.org/10.1016/S0344-0338(88)80057-8
- Stoop
JW, Ballieux RE, Hijmans W, Zegers BJ. Alpha-chain disease with
involvement of the respiratory tract in a Dutch child. Clin Exp
Immunol. 1971;9:625-35.
- Franklin
EC, Lowenstein J, Bigelow B, Meltzer M. Heavy chain disease—a new
disorder of serum gamma-globulins: report of the first case. Am J Med.
1964;37:332-50. https://doi.org/10.1016/0002-9343(64)90191-3
- Dammacco F, Rigoli E, Ferrarese M, Bonomo L. Gamma heavy chain disease in a young girl. Haematologica. 1976;61:278-290.
- Wahner-Roedler
DL, Witzig TE, Loehrer LL, Kyle RA. Gamma-heavy chain disease: review
of 23 cases. Medicine (Baltimore). 2003;82:236-50. https://doi.org/10.1097/01.md.0000085058.63483.7f
- Bieliauskas
S, Tubbs RR, Bacon CM, Eshoa C, Foucar K, Gibson SE, Kroft SH, Sohani
AR, Swerdlow SH, Cook JR. Gamma heavy-chain disease: defining the
spectrum of associated lymphoproliferative disorders through analysis
of 13 cases. Am J Surg Pathol. 2012;36:534-43. https://doi.org/10.1097/PAS.0b013e318240590a
- Fermand
JP, Brouet JC, Danon F, Seligmann M. Gamma heavy chain disease”:
heterogeneity of the clinicopathologic features. Report of 16 cases and
review of the literature. Medicine (Baltimore). 1989;68:321-35. https://doi.org/10.1097/00005792-198911000-00001
- Forte
FA, Prelli F, Yount WJ, Jerry LM, Kochwa S, Franklin EC, Kunkel HG.
Heavy chain disease of the gamma (gamma M) type: report of the first
case. Blood. 1970;36:137-44.
- Ballard
HS, Hamilton LM, Marcus AJ, Illes CH. A new variant of heavy-chain
disease (mu-chain disease). N Engl J Med. 1970;282:1060-2. https://doi.org/10.1056/NEJM197005072821902
- Dammacco
F, Bonomo L, Franklin EC. A new case of mu heavy chain disease:
clinical and immunochemical studies. Blood. 1974;43:713-719.
- Witzens
M, Egerer G, Stahl D, Werle E, Goldschmidt H, Haas R. A case of mu
heavy-chain disease associated with hyperglobulinemia, anemia, and a
positive Coombs test. Ann Hematol. 1998;77:231-4. https://doi.org/10.1007/s002770050448
- Iwasaki
T, Hamano T, Kobayashi K, Kakishita E. A case of mu-heavy chain
disease: combined features of mu-chain disease and macroglobulinemia.
Int J Hematol. 1997;66:359-65. https://doi.org/10.1016/S0925-5710(97)00039-X
- Maeda
A, Mori M, Torii S, Nagai Y, Togami K, Fujita H, Kurata M, Matsushita
A, Nagai K, Imai Y, Takahashi T. Multiple extranodal tumors in mu-heavy
chain disease. Int J Hematol. 2006;84:286-7. https://doi.org/10.1532/IJH97.06124
- Kinoshita
K, Yamagata T, Nozaki Y, Sugiyama M, Ikoma S, Funauchi M, Kanamaru A.
Mu-heavy chain disease associated with systemic amyloidosis.
Hematology. 2004;9:135-7. https://doi.org/10.1080/10245330410001671561
- Dammacco
F, Antonaci S, Miglietta A. Two-dimensional immunoelectrophoresis as a
diagnostic tool for heavy chain disease. Boll Ist Sieroter Milan.
1975;54:460-465.
- Seligmann
M. Alpha chain disease: immunoglobulin abnormalities, pathogenesis and
current concepts. Br J Cancer Suppl. 1975;2:356-61. https://doi.org/10.1136/jcp.s1-6.1.72
- Al-Saleem
T, Al-Mondhiry H. Immunoproliferative small intestinal disease (IPSID):
a model for mature B-cell neoplasms. Blood. 2005;105:2274-80. https://doi.org/10.1182/blood-2004-07-2755
- Lecuit
M, Abachin E, Martin A, Poyart C, Pochart P, Suarez F, Bengoufa D,
Feuillard J, Lavergne A, Gordon JI, Berche P, Guillevin L, Lortholary
O. Immunoproliferative small intestinal disease associated with
Campylobacter jejuni. N Engl J Med. 2004;350:239-48. https://doi.org/10.1056/NEJMoa031887
- Peterson MC. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. Engl J Med. 2004;350:1685-6. https://doi.org/10.1056/NEJM200404153501619
- Isaacson
PG. Extranodal marginal zone lymphoma: MALT lymphoma. In: Jaffe ES,
Harris NL, Vardiman JW, et al, editors. Hematopathology. St. Louis:
Elsevier; 2011. p. 291-305. https://doi.org/10.1016/B978-0-7216-0040-6.00018-6
- Halphen
M, Najjar T, Jaafoura H, Cammoun M, Tufrali G. Diagnostic value of
upper intestinal fiber endoscopy in primary small intestinal lymphoma.
A prospective study by the Tunisian-French Intestinal Lymphoma Group.
Cancer. 1986;58:2140-5. https://doi.org/10.1002/1097-0142(19861101)58:9<2140::AID-CNCR2820580930>3.0.CO;2-P
- Isaacson
PG, Dogan A, Price SK, Spencer J. Immunoproliferative small-intestinal
disease. An immunohistochemical study. Am J Surg Pathol.
1989;13:1023-33. https://doi.org/10.1097/00000478-198912000-00004
- Parsonnet J, Isaacson PG. Bacterial infection and MALT lymphoma. N Engl J Med. 2004;350:213-5. https://doi.org/10.1056/NEJMp038200
- Presti
BC, Sciotto CG, Marsh SG. Lymphocytic lymphoma with associated gamma
heavy chain and IgM-lambda paraproteins. An unusual biclonal
gammopathy. Am J Clin Pathol. 1990;93:137-41. https://doi.org/10.1093/ajcp/93.1.137
- Zhang
L, Sotomayor EM, Papenhausen PR, Shao H, Moscinski LC, Sandin RL,
Caceres G, Valenica H, Malafa M, List AF, Sokol L. Unusual concurrence
of T-cell large granular lymphocytic leukemia with Franklin disease
(gamma heavy chain disease) manifested with massive splenomegaly. Leuk
Lymphoma. 2013;54:205-8. https://doi.org/10.3109/10428194.2012.697561
- Wahbi
A, Neel A, Perrin F, Graveleau J, Mahe B, Dejoie T, Hamidou M. Gamma
heavy chain disease associated with large granular lymphocytic
leukemia: A report of two cases and review of the literature.
Hematology. 2016;21:92-4.
- Maisnar
V, Tichy M, Stulik J, Urban P, Adam Z, Kadlckova E, Vavrova J, Palicka
V, Jebavy L, Kodet R, Buchler T, Hajek R. Capillary immunotyping
electrophoresis and high-resolution two-dimensional electrophoresis for
the detection of mu-heavy chain disease. Clin Chim Acta.
2008;389:171-3. https://doi.org/10.1016/j.cca.2007.10.035
- Preud’homme
JL, Bauwens M, Dumont G, Goujon JM, Dreyfus B, Touchard G. Cast
nephropathy in mu heavy chain disease. Clin Nephrol. 1997;48:118-21.
- Courtois L, Sujobert P. Morphologic features of µ-heavy-chain disease. Blood. 2017;130:558. https://doi.org/10.1182/blood-2017-04-781344
- Yanai
M, Maeda A, Watanabe N, Sugimoto N, Matsushita A, Nagai K, Oida T,
Takahashi T. Successful treatment of mu-heavy chain disease with
fludarabine monophosphate: a case report. Int J Hematol. 2004;79:174-7.
https://doi.org/10.1532/IJH97.03053
- Bonomo
L, Dammacco F, Marano R, Bonomo GM. Abdominal lymphoma and alpha chain
disease. Report of three cases. Am J Med. 1972;52:73-86.
https://doi.org/10.1016/0002-9343(72)90009-5
- Alpha-chain disease and related small-intestinal lymphoma: a memorandum. Bull World Health Organ. 1976;54:615-24.
- Fine
KD, Stone MJ. Alpha-heavy chain disease, Mediterranean lymphoma, and
immunoproliferative small intestinal disease: a review of
clinicopathological features, pathogenesis, and differential diagnosis.
Am J Gastroenterol. 1999;94:1139-52. https://doi.org/10.1111/j.1572-0241.1999.01057.x
- Ben-Ayed
F, Halphen M, Najjar T, Boussene H, Jaafoura H, Bouguerra A, Ben Salah
N, Mourali N, Ayed K, Ben Khalifa H, Garoui H., Gargouri M., Tufrali G.
Treatment of alpha chain disease. Results of a prospective study in 21
Tunisian patients by the Tunisian-French intestinal Lymphoma Study
Group. Cancer. 1989;63:1251-6. https://doi.org/10.1002/1097-0142(19890401)63:7<1251::AID-CNCR2820630704>3.0.CO;2-H
- Salem PA, Estephan FF. Immunoproliferative small intestinal disease: current concepts. Cancer J. 2005;11:374-82. https://doi.org/10.1097/00130404-200509000-00003
- Akbulut
H, Soykan I, Yakaryilmaz F, Icii F, Aksoy F, Haznedaroglu S, Yildirim
S. Five-year results of the treatment of 23 patients with
immunoproliferative small intestinal disease: a Turkish experience.
Cancer. 1997;80:8-14. https://doi.org/10.1002/(SICI)1097-0142(19970701)80:1<8::AID-CNCR2>3.0.CO;2-T
- Martin
IG, Aldoori MI. Immunoproliferative small intestinal disease:
Mediterranean lymphoma and alpha heavy chain disease. Br J Surg.
1994;81:20-4. https://doi.org/10.1002/bjs.1800810107
- Inoue
D, Matsushita A, Kiuchi M, Takiuchi Y, Nagano S, Arima H, Mori M,
Tabata S, Yamashiro A, Maruoka H, Oita T, Imai Y, Takahashi T.
Successful treatment of gamma-heavy-chain disease with rituximab and
fludarabine. Acta Haematol. 2012;128:139-43. https://doi.org/10.1159/000339097
- Witzig TE, Wahner-Roedler DL. Heavy chain disease. Curr Treat Options Oncol. 2002;3:247-54. https://doi.org/10.1007/s11864-002-0014-3
- Lee
DB, Drinkard JP, Rosen VJ, Gonick HC. The adult Fanconi syndrome:
observations on etiology, morphology, renal function and mineral
metabolism in three patients. Medicine (Baltimore). 1972;51:107-138, https://doi.org/10.1097/00005792-197203000-00003
- Aucouturier
P, Bauwens M, Khamlichi AA, Denoroy L, Spinelli S, Touchard G,
Preud'homme JL, Cogné M. Monoclonal Ig L chain and L chain V domain
fragment crystallization in myeloma-associated Fanconi's syndrome. J.
Immunol. 1993;150:3561-3568.
- Engle
RL, Wallis LA. Multiple myeloma and the adult Fanconi syndrome. I.
Report of a case with crystal-like deposits in the tumor cells and in
the epithelial cells of the kidney. Am. J. Med. 1957;22:5-12 https://doi.org/10.1016/0002-9343(57)90333-9
- Costanza
DJ, Smoller M. Multiple myeloma with the Fanconi syndrome: study of a
case, with electron microscopy of the kidney. Am J Med. 1963;34:125-133
https://doi.org/10.1016/0002-9343(63)90046-9
- Deret
S, Denoroy L, Lamarine M Vidal R, Mougenot B, Frangione B, Stevens FJ,
Ronco PM, Aucouturier P. Kappa light chain-associated Fanconi’s
syndrome: molecular analysis of monoclonal immunoglobulin light chains
from patients with and without intracellular crystals. Protein Eng
1999; 12: 363-369. https://doi.org/10.1093/protein/12.4.363
- Messiaen
T, Deret S, Mougenot B, Bridoux F, Dequiedt P, Dion JJ, Makdassi R,
Meeus F, Pourrat J, Touchard G, Vanhille P, Zaoui P, Aucouturier P,
Ronco PM: Adult Fanconi syndrome secondary to light chain gammopathy.
Clinicopathologic heterogeneity and unusual features in 11 patients.
Medicine (Baltimore) 2000; 79: 135-154. https://doi.org/10.1097/00005792-200005000-00002
- Rocca
A, Khamlichi AA, Touchard G, Mougenot B, Ronco P, Denoroy L, Déret S,
Preud'homme JL, Aucouturier P, Cogné M. Sequences of V kappa I subgroup
light chains in Fanconi’s syndrome. Light chain V region gene usage
restriction and peculiarities in myeloma-associated Fanconi’s syndrome.
J Immunol 1995; 155: 3245-3252.
- Leboulleux
M, Lelongt B, Mougenot B, Touchard G, Makdassi R, Rocca A, Noël LH,
Ronco P, Aucouturier P. Protease resistance and binding of Ig light
chains in myeloma-associated tubulopathies. Kidney Int. 1995;48:72-79. https://doi.org/10.1038/ki.1995.269
- Rao
DS, Parfitt AM, Villanueva AR, Dorman PJ, Kleerekoper M.
Hypophosphatemic osteomalacia and adult Fanconi syndrome due to
light-chain nephropathy. Another form of oncogenous osteomalacia. Am.
J. Med. 1987; 82:333-338. https://doi.org/10.1016/0002-9343(87)90081-7
- Clarke
BL, Wynne AG, Wilson DM, Fitzpatrick LA. Osteomalacia associated with
adult Fanconi's syndrome: clinical and diagnostic features. Clin.
Endocrinol. 1995;43:479-490. https://doi.org/10.1111/j.1365-2265.1995.tb02621.x
- Levine SB, Bernstein LD. Crystalline inclusions in multiple myeloma. JAMA. 1985;254:1985. https://doi.org/10.1001/jama.1985.03360140143043
- DeFronzo
RA, Cooke CR, Wright JR, Humphrey RL. Renal function in patients with
multiple myeloma. Medicine (Baltimore) 1978; 57: 151-166. https://doi.org/10.1097/00005792-197803000-00003
- Batuman
V, Guan S, O’Donovan R Puschett JB. Effect of myeloma light chains on
phosphate and glucose transport in renal proximal tubule cells. Ren
Physiol Biochem 1994; 17: 294-300 https://doi.org/10.1159/000173861
- Batuman
V, Sastrasinh M, Sastrasinh S. Light chain effects on alanine and
glucose uptake by renal brush border membranes. Kidney Int 1986; 30:
662-665. https://doi.org/10.1038/ki.1986.237
- Smithline
N, Kassirer JP, Cohen JJ. Light-chain nephropathy. Renal tubular
dysfunction associated with light-chain proteinuria. N Engl J Med 1976;
294: 71-74 https://doi.org/10.1056/NEJM197601082940202
- Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 108: 2520-2530, 2006 https://doi.org/10.1182/blood-2006-03-001164
- Thorner
PS, Bedard YC, Fernandes BJ. Lambda-light-chain nephropathy with
Fanconi’s syndrome. Arch Pathol Lab Med 1983; 107: 654-657
- Terashima
K, Takahashi K, Kojima M, Imai Y, Tsuchida S, Migita S, Ebina S, Itoh
C. Kappa-type light chain crystal storage histiocytosis. Acta Pathol
Jpn. 1978;28: 111-138. https://doi.org/10.1111/j.1440-1827.1978.tb01254.x
- Lebeau
A, Zeindl-Eberhart E, Muller EC, Müller-Höcker J, Jungblut PR, Emmerich
B, Löhrs U. Generalized crystal-storing histiocytosis associated with
monoclonal gammopathy: molecular analysis of a disorder with rapid
clinical course and review of the literature. Blood. 2002;100:
1817-1827
- Ma
CX, Lacy MQ, Rompala JF, Dispenzieri A, Rajkumar SV, Greipp PR, Fonseca
R, Kyle RA, Gertz MA. Acquired Fanconi syndrome is an indolent disorder
in the absence of overt multiple myeloma. Blood 2004; 104(1): 40-42 https://doi.org/10.1182/blood-2003-10-3400
- Alter BP. Fanconi anemia and the development of leukemia. Best Practice & Research Clinical Haematology 2014;27:214-221 https://doi.org/10.1016/j.beha.2014.10.002
- Gavriatopoulou
M, Terpos E, Kastritis E, Dimopoulos MA. Current treatments for renal
failure due to multiple myeloma. Expert Opin Pharmacother.
2016;17:2165-2177. https://doi.org/10.1080/14656566.2016.1236915
[TOP]