Valentina Brancaleoni1‡, Isabella Nava1‡, Paola Delbini2, Lorena Duca1 and Irene Motta 1-2*.
1 Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, UOC General Medicine, Milan, Italy.
2 Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy.
‡ The authors equally contributed to this work.
Correspondence to: Irene Motta, MD. Department of Clinical
Sciences and Community Health Università degli Studi di Milano, UOC
General Medicine, Fondazione IRCCS Ca' Granda Ospedale Maggiore
Policlinico, Via F. Sforza 35, 20122 – Milan, Italy. E-mail:
irene.motta@unimi.it
Published: November 1, 2020
Received: August 10, 2020
Accepted: October 9, 2020
Mediterr J Hematol Infect Dis 2020, 12(1): e2020075 DOI
10.4084/MJHID.2020.075
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
β-thalassemia is a hereditary disorder caused by defective production of β-globin chains of hemoglobin (Hb) that leads to an increased α/β globins ratio with subsequent free α-globins.
Alpha globin excess causes oxidative stress, red blood cells membrane
damage, premature death of late-stage erythroid precursors, resulting
in ineffective erythropoiesis.
The transforming growth factor β (TGF-β)
superfamily signaling acts on biological processes, such as cell
quiescence, apoptosis, proliferation, differentiation, and migration,
and plays an essential role in regulating the hematopoiesis. This
pathway can lose its physiologic regulation in pathologic conditions,
leading to anemia and ineffective erythropoiesis. Activin
receptor-ligand trap molecules such as Sotatercept and Luspatercept
downregulate the TGF-β pathway, thus inhibiting the Smad2/3 cascade and
alleviating anemia in patients with β-thalassemia and myelodysplastic syndromes.
In this review, we describe in extenso the TGF-β
pathway, as well as the molecular and biological basis of activin
receptors ligand traps, focusing on their role in various β-thalassemia
experimental models. The most recent results from clinical trials on
sotatercept and luspatercept will also be reviewed.
|
Introduction
β-thalassemia is a hereditary disorder caused by defective production of β-globin chains of hemoglobin (Hb)[1] that leads to an increased α/β globins ratio with subsequent free α-globins that precipitate in the red blood cells (RBCs). Excess of α-globin
aggregates in erythroblasts lead to maturation arrest, oxidative
stress, membrane damage, and premature death of late-stage erythroid
precursors and, in turn, to a reduced RBCs half-life.[2]
Transforming growth factor β (TGF-β)
superfamily signaling acts on cell quiescence, apoptosis,
proliferation, differentiation, and migration and plays a crucial role
in the regulation of hematopoiesis.[3] In selected pathological conditions, including β-thalassemia,
this pathway can lose its physiologic regulation leading to anemia and
ineffective erythropoiesis. Activin receptors ligand traps such as
Sotatercept and Luspatercept downregulate the TGF-β pathway, thus
inhibiting the Smad2/3 cascade and alleviating anemia in patients
with β-thalassemia and also myelodysplastic syndromes.[3]
In this review, we describe in extenso the TGF-β
pathway, starting from the role of activins and their cognate receptors
to get to the description of signal effectors. We also summarize the
molecular and biological basis of activin receptors ligand traps,
focusing on their role in various β-thalassemia
experimental models. The most recent results from clinical trials on
sotatercept and luspatercept will also be reviewed.
Activins and Activin Receptors
Activins are typical proteins members of transforming growth factor β (TGF-β)
superfamily and control many cellular processes involved in cell
proliferation, death, metabolism, homeostasis, differentiation, immune
response, and endocrine function.[4,5]
Activin A,
initially described as a regulator of reproductive processes, is an
erythroid differentiation factor of hematopoietic progenitor cells in vitro and in vivo.[6]
Activins are biologically active in all tissues as βA and βB homodimers, or βA and βB heterodimers, whereas βC and βE
are predominantly expressed in the liver. Activins are synthesized as
larger precursor proteins containing a prodomain and a mature region.
Pro-domains, non-covalently bound to their mature regions, are
essential for the dimer stabilization, its secretion, and association
with heparan sulfate residues of proteoglycans of target cells,
allowing activins to concentrate near their receptors and protecting
themselves from proteolysis. Activin B lacks residues fundamental for
binding heparan sulfate and shows a lower affinity for their receptors.
The mature regions contain a cysteine-rich domain which forms intra-
and inter- disulfide bonds, well conserved in other members of TGF-β family, including TGF-β1, TGF-β2 e TGF-β3, BMP2 e BMP7 and probably responsible for the characteristic open hand configuration of Activin.[7]
Activins
initiate their biological signaling by binding specific type II A
(ActRIIA) or B (ActRIIB) receptors (for activin A or B respectively)
serine/threonine kinases on the surface of target cells. Both type II
receptors present an extracellular domain, the transmembrane domain,
and the cytoplasmic domain carrying kinase activity. The interaction of
Activin with a dimer of type II receptors is necessary for recruitment
and phosphorylation of the activin type I receptor-like kinase dimers
(ALK4 or ALK7) at their glycine-serine-rich domain. Once activated, ALK
4/ALK7 binds and phosphorylates the cytoplasmic Smad (Smad2 and Smad3)
proteins, thus allowing signal transduction to the nucleus.[8]
Smad
proteins are a family of transcription factors that regulate more than
500 target genes in a cell-specific and dose-dependent manner. They are
divided into three groups: (i) receptor-regulated Smad (R-Smad)
consisting of Smad2 and Smad3 activated by TGF-β
and activins and Smad1, Smad5 and Smad8 activated by BMP, (ii)
inhibitory Smad, i.e., Smad6 and Smad7, (iii) and the common mediator
Smad4. Once phosphorylated, R-Smad binds another R-Smad or Smad4, and
then the complex translocates into the nucleus where it binds the
target DNA consensus sequence triggering activin signaling dependent
transcription (Figure 1A).[9]
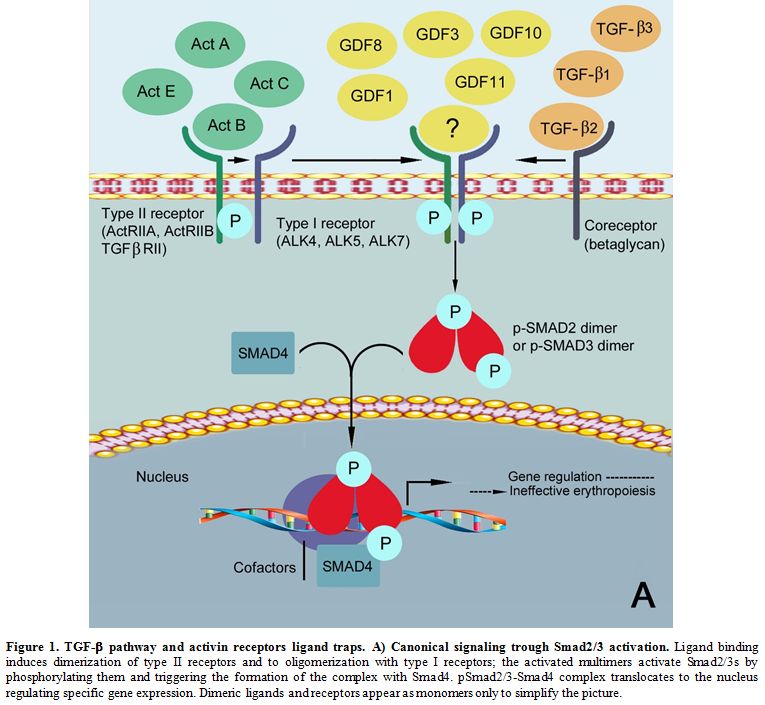 |
Figure 1. TGF-β pathway and activin receptors ligand traps. A) Canonical signaling trough Smad2/3 activation.
Ligand binding induces dimerization of type II receptors and to
oligomerization with type I receptors; the activated multimers activate
Smad2/3s by phosphorylating them and triggering the formation of the
complex with Smad4. pSmad2/3-Smad4 complex translocates to the nucleus
regulating specific gene expression. Dimeric ligands and receptors
appear as monomers only to simplify the picture.
|
Modulation of Activin Signaling.
The activin pathway is regulated and modulated at various levels,
including extracellular binding proteins (follistatin and protein
encoded by FLRG), molecules which antagonize their bind with the
receptor (inhibins), co-receptors of activin antagonists (beta-glycan),
inhibitory Smad proteins, and other proteins ligands of activin
receptors (myostatin -or GDF8- and GDF11).
The glycoprotein
follistatin exists in two isoforms of 288 (FS288) and 315 (FS315)
aminoacid residues due to alternative RNA splicing. FS288 is a potent
extracellular negative regulator of Activin; in particular, its FS-1
domain, consisting of ten cysteine residues involved in binding the
cell surface, is essential to abolish activin biological activity.
Follistatin acts by masking the activin regions crucial for the
interaction with ActRII and ALK4.[10]
Another
follistatin family member is the follistatin-related gene FLRG,
differing from follistatin because it lacks the FS-3 domain, and it is
considered a homolog, not totally functional, of the alternative
spliced circulating isoform of follistatin (FS315). FLRG is highly
expressed in the placenta, testis, and cardiovascular tissue, while
follistatin expression is higher in the pituitary and ovary.[11]
Nevertheless, the protein encoded by FLRG shows functional similarity
to follistatin in inhibiting erythropoiesis, being follistatin a
repressor of Activin A-induced erythroid differentiation.[12] Follistatin and the protein encoded by FLRG inhibit not only activins but also GDF11 and GDF8.[13]
Activins beta A and beta B subunits can also heterodimerize with inhibin α-subunit
to form inhibin A or B. Inhibins share the same binding site of ActRII;
however, their affinity is tenfold lower than activins. Beta glycan is
the co-receptor for inhibins, which increases their affinity by 30
times for ActRII, forming a complex, which prevents ActRII from binding
to ALK4.[14] Beta glycan is expressed in rat brain, pituitary gland, and gonads confirming its modulatory role on inhibins.[15,16] Betaglycan is also an accessory receptor for TGF-β ligands 1, 2, and 3.
Inhibitory Smads bind stably with type I and type II receptors. Smad7 strongly inhibits Activin, BMP, and TGF-β signaling,[17] but Activin upregulates its expression, BMP, and TGF-β representing a feedback control mechanism of extracellular signaling.
Activin
signaling can also be modulated via post-translational degradation of
Smads when Smad-ubiquitination-regulatory factors Smurf interacts with
Smad, targeting their ubiquitin proteasome-mediated degradation. While
Smurf1 modulates the BMP pathway targeting Smad1 and Smad5, Smurf2
interferes with all BMP, TGF-β
and activin signaling because of its broad interaction with Smad. It is
noteworthy that Smad4, regulated by BMP and activin/TGF-β competing with each other, is free from ubiquitination regulated by Smurf.[18]
GDF8,
a negative regulator cytokine of muscle mass, expressed in cardiac and
smooth cells, adipose tissue, mammary gland, and placenta, shares
ActRII and ALK4 for intracellular signaling with Activin. GDF8 is
expressed in blood cells and promotes differentiation in hematopoietic
cell lines, supporting the idea that it may be an autocrine/paracrine
factor involved in hematopoiesis control. Its activity could be
partially redundant with the activin pathway.[19]
GDF11,
whose amino acid sequence is 90% identical to GDF8, is another
essential ligand for ALK4 and ActRII involved in neurogenesis, kidney,
endocrine pancreas development, and heart. It has been recently
identified as a negative regulator of late-stage maturation of
erythroid precursors. GDF11 is overexpressed in β-thalassemia,
and it has been hypothesized to lead to ineffective erythropoiesis
through a deleterious autocrine amplification loop involving oxidative
stress and alpha-globin precipitation.[20]
TGF-β and TGF-β Receptors
TGF-β, the founding member of the complex TGF-β superfamily signaling, is a critical crucial and multifunctional cytokine existing in three mammalian isoforms (TGF-β1, TGF-β2 and TGF-β3). Each isoform is produced in an inactive form made by a latency-associated peptide (LAP) and the active TGF-β fraction which is covalently associated with latent TGF-β binding protein (LTBP) with significant consequences on TGF-β
localization and activation. Although various stimuli have been
proposed (heat, acid pH, wound, reactive oxygen species), the key
activators of TGF-β are integrins. Integrins are cell adhesion and signaling receptors formed by one of 18 α and one of eight β
subunits of a transmembrane receptor, thus creating 24 types of
different integrins. Only four have been shown to liberate active TGF-β. It is noteworthy that TGF-β2 lacks the integrin-binding motif; consequently, another mechanism might be involved in TGF-β2 activation.[21]
The pivotal role of TGF-β in regulating proliferation of hematopoietic stem cells (HSCs) has been well demonstrated. Indeed, TGF-β1 is essential for controlling the quiescence of HSCs, and the production of TGF-β by both HSCs and niche stromal cells can contribute to the maintenance of the stem cell compartment. Opposite to TGF-β1, TGF-β2 is a positive regulator of hematopoietic stem cells, while TGF-β3 only functions as an inhibitor on primitive hematopoietic cells. Generally, TGF-β
inhibits growth in vitro by transcriptional downregulating the
growth-stimulating protein c-Myc and receptors for hematopoietic
cytokines (GM-CSF, IL-3, IL-1), and inducing the cyclin-dependent
kinase inhibitors (CDKIs) p15, p21, p27 and p57, and stem cell factor.
Indeed, TGF-β has been considered dispensable for regulating hematopoietic stem cells in vivo, probably because of the existence of redundant signals in the whole system.[22]
TGF-β isoforms signaling acts via different expression functions by sharing the same receptors, TGF-β type II receptor (TGF-βRII)
and type I receptors (ALK1 and ALK5), and it is sustained by Smad2 and
Smad3 at the cytoplasmic level. As for activins, also in the case of
TGF-β
the binding of R-Smad with Smad4 stimulate the transcription of target
genes. Otherwise, it has been shown that the ubiquitous nuclear protein
Transcriptional Intermediary Factor 1 gamma (TIF1-γ)
binds Smad2/3 selectively competing with Smad4. Whereas the association
of Smad2/3 with Smad4 inhibits the proliferation of hematopoietic stem
cells, the interaction of TIF1-γ
with Smad2/3 stimulates erythroid differentiation, suggesting that
hematopoiesis could be controlled by these two distinct branches of TGF-β pathway.[23]
Finally, TGF-β can modulate other signaling pathways without engaging the canonical Smad system: TGF-β
activated kinase 1 (TAK1), a component of mitogen-activated protein
kinase (MAPK), activates c-Jun-N terminal kinase (JNK) and p38. Other
downstream transducers directly activated by TGF-β
are the extracellular signal-regulated kinase (ERK),
phosphatidylinositol three kinases (PI3K), and RHO-like small guanosine
triphosphatases. The role of these systems is not yet well defined in
hematopoietic stem cells.[24]
Activin Receptor II Ligand Traps
Misregulation
of the activin pathway has been implicated in anemia,
myeloma-associated osteolysis, metastatic bone disease, and
carcinogenesis.[25] Activin and activin receptors,
because of their diverse biological processes, have been extensively
studied as potential therapeutic targets in several pathological
conditions.[26] In particular, the inhibition of the TGF-β
pathway is the mechanism by which a therapeutic effect can be achieved,
acting on the Smad2/3 cascade. Two such agents are Sotatercept
(ACE-011) and Luspatercept (ACE-536), ligand-trapping fusion proteins
containing the modified extracellular domain of activin receptor type
IIA (ActRIIA) or IIB (ActRIIB), respectively, fused to the Fc domain of
IgG1.[27,28] Both of them sequestrate the ligand before it can interact with the receptor, thus inhibiting the signal transduction cascade.
It has been demonstrated that in ex vivo
conditions, ACE-011 binds Activin A and B, GDF8, GDF11, and other BMPs
(such as BMP6, BMP7, and BMP10) with different affinities.[27] Luspatercept, on the other hand, has shown high binding capacity with activin B, GDF8, and GDF11, but not with Activin A.[28,32] Both of them do not interact with TGF-β1, TGF-β2 nor TGF-β1.
These activin ligand traps show similar binding ligand-binding
profiles, but ACE-536 shows a higher ligand selectivity making this
specific molecule more suitable for treating anemia and ineffective
erythropoiesis.
Preclinical Studies on the Effect on Hematological Parameters.
The murine counterpart of Sotatercept (namely RAP-011) was firstly
studied to evaluate the role of Activin and its related ligands in bone
metabolism,[29] and ACE-011 showed an unexpected effect in increasing hematocrit and Hb levels[30,31]
during clinical trials for post-menopausal osteoporosis. Different
studies are ongoing in the attempt to elucidate the molecular
mechanisms underlying Hb elevation. Wildtype mice treated with RAP-011
showed a rapid increase of hematocrit, Hb, and RBC count.[33] Similarly, in Hbbth1/th1 mouse model of β-thalassemia
intermedia, RAP-011, or RAP-536 treatment resulted in higher RBC count,
hematocrit, total Hb concentration, as well as reduced reticulocytosis.[34]
Also, thalassemia RBC morphology ameliorates under RAP-011 treatment,
confirming this molecule's effect on erythropoiesis and the effect
observed in clinical studies.[35] In treated Hbbth1/th1
mice, physiologic erythroid differentiation was partly restored,
showing a decrease in the number of basophilic erythroblasts and an
increase in orthochromatic erythroblasts and reticulocytes, both in
bone marrow and spleen. Also, the degree of splenomegaly improved, thus
acting on ineffective erythropoiesis.[34,35] These
results indicated that these molecules promote erythropoiesis,
augmenting the terminal erythroid differentiation by inhibiting the
ActRII pathway.
In the attempt to identify the specific
precursor targeted by activin ligand traps, the numbers of BFU-E and
CFU-E were also assessed in wild type mice treated with RAP-011 and
ACE-536.[32,33] The results were controversial since
RAP-011 was able to increase the percentage of bone marrow BFU-E in
bone marrow, inducing the formation of larger colonies.[33]
On the other side, by 48h, ACE-536 reduced both bone marrow and spleen
BFU-E and CFU-E, followed seven days after by an increased number of
these progenitors. EPO acts synergically with these molecules since
studies have shown that the EPO antibodies can decrease hematological
parameters such as hematocrit, Hb level, and total red blood cells
while cotreatment with ACE-536 rescued this blockage. EPO and ACE-536
acted additively since their combined effect was even more significant
than the sum of the single-agent' effects, and that suggests that
activin ligand traps act on later stage differentiation while EPO
sustains RBC production by increasing the availability of early-stage
progenitors.[32]
Despite the proven effects in
mouse models and clinical trials, the target of these molecules remains
unknown. Some works have shown, with the use of ex-vivo models, such as
CD34+ cells treated with the molecule
in liquid cultures, that there is not a measurable effect of these
agents on the stimulation of growth or differentiation of erythroid
progenitors or precursors, thereby suggesting that the surrounding
environment could mediate the effect. Thus, the use of co-cultures or
conditioned media obtained by administration of the RAP-011 or ACE-011
to stromal or long term bone marrow cultures[33,36,37]
could be a potential model. In these settings, factors secreted by bone
marrow and/or stromal cells probably mediate the effects of RAP or
ACE-011 on erythroid differentiation, thus creating a microenvironment
that is more permissive for erythropoiesis.
Modulation of Iron Homeostasis and ROS. It was also observed that RAP-011 and RAP-536 modulate iron homeostasis in the Hbbth1/th1 mouse model since treated mice did show a reduction in serum iron, transferrin saturation, and ferritin,[34,35]
as well as a restored splenic architecture. Since iron parameters are
not affected in wildtype animals, it is conceivable that the ligand
traps act indirectly on iron homeostasis. Suragani et al. did
investigate the expression of two essential genes in the regulation of
iron homeostasis: Hepcidin (Hamp) and Bmp6. RAP-536 did increase Hamp
liver expression in treated versus untreated Hbbth1/th1
mice, while Bmp6 expression was elevated versus wild type mice and
unchanged in both of them. The authors hypothesized that Bmp6
expression could remain elevated since it enhances Hamp expression
until liver iron reaches average values.
Further information on the mechanism of action was obtained by evaluating α-globin aggregates, ROS, and hemolysis since erythrocytic damage and hemolysis caused by unpaired α-chain aggregates and oxidative stress are key features of β-thalassemia. It was seen that both RAP-011 and RAP-536 reduced ROS and α membrane-associated aggregates levels.[34,35] α-globin gene expression was also decreased[35]
(personal data). Also, treated mice display improved hemolysis
parameters such as a reduction in the mean concentration of total
bilirubin and increased erythrocyte life span.[34]
The Identification of a Specific Ligand.
Further studies have investigated the molecular mechanisms aiming to
identify the potential candidates that mediate activin receptor ligand
traps' action. Most of these studies focused their attention on known
ligands, such as Activins A and B, GDF8, and GDF11, involved in the TGF-α
pathway and mediating the pathway activation through Smad2/3
phosphorylation. RAP-011 is able to block phosphorylation of Smad 2/3
induced by activin A and GDF11.[33] ACE-536 displayed
quite the same activity also towards GDF8 but did not inhibit signaling
induced by activins. Among these ligands, the role of GDF11 as a
potential target of activin receptors ligand traps and as a new
regulator of erythropoiesis has been studied in different disease
models. In a β-thalassemia
mouse model, GDF11 expression is prominent in the red pulp area, the
spleen's erythropoietic niche, and it was abundant in the sera
from β-thalassemia
as well as myelodysplastic syndrome (MDS) patients. GDF11 expression
seems to be higher in immature erythroid precursors and then
progressively decreases as maturation proceeds. Wild type mice treated
with GDF11 developed mild anemia with reduced RBC parameters and
increased the spleen weight, which is indicative of ineffective
erythropoiesis. Ex vivo treatment with GDF11 caused a reduction of mature erythroblasts.[32] At the same time, treatment with GDF11 antibodies (Ab) promoted terminal erythroblast maturation in the Hbbth1/th1 β-thal mice model, suggesting that GDF11 negatively regulates erythroid maturation.[35]
Thus, GDF11 seems to inhibit differentiation and maintain the survival
of immature progenitors, and as it decreases, erythroid maturation
proceeds. The concomitant use of ACE-536 also reduced the effect
mediated by GDF11,[32] and RAP-011 in Hbbth1/th1 β-thal mice model also reduced GDF11 expression and reduced ROS levels indicating that in Hbbth1/th1 β-thal mouse GDF11 is overexpressed, and it represents a characteristic of ineffective erythropoiesis.[35] The treatment with RAP-011, RAP-536, or ACE-536 in wild type and Hbbth1/th1
mouse models and, also in ex-vivo cellular models, inhibited the
Smad2/3 phosphorylation promoting terminal erythroid differentiation
and the effect on RBC indices.[32-34] Thus, these studies identified GDF11 as a putative key member of TGF-β family directly implicated in late-stage differentiation inhibition through increased oxidative stress.
Nevertheless,
this proposed model is controversial, since ACE- and RAP-536 have shown
their effectiveness in healthy humans and mice; although GDF11 is not
overexpressed,[34,38] GDF11 could
be a player, but not the principal one, in this game. A recent study by
Guerra et al. excluded GDF11 as the master target of RAP-536, using
different genetic approaches. They evaluated hematological parameters
in tamoxifen-inducible Cre/Lox recombinase conditional GDF11-knock-out
Hbbth3/+ mice compared to those of a GDF11-KO-Hbb+/+ control mouse. The absence of GDF11 did not improve anemia: GDF11-knock-out Hbbth3/+
mice did not show an improvement of hematological parameters, since no
change in RBC numbers, Hb, hematocrit, and the percentage of
reticulocytes was noted versus Hbb+/+ control mice. Testing the efficacy of RAP-536, Guerra et al. also reported that GDF11- knock -out Hbbth3/+
and the wild type mice responded in the same way, increasing RBC, Hb,
and Hct parameters. Also, GDF11 expression was evaluated in erythroid
progenitors derived from healthy and β-thal CD34+ cells, and although GDF11 was very low in both conditions, it was higher in β-thal cells than control cells, even though this difference was not significant.
Similarly, low expression was observed in Ter119+ spleen late erythroblasts and also after RAP-536 treatment.[39] Likewise, in our unpublished data, GDF11 mRNA levels were low in CD34+-derived
erythroid cultures from β-NTDT patients, but at the same time, the
relative levels of GDF11 were higher compared with healthy cultures.
RAP-011 treatment did not induce any difference, consistent with Guerra
et al. results. Altogether, the results from Guerra et al. excluded
that RAP-536 exerts its action exclusively through GDF11 blockage and
that this latter is the master effector of TGF-B inhibition of late
erythropoiesis.
Modulation of GATA1 and TIF1-γ by ACE-536.
Another work addressing the molecular mechanism beyond ACE-536 action
focused on pSmad2/3-mediated inhibition of erythroid differentiation.
Martinez et al., using differentiating MEL cells and GDF11, induced
overactivation of the smad2/3 pathway saw a higher nuclear localization
of pSmad2/3 and Smad4 and a concomitant reduced nuclear localization
and expression of GATA1 and TIF1-γ.
GDF11-induced overactivation of Smad2/3 led to an increase in cells
with a wider range of cell size, suggesting an accumulation of more
immature cells, increased ROS levels, and reduced cell viability and
hemoglobin levels. ACE-536 was able to revert these effects and
increased nuclear localization of TIF1-γ and expression of GATA1. Moreover, the transcriptome analysis of splenic erythroblasts from Hbbth3/+
mice treated with RAP-536 revealed that different genes were
differentially expressed; among them, GATA1 was found. A gene set
enrichment analysis of GATA1 activator against RAP-536 data identified
other genes that were significantly upregulated. Specifically, specific
GATA 1 target genes, regulated by RAP-536, were involved in erythroid
differentiation, heme biosynthesis protein quality control, and
proliferation, and cell death. At present, it is not still clear if
increased GATA1 expression and availability could be a direct effect of
RAP-536 or an induced effect due to reduced oxidative stress and cell
death.[40] Our unpublished data are consistent with
this result, since in RAP-011-treated cultures, at day seven, we
observed a higher expression of GATA1 and α-globin compared to
untreated cells (p<0.01 and p<0.05, respectively), which was
associated with a transitory reduction in β/a globin ratio.
Interestingly, on day 14, GATA1 and α-globin expression were reduced in
RAP-011-treated cells, associated with a re-balance of β/a globin ratio
compared with untreated cultures (p<0.005) (unpublished data).
Martinez
and coauthors conclude that higher pSmad2/3 facilitates the complex
with Smad4. The treatment with RAP-536 lowers of pSmad2/3 and favors
the formation of pSmad2/3-TIF1-γ complexes, influencing GATA1 nuclear availability and expression and other key genes (Figure 1B).[40]
Thus, they indicate GATA1 as a possible key effector of ACE-536 action
in alleviating the ineffective erythropoiesis in MDS cellular and β-thal mouse models.
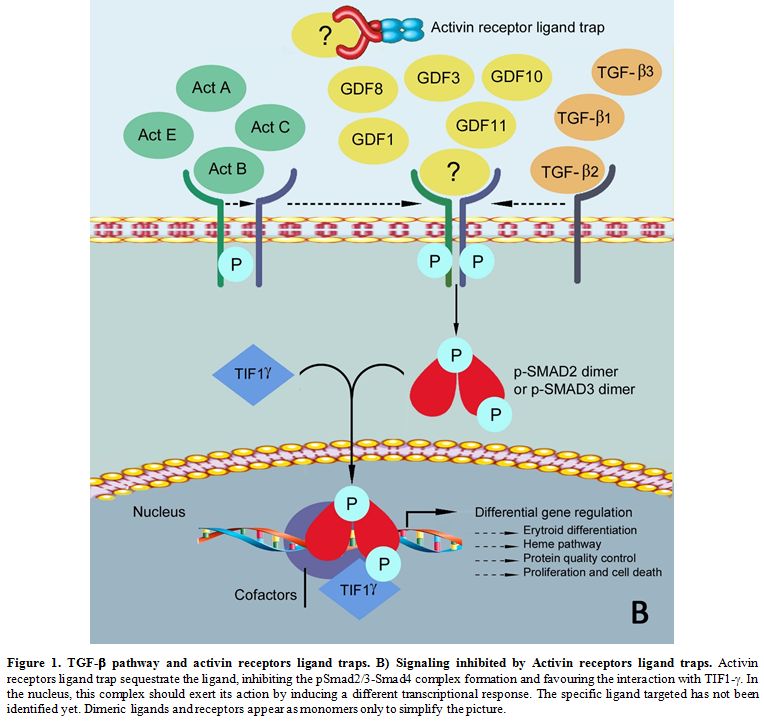 |
Figure 1. TGF-β pathway and activin receptors ligand traps. B) Signaling inhibited by Activin receptors ligand traps.
Activin receptors ligand trap sequestrate the ligand, inhibiting the
pSmad2/3-Smad4 complex formation and favouring the interaction with
TIF1-γ. In
the nucleus, this complex should exert its action by inducing a
different transcriptional response. The specific ligand targeted has
not been identified yet. Dimeric ligands and receptors appear as
monomers only to simplify the picture.
|
Activin Receptor-Ligand Traps in Clinical Trials. Both sotatercept and luspatercept have been tested in clinical trials.
Sotatercept
was first tested in single and multiple doses in post-menopausal
healthy women volunteers, inducing an increase in bone mineral density
and bone formation biomarkers. Moreover, clinically significant,
dose‐dependent increases in Hb, hematocrit, red blood cells (RBCs), and
reticulocytes were observed.[30,41]
Similarly, luspatercept was evaluated in post-menopausal female healthy
volunteers, and they showed an increase in Hb, RBCs, and reticulocytes
in a dose- related manner.[38] A phase IIa study
evaluated the safety and tolerability of sotatercept and its effects on
bone metabolism and hematopoiesis in newly diagnosed and relapsed
multiple myeloma patients. Anabolic improvements in bone mineral
density and bone formation were observed, whereas bone resorption was
minimally affected. Among sotatercept-treated patients, 71% had at
least one dose interruption, mainly due to increases in Hb levels,
which was dose-dependent.[42]
Activin receptor-ligand trap for hematological disorders.
Given the increase in Hb levels, these compounds have been tested in
the hematological setting as a potential treatment for patients with
ineffective erythropoiesis. Sotatercept was tested for
chemotherapy-induced anemia in breast and lung cancer; the studies were
terminated early because of slow accrual, but approximately half of the
patients receiving sotatercept achieved Hb increase of more than 1
g/dL.[43] Moreover, both were tested in either
thalassemia or myelodysplastic syndrome (MDS), resulting in the
approval of luspatercept by the Food and Drug Administration (FDA) in
November 2019 for thalassemia, April 2020 for MDS, and in July 2020 by
European Medicines Agency (EMA).
Sotatercept and luspatercept for β-thalassemia. A multicenter international phase 2 study using sotatercept was conducted on 16 TDT and 30 NTDT patients (NCT01571635).[2]
They were treated with sotatercept at doses ranging from 0.1 to 1.0
mg/kg subcutaneously every three weeks. In the TDT group, 63% of the
patients achieved a transfusion burden reduction of ≥ 20% sustained for
≥ 24 weeks, 44% a reduction of ≥33%, and 13% a reduction of ≥50%. In
these patients, the mean change in Hb level from baseline to the end of
treatment was 0.7 g/dL, and the effective dose of sotatercept was ≥ 0.5
mg/kg. In the NTDT group, 60% achieved a mean Hb increase of ≥1.0 g/dL,
and 37% of ≥1.5 g/dL sustained for ≥12 weeks.[2] Sotatercept exhibited an overall good safety profile and was tolerated by most patients.
Luspatercept
has been first approved by the FDA to treat TDT patients at the
recommended starting dose of 1 mg/kg every three weeks by subcutaneous
injection.[44] In a multicenter international phase 2
dose-finding study in β-thalassemia patients (NCT01749540),
luspatercept was administered subcutaneously every three weeks at doses
ranging from 0.2 to 1.25 mg/kg. In the NTDT group (n=33), 58% of the
patients achieved a mean Hb increase from baseline of ≥1.5 g/dL over 14
consecutive days. In the TDT group (n=31), 81% achieved a transfusion
burden reduction of ≥20% over any 12 weeks on study compared with the
12 weeks before baseline.[45] These findings paved the way to a randomized Phase 3 clinical trial.
The
BELIEVE study is a phase 3 multicenter international randomized,
double-blind, placebo-controlled trial that enrolled 336 adult TDT
patients randomized in a 2:1 ratio to receive luspatercept plus best
supportive care (BSC) versus placebo plus BSC every three weeks for at
least 48 weeks (NCT02604433). The primary endpoint was the percentage
of patients who had an erythroid response, defined as a reduction in
the transfusion burden of at least 33% from baseline (the 12 weeks
before the first dose of luspatercept or placebo) during weeks 13
through 24 plus a reduction of at least two red cell units over this
12-week interval. Forty-eight out of 224 (21.4%) in the luspatercept
group achieved the primary endpoints compared to the placebo group
(4.5%) (P<0,001). Also, 75% had at least a 33% reduction in
transfusion burden during any rolling 12-week interval with
luspatercept. Transfusion independence was achieved by 11% of the
patients in the luspatercept group during any 8-week interval. Adverse
events of transient bone pain, arthralgia, dizziness, hypertension, and
hyperuricemia were more frequent with luspatercept than placebo.[46] A 5-year open-label extension phase is ongoing to provide long-term efficacy and safety data.
A
phase 2 trial (BEYOND) in adults with NTDT is ongoing (NCT03342404).
The primary endpoint is the proportion of patients with an increase in
mean Hb concentration of ≥ 1 g/dL in the absence of RBC transfusion
from weeks 13-24 vs. baseline.
Luspatercept for myelodysplastic syndrome.
A phase II multicenter dose-finding study was conducted with
sotatercept to treat anemia in low- or intermediate-1-risk MDS and
transfusion-dependent anemia failing anemia erythropoiesis-stimulating
agent (ESA) (NCT01736683). Sotatercept was administered once every
three weeks at a dose ranging from 0.1 to 2.0 mg/kg. Approximately half
of the patients achieved hematological improvement-erythroid (HI-E),
according to the International Working Group 2006 criteria. Treatment
was well tolerated.[47]
Luspatercept has been
recently approved by the FDA for the treatment of anemia failing an ESA
and requiring two or more RBC units over eight weeks in adult patients
with very low- to intermediate-risk MDS with ring sideroblasts (MDS-RS)
or with myelodysplastic/myeloproliferative neoplasm with ring
sideroblasts and thrombocytosis (MDS/MPN-RS-T).[44]
In phase 2, multicenter, dose-finding study (PACE-MDS) (NCT01749514,
extension study NCT02268383), patients with low or intermediate 1 risk
MDS or non-proliferative chronic myelomonocytic leukemia received
luspatercept subcutaneously once every three weeks at dose
concentrations ranging from 0.125 mg/kg to 1.75 mg/kg. An erythroid
response (defined as a reduction in red-cell transfusions of ≥4 units
per eight weeks in patients with a baseline transfusion burden of ≥4
units per 8 weeks or as an increase in the hemoglobin level of ≥1.5
g/dL per deciliter over eight weeks in patients with a baseline
transfusion burden of <4 units per eight weeks) was observed in 63%
of luspatercept-treated patients and 38% had transfusion independence
for eight weeks or longer.[48] Since the overall
erythroid response rate was higher among patients with ringed
sideroblasts (≥15% ring sideroblasts) than other subtypes of MDS, the
phase 3 enrolled patients with lower-risk MDS with ring sideroblasts
who had been receiving regular RBCs transfusions and were refractory or
unlikely to respond to ESA.
The MEDALIST (NCT02631070) is a
multicenter randomized, double-blind, placebo-controlled trial that
enrolled 229 patients randomly assigned in a 2:1 ratio to receive
luspatercept or placebo, administered subcutaneously every three weeks
for 24 weeks. For at least eight weeks, transfusion independence was
observed in 38% of the luspatercept group patients, compared with 13%
of the placebo group (P<0.001). During the first 24 weeks, 28% in
the luspatercept group had transfusion independence for 12 weeks or
longer, as compared with 8% in the placebo group, and the corresponding
values during weeks 1 through 48 were 33% and 12% (P<0.001). Also, a
higher percentage of patients in the luspatercept group than in the
placebo group had transfusion independence for 16 weeks or longer. The
most common luspatercept-associated adverse events (of any grade)
included fatigue, diarrhea, asthenia, nausea, and dizziness. The
incidence of adverse events decreased over time.
Conclusions
Activin
receptor ligand traps are the first pharmacological treatment approved
for TDT. Its introduction in clinical practice has the potential to
dramatically impact on TDT management; however, further studies are
needed to elucidate its mechanism of action.
Author Contributions
:V.B,
I.N., I.M drafted and wrote the paper and prepared figures; P.D. and
L.D. performed experiments. All authors have read and agreed to the
final version of the manuscript.
Funding
This
research was partly supported by Italian ministry of health and
Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico (RC2020)..
Acknowledgments
:We thank Daniela Canali for the artwork.
References
- Galanello R, Origa R. b-thalassemia. Orphanet J Rare Dis. 2010;5:11. https://doi.org/10.1186/1750-1172-5-11 PMid:20492708 PMCid:PMC2893117
- Cappellini
MD, Porter J, Origa R, Forni GL, Voskaridou E, Galacteros F, et al.
Sotatercept, a novel transforming growth factor b ligand trap, improves
anemia in b-thalassemia: a phase II, open-label, dose-finding study.
Haematologica. 2019;104(3):477-84. https://doi.org/10.3324/haematol.2018.198887 PMid:30337358 PMCid:PMC6395345
- Verma
A, Suragani RN, Aluri S, Shah N, Bhagat TD, Alexander MJ, et al.
Biological basis for efficacy of activin receptor ligand traps in
myelodysplastic syndromes. J Clin Invest. 2020;130(2):582-9. https://doi.org/10.1172/JCI133678 PMid:31961337
- Aleman-Muench
GR, Soldevila G. When versatility matters: activins/inhibins as key
regulators of immunity. Immunol Cell Biol. 2012;90(2):137-48. https://doi.org/10.1038/icb.2011.32 PMid:21537340
- Chen
YG, Wang Q, Lin SL, Chang CD, Chuang J, Chung J, et al. Activin
signaling and its role in regulation of cell proliferation, apoptosis,
and carcinogenesis. Exp Biol Med (Maywood). 2006;231(5):534-44. https://doi.org/10.1177/153537020623100507 PMid:16636301
- Shiozaki
M, Sakai R, Tabuchi M, Nakamura T, Sugino K, Sugino H, et al. Evidence
for the participation of endogenous activin A/erythroid differentiation
factor in the regulation of erythropoiesis. Proc Natl Acad Sci U S A.
1992;89(5):1553-6. https://doi.org/10.1073/pnas.89.5.1553 PMid:1542647 PMCid:PMC48490
- Greenwald
J, Vega ME, Allendorph GP, Fischer WH, Vale W, Choe S. A flexible
activin explains the membrane-dependent cooperative assembly of
TGF-beta family receptors. Mol Cell. 2004;15(3):485-9. https://doi.org/10.1016/j.molcel.2004.07.011 PMid:15304227
- Walton
KL, Makanji Y, Harrison CA. New insights into the mechanisms of activin
action and inhibition. Mol Cell Endocrinol. 2012;359(1-2):2-12. https://doi.org/10.1016/j.mce.2011.06.030 PMid:21763751
- Abe Y, Minegishi T, Leung PC. Activin receptor signaling. Growth Factors. 2004;22(2):105-10. https://doi.org/10.1080/08977190410001704688 PMid:15253386
- Keutmann
HT, Schneyer AL, Sidis Y. The role of follistatin domains in
follistatin biological action. Mol Endocrinol. 2004;18(1):228-40. https://doi.org/10.1210/me.2003-0112 PMid:14563935
- Welt
C, Sidis Y, Keutmann H, Schneyer A. Activins, inhibins, and
follistatins: from endocrinology to signaling. A paradigm for the new
millennium. Exp Biol Med (Maywood). 2002;227(9):724-52. https://doi.org/10.1177/153537020222700905 PMid:12324653
- Wu
J, Dong Y, Teng X, Cheng M, Shen Z, Chen W. Follistatin-like 1
attenuates differentiation and survival of erythroid cells through
Smad2/3 signaling. Biochem Biophys Res Commun. 2015;466(4):711-6. https://doi.org/10.1016/j.bbrc.2015.09.044 PMid:26365350
- Hill
JJ, Davies MV, Pearson AA, Wang JH, Hewick RM, Wolfman NM, et al. The
myostatin propeptide and the follistatin-related gene are inhibitory
binding proteins of myostatin in normal serum. J Biol Chem.
2002;277(43):40735-41. https://doi.org/10.1074/jbc.M206379200 PMid:12194980
- Lewis
KA, Gray PC, Blount AL, MacConell LA, Wiater E, Bilezikjian LM, et al.
Betaglycan binds inhibin and can mediate functional antagonism of
activin signalling. Nature. 2000;404(6776):411-4. https://doi.org/10.1038/35006129 PMid:10746731
- MacConell
LA, Leal AM, Vale WW. The distribution of betaglycan protein and mRNA
in rat brain, pituitary, and gonads: implications for a role for
betaglycan in inhibin-mediated reproductive functions. Endocrinology.
2002;143(3):1066-75. https://doi.org/10.1210/endo.143.3.8707 PMid:11861534
- Chapman
SC, Woodruff TK. Betaglycan localization in the female rat pituitary:
implications for the regulation of follicle-stimulating hormone by
inhibin. Endocrinology. 2003;144(12):5640-9. https://doi.org/10.1210/en.2003-0670 PMid:14500575
- Miyazono K. Positive and negative regulation of TGF-beta signaling. J Cell Sci. 2000;113 ( Pt 7):1101-9.
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-b family signalling. Nature. 2003;425(6958):577-84. https://doi.org/10.1038/nature02006 PMid:14534577
- Fernandez-Nocelo
S, Gallego R, Costoya JA, Arce VM. Expression of myostatin in human
hematopoietic cells unveils novel autocrine/paracrine actions for the
hormone. J Cell Physiol. 2019;234(5):7236-46. https://doi.org/10.1002/jcp.27494 PMid:30370618
- Rochette
L, Zeller M, Cottin Y, Vergely C. Growth and differentiation factor 11
(GDF11): Functions in the regulation of erythropoiesis and cardiac
regeneration. Pharmacology and Therapeutics. 2015;156:26-33. https://doi.org/10.1016/j.pharmthera.2015.10.006 PMid:26523637
- Worthington JJ, Klementowicz JE, Travis MA. TGF-β: a sleeping giant awoken by integrins. Trends Biochem Sci. 2011;36(1):47-54. https://doi.org/10.1016/j.tibs.2010.08.002 PMid:20870411
- Soderberg
SS, Karlsson G, Karlsson S. Complex and context dependent regulation of
hematopoiesis by TGF-beta superfamily signaling. Ann N Y Acad Sci.
2009;1176:55-69. https://doi.org/10.1111/j.1749-6632.2009.04569.x PMid:19796233
- He
W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massague J.
Hematopoiesis controlled by distinct TIF-1g and Smad4 branches of the
TGF-b pathway. Cell. 2006;125(5):929-41. https://doi.org/10.1016/j.cell.2006.03.045 PMid:16751102
- Blank U, Karlsson S. TGF-b signaling in the control of hematopoietic stem cells. Blood. 2015;125(23):3542-50. https://doi.org/10.1182/blood-2014-12-618090 PMid:25833962
- Fields
SZ, Parshad S, Anne M, Raftopoulos H, Alexander MJ, Sherman ML, et al.
Activin receptor antagonists for cancer-related anemia and bone
disease. Expert Opinion on Investigational Drugs. 2013;22(1):87-101. https://doi.org/10.1517/13543784.2013.738666 PMid:23127248
- Tsuchida
K, Nakatani M, Hitachi K, Uezumi A, Sunada Y, Ageta H, et al. Activin
signaling as an emerging target for therapeutic interventions. Cell
Commun Signal. 2009;7:15. doi: 10.1186/1478-811X-7-15. https://doi.org/10.1186/1478-811X-7-15 PMid:19538713 PMCid:PMC2713245
- Aykul
S, Martinez-Hackert E. Transforming Growth Factor-β Family Ligands Can
Function as Antagonists by Competing for Type II Receptor Binding. J
Biol Chem. 2016;291(20):10792-804. https://doi.org/10.1074/jbc.M115.713487 PMid:26961869 PMCid:PMC4865925
- Sako
D, Grinberg AV, Liu J, Davies MV, Castonguay R, Maniatis S, et al.
Characterization of the ligand binding functionality of the
extracellular domain of activin receptor type IIB. J Biol Chem.
2010;285(27):21037-48. https://doi.org/10.1074/jbc.M110.114959 PMid:20385559 PMCid:PMC2898293
- Pearsall
R, Canalis E, Cornwall-Brady M, Underwood K, Haigis B, Ucran J, et al.
A soluble activin type IIA receptor induces bone formation and improves
skeletal integrity. Proceedings of the National Academy of Sciences of
the United States of America. 2008;105(19):7082-7. https://doi.org/10.1073/pnas.0711263105 PMid:18460605 PMCid:PMC2383948
- Ruckle
J, Jacobs M, Kramer W, Pearsall AE, Kumar R, Underwood KW, et al.
Single-dose, randomized, double-blind, placebo-controlled study of
ACE-011 (ActRIIA-IgG1) in post-menopausal women. J Bone Miner Res.
2009;24(4):744-52. https://doi.org/10.1359/jbmr.081208 PMid:19049340
- Sherman
ML, Borgstein NG, Mook L, Wilson D, Yang Y, Chen N, et al.
Multiple-dose, safety, pharmacokinetic, and pharmacodynamic study of
sotatercept (ActRIIA-IgG1), a Novel erythropoietic agent, in healthy
post-menopausal women. Journal of Clinical Pharmacology.
2013;53(11):1121-30. https://doi.org/10.1002/jcph.160 PMid:23939631
- Suragani
RNVS, Cadena SM, Cawley SM, Sako D, Mitchell D, Li R, et al.
Transforming growth factor-β superfamily ligand trap ACE-536 corrects
anemia by promoting late-stage erythropoiesis. Nature Medicine.
2014;20(4):408-14. https://doi.org/10.1038/nm.3512 PMid:24658078
- Carrancio
S, Markovics J, Wong P, Leisten J, Castiglioni P, Groza MC, et al. An
activin receptor IIA ligand trap promotes erythropoiesis resulting in a
rapid induction of red blood cells and haemoglobin. British Journal of
Haematology. 2014;165(6):870-82. https://doi.org/10.1111/bjh.12838 PMid:24635723 PMCid:PMC4282119
- Suragani
RN, Cawley SM, Li R, Wallner S, Alexander MJ, Mulivor AW, et al.
Modified activin receptor IIB ligand trap mitigates ineffective
erythropoiesis and disease complications in murine b -thalassemia.
Blood. 2014;123(25):3864-72. https://doi.org/10.1182/blood-2013-06-511238 PMid:24795345 PMCid:PMC4064330
- Dussiot
M, Maciel TT, Fricot A, Chartier C, Negre O, Veiga J, et al. An activin
receptor IIA ligand trap corrects ineffective erythropoiesis in
β-thalassemia. Nature Medicine. 2014;20(4):398-407. https://doi.org/10.1038/nm.3468 PMid:24658077
- Iancu-Rubin
C, Mosoyan G, Wang J, Kraus T, Sung V, Hoffman R. Stromal cell-mediated
inhibition of erythropoiesis can be attenuated by Sotatercept
(ACE-011), an activin receptor type II ligand trap. Experimental
Hematology. 2013;41(2):155-66. https://doi.org/10.1016/j.exphem.2012.12.002 PMid:23261964
- Flotta
S, Delbini P, Graziadei G, Marcon A, Sung V, Cappellini MD.
Erythropoietic response to a ligand trap of activin receptor II in
cultures from b-thalassemia patients. Haematologica. 2015;100:766.
- Attie
KM, Allison MJ, McClure T, Boyd IE, Wilson DM, Pearsall AE, et al. A
phase 1 study of ACE-536, a regulator of erythroid differentiation, in
healthy volunteers. Am J Hematol. 2014;89(7):766-70. https://doi.org/10.1002/ajh.23732 PMid:24715706 PMCid:PMC4173124
- Guerra
A, Oikonomidou PR, Sinha S, Zhang J, Presti VL, Hamilton CR, et al.
Lack of GDF11 does not improve anemia or prevent the activity of
RAP-536 in a mouse model of b-thalassemia. Blood. 2019;134(6):568-72. https://doi.org/10.1182/blood.2019001057 PMid:31151988 PMCid:PMC6688431
- Martinez
PA, Li R, Ramanathan HN, Bhasin M, Pearsall RS, Kumar R, et al.
Smad2/3-pathway ligand trap luspatercept enhances erythroid
differentiation in murine β-thalassaemia by increasing GATA-1
availability. Journal of Cellular and Molecular Medicine. 2020. https://doi.org/10.1111/jcmm.15243 PMid:32351032 PMCid:PMC7294138
- Sherman
ML, Borgstein NG, Mook L, Wilson D, Yang Y, Chen N, et al.
Multiple-dose, safety, pharmacokinetic, and pharmacodynamic study of
sotatercept (ActRIIA-IgG1), a novel erythropoietic agent, in healthy
post-menopausal women. J Clin Pharmacol. 2013;53(11):1121-30. https://doi.org/10.1002/jcph.160 PMid:23939631
- Abdulkadyrov
KM, Salogub GN, Khuazheva NK, Sherman ML, Laadem A, Barger R, et al.
Sotatercept in patients with osteolytic lesions of multiple myeloma. Br
J Haematol. 2014;165(6):814-23. https://doi.org/10.1111/bjh.12835 PMid:24650009 PMCid:PMC4312883
- Raftopoulos
H, Laadem A, Hesketh PJ, Goldschmidt J, Gabrail N, Osborne C, et al.
Sotatercept (ACE-011) for the treatment of chemotherapy-induced anemia
in patients with metastatic breast cancer or advanced or metastatic
solid tumors treated with platinum-based chemotherapeutic regimens:
results from two phase 2 studies. Support Care Cancer.
2016;24(4):1517-25. https://doi.org/10.1007/s00520-015-2929-9 PMid:26370220 PMCid:PMC4766217
- Reblozyl. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761136orig2lbl.pdf Accessed June 06, 2020.
- Piga
A, Perrotta S, Gamberini MR, Voskaridou E, Melpignano A, Filosa A, et
al. Luspatercept improves hemoglobin levels and blood transfusion
requirements in a study of patients with b-thalassemia. Blood. 2019. https://doi.org/10.1182/blood-2018-10-879247 PMid:30617198 PMCid:PMC6440118
- Cappellini
MD, Cohen A, Piga A, Bejaoui M, Perrotta S, Agaoglu L, et al. A phase 3
study of deferasirox (ICL670), a once-daily oral iron chelator, in
patients with b-thalassemia. Blood. 2006;107(9):3455-62. https://doi.org/10.1182/blood-2005-08-3430 PMid:16352812
- Komrokji
R, Garcia-Manero G, Ades L, Prebet T, Steensma DP, Jurcic JG, et al.
Sotatercept with long-term extension for the treatment of anaemia in
patients with lower-risk myelodysplastic syndromes: a phase 2,
dose-ranging trial. Lancet Haematol. 2018;5(2):e63-e72. https://doi.org/10.1016/S2352-3026(18)30002-4
- Platzbecker
U, Germing U, Gotze KS, Kiewe P, Mayer K, Chromik J, et al.
Luspatercept for the treatment of anaemia in patients with lower-risk
myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2
dose-finding study with long-term extension study. Lancet Oncol.
2017;18(10):1338-47. https://doi.org/10.1016/S1470-2045(17)30615-0
[TOP]