Guiping Liao2,*, Yali Zhou2,*, Xiaolin Yin2, Sheng He3, Yi Wu4, Jian Xiao1, Zhili Geng2, Qiuying Huang2, Ganghui Luo5 and Kun Yang1.
1 Department of Hematology, Zigong First People's Hospital, Zigong, China.
2
Department of Hematology, The 923rd Hospital of the Joint Logistics
Support Force of the People's Liberation Army, Nanning, China.
3
Department of Genetic and Metabolic Laboratory, Guangxi Zhuang
Autonomous Region Women and Children Health Care Hospital, Nanning,
China.
4 Department of Hematology, Guangan People's Hospital, Guangan, China
5
Department of Laboratory Medicine, The 923rd Hospital of the Joint
Logistics Support Force of the People's Liberation Army, Nanning, China.
* These authors contributed equally to this work.
Correspondence to: Kun
Yang. Department of Hematology, Zigong First People's Hospital, Zigong,
China. Tel: +86-15181977603. E-mail:
1759874951@qq.com
Published: May 1, 2022
Received: January 16, 2022
Accepted: April 4, 2022
Mediterr J Hematol Infect Dis 2022, 14(1): e2022034 DOI
10.4084/MJHID.2022.034
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background: IVS-II-5 G>C (HBB: c.315+5 G>C)
is a rare β-thalassemia mutation. However, there is no clear evidence
regarding the effect of this defect or co-inheritance of other
β-thalassemia mutations on phenotypes.
Methods:
The clinical phenotypes associated with compound heterozygosity for the
IVS-II-5 G>C mutation and other β-thalassemia mutations, together
with the genetic modifiers' potential effect of the genetic modifiers
α-thalassemia, were studied in 13 patients. In addition, analyses of
red cell indices, hemoglobin component, iron status, and α-globin genes
were carried out in 19 heterozygotes.
Results:
Next-generation sequencing of 24 undiagnosed patients with
transfusion-dependent thalassemia (TDT) or non-transfusion-dependent
thalassemia (NTDT) identified 13 carriers of the IVS-II-5 G>C
mutation. There was a wide spectrum of phenotypic severity in compound
heterozygotes and 6 (46.2%) of 13 were transfusion dependent. Analysis
of 19 heterozygotes indicated that most were hematologically normal
without appreciable microcytosis or hypochromia, and approximately half
had normal hemoglobin A2 levels at the same time.
Conclusion:
Compound heterozygotes for IVS-II-5 G>C and other severe
β-thalassemia mutations are phenotypically severe enough to necessitate
appropriate therapy and counselling. Co-inheritance of this nucleotide
substitution with other β-thalassemia mutations may account for a
considerable portion of the incidence of undiagnosed patients with NTDT
and TDT in Guangxi. Therefore, the IVS-II-5 G>C mutation can pose
serious difficulties in screening and counselling.
|
Introduction
β-Thalassemia is a genetic hemolytic disease caused by a reduction (β+ allele) or deletion (β0
allele) of the beta globin gene. So far, more than 350 pathogenic
genetic mutations with a high degree of genetic heterogeneity based on
geographical location and race have been associated with β-thalassemia.[1]
Further elucidation of the genotype/phenotype relationship could
benefit phenotype prediction for genetic counseling of at-risk couples
and appropriate clinical treatment for homozygous or compound
heterozygous patients. Usually, in the heterozygous state,
β-thalassemia is characterized by mild microcytic hypochromic anemia
and increased hemoglobin A2 (HbA2)
levels. However, some β-globin mutations are very mild or silent. Thus,
heterozygote carriers have normal hematological indices and
electrophoretic fractions. These specific defects to the β-globin gene
can be identified by genetic and molecular analyses.[2,3]
The rare IVS-II-5 G>C (HBB: c.315+5 G>C)
mutation of the β-globin gene was first observed in a family in
Guangxi, China. The proband and her two brothers were compound
heterozygous for two mutations: the IVS-II-5 G>C substitution and
the deletion -TCTT from codons 41–42 (HBB: c.126_129delCTTT). These mutations resulted in severe anemia, requiring regular blood transfusions.[4] In addition, Zhao et al.[5] reported a patient with normal red blood cell indices and borderline HbA2 levels, which was found to be compound heterozygous for the IVS-II-5 G>C and IVS-II-672 A>C (HBB: c.316-179 A>C)
mutations. There is no clear evidence regarding the effect of this
defect or co-inheritance of other β-globin gene mutations on
phenotypes. This study describes 32 individuals with the IVS-II-5
G>C mutation, including 19 heterozygous and 13 with co-inheritance
of the β0-thalassemia mutation. In
addition, the clinical and hematological phenotypes of heterozygotes
with the very rare IVS-II-5 G>C variant or compound heterozygotes
with β0-thalassemia mutations and the importance of genetic counseling are discussed.
Subjects and Methods
Subjects.
The subjects were selected from undiagnosed patients presenting with
transfusion dependent thalassemia (TDT) or non-transfusion dependent
thalassemia (NTDT) and relatives. The gap-polymerase chain reaction
(Gap-PCR) and reverse dot-blot (RDB) technique were previously
performed to investigate the incidences of 17 common types of Chinese
β-thalassemia mutations [-28 A>G (HBB: c.-78 A>G), -29 A>G
(HBB: c.-79 A>G), -30 T>C (HBB: c.-80 T>C), -32 C>A (HBB: c.-82 C>A), codons 14/15 +G (HBB: c.45_46insG), codon 17 A>T (HBB: c.52 A>T), codon 26 G>A (HbE or HBB: c.79 G>A), codons 27–28 +C (HBB: c.84_85G), codon 31 -C (HBB: c.94delC), codons 41–42 -TCTT (HBB: c.126_129delCTTT), codon 43 G>T (HBB: c.130 G>T), codons 71-72 +A (HBB: c.216_217insA), IVS-I-1 G>T (HBB: c(0).92+1 G>T), IVS-I-5 G>C (HBB: c.92+5 G>C), IVS-II-654 C>T (HBB: c.316-197 C>T), Cap+40-43 -AAAC (HBB: c.11_-8delAAAC), and codon ATG>AGG (HBB: c.2 T>G)] and six common Chinese α-thalassemia mutations [– –SEA (South East Asian) deletion, –α3.7 (rightward) and –α4.2 (leftward) deletions and Hb Constant Spring (Hb CS; HBA2: c.427 T>C), Hb Quong Sze (Hb QS; HBA2: c.377 T>C) and Hb Westmead (HBA2: c.369 C>G) point mutations] in the undiagnosed patients.[6]
Genotypic analysis using traditional methods.
The family members were analyzed with Gap-PCR and RDB methods on the
common types of Chinese thalassemia mutations. In addition, the.
Multiplex ligation-dependent probe amplification was conducted to
identify variations in α and β gene copy numbers for the undiagnosed
patients and their family members.
Genetic sequencing.
Genomic DNA was extracted from peripheral blood samples using the
MagCore® Genomic DNA Whole Blood Kit (ATRiDA B.V., Amersfoort,
Netherlands). A library was constructed using the Twist Library
Preparation Kit Twist Comprehensive Exome 96 Reactions Kit (Twist
Bioscience, South San Francisco, CA, USA). Next-generation sequencing
(NGS) was performed to screen potential variants using the Illumina
NovaSeq sequencing platform (Illumina, Inc., San Diego, CA, USA) with a
sequencing read length of PE150. This procedure included whole-exome
sequencing for nearly 700 genes related to hematological hereditary and
immunodeficiency disorders based on NGS. In addition, Sanger sequencing
was performed to confirm the presence or absence of these mutations.
Hematological analysis.
Complete blood counts were analyzed using an XE 5000 automatic blood
cell analyzer (Sysmex Corporation, Kobe, Japan). Fetal hemoglobin (HbF)
and HbA2 levels were quantified by
high-pressure liquid chromatography (HPLC) (VARIANT II Hemoglobin
Testing System; Bio-Rad Laboratories, Hercules, CA, USA) and capillary
electrophoresis (Sebia, Lisses, France). Serum iron and ferritin levels
were measured using a Cobas 6000 chemistry analyzer (Roche Diagnostics,
Mannheim, Germany). Total iron-binding capacity was determined using a
BN ProSpec® System (Siemens Healthineers, Erlangen, Germany).
Results
Of
the 24 undiagnosed patients with TDT or NTDT, 13 (54.2%) were compound
heterozygous for the IVS-II-5 G>C mutation and a second β0
gene mutation (-TCTT at codons 41–42 in seven, A>T at codon 17 in
five, and +A at codons 71–72 in one). The clinical phenotype was TDT in
six patients and NTDT in seven. The details of the genotypes and
phenotypes are described in Table 1.
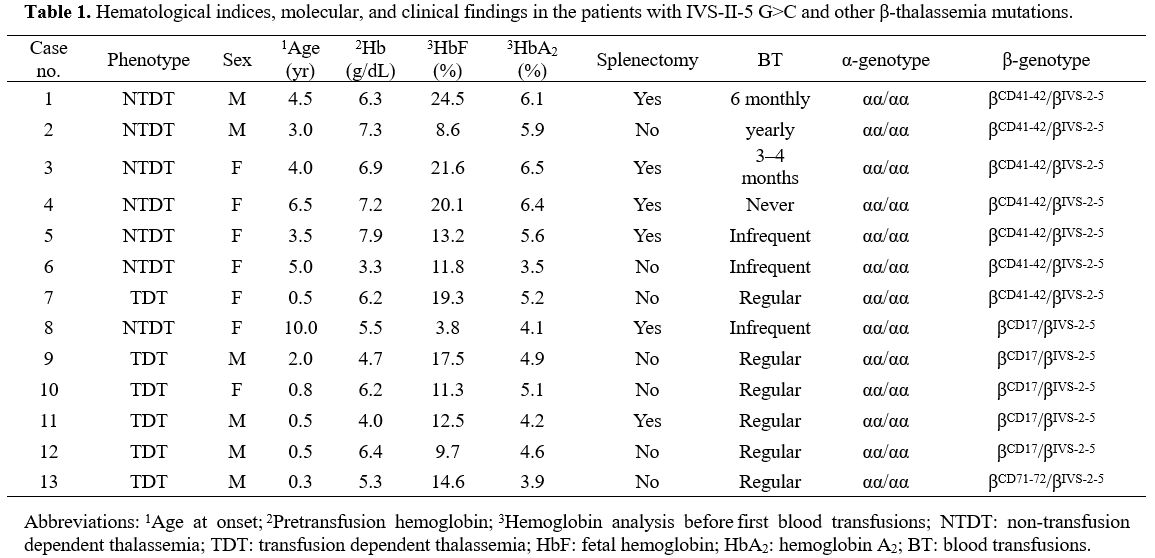 |
Table 1. Hematological
indices, molecular, and clinical findings in the patients with IVS-II-5
G>C and other β-thalassemia mutations. |
The
age of onset, pretransfusion hemoglobin (Hb) levels, and number and
frequency of transfusions were obtained from the patient's records. All
patients were compound heterozygotes and at diagnosis before the age of
5 years, except two at ages of 6.5 and 10. Seven patients had received
variable numbers of blood transfusions before being referred.
Hemoglobin analysis before the first transfusion found that these
patients had increased levels of HbF (range, 3.8%–24.5%) and HbA2
(3.5%–6.5%) to various degrees. The mean pretransfusion Hb
concentration of the six patients with the TDT phenotype was 5.5
(range, 4.0–6.4) g/dL. All patients had mild to moderate splenomegaly
at the time of presentation. During the follow-up period, six patients
underwent splenectomy (Table 1).
Compound heterozygotes for the IVS-II-5 G>C and -TCTT at codons 41–42. Of
the seven patients with the IIS-II-5 G>C and -TCTT at codons 41–42
genotype, 6 (85.7%) had the NTDT phenotype. One had never received a
transfusion, three required occasional blood transfusions, and one
received 3-4 blood transfusions each year after disease onset, but
transfusions were no longer necessary after splenectomy. Another
patient with TDT received 1–2 transfusions per month.
Compound heterozygotes for the IVS-II-5 G>C and A>T at codon 17 mutations. Of
the five patients with the IVS-II-5 G>Cand A>T at codon 17
genotype, 4 (80.0%) had the TDT phenotype, including three who required
monthly blood transfusions, while the fourth died from anemic heart
disease. The remaining patient had the NTDT phenotype and rarely
required transfusion at the disease onset.
Compound heterozygotes for the IVS-II-5 G>C and +A at codons 71–72.
One case with the IVS-II-5 G>C and +A at codons 71–72 genotype
required one blood transfusion per month at disease onset. This patient
received hematopoietic stem cell transplantation, and Hb levels
remained at >7 g/dL afterwards.
IVS-II-5 G>C in a heterozygous state. Nineteen
individuals (six men and 13 women; age range, 6–68 years) heterozygous
for the IVS-II-5 G>C mutation were screened from 42 relatives. None
had co-inherited α-thalassemia. Iron deficiency anemia was ruled out in
15 carriers, as Hb levels were 11.0–15.0 g/dL. The mean corpuscular
volume (MCV) was > 80 (reference range, 80–100) fL in 12 (63.2%)
individuals and the mean corpuscular hemoglobin (MCH) was > 27
(reference range, 27–34) pg in 11 (57.9%). HbA2 levels measured by HPLC ranged from 2.7% to 3.9%. When defining HbA2 ≥ 4.0% as abnormal, 3.5%–4.0% as critical, and <3.5% as normal, 13 (68.4%) of the 19 patients had normal HbA2 levels, while six were considered critical. Eight patients with MCV and HbA2
were completely normal (cases 1, 4, 5, 8, 12, 13, 16, and 17), which
included five who received capillary electrophoresis, suggested that HbA2 levels were in the normal range (<3.5%) (Table 2).
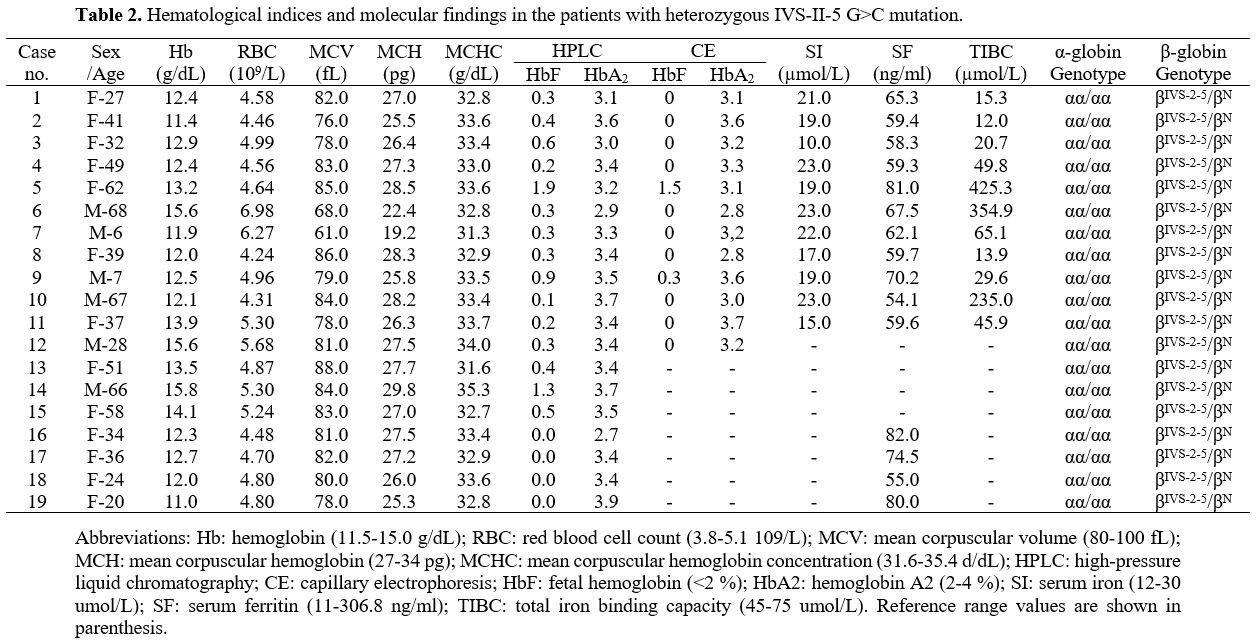 |
Table
2. Hematological indices and molecular findings in the patients with heterozygous IVS-II-5 G>C mutation. |
Discussion
Molecular
defects causing β-thalassemia are mainly due to point mutations, few
deletions, dysregulation of the globin gene, or an insert in the coding
region. A previous study conducted in Guangxi province, China, revealed
the presence of few frequent mutations and a large number of rare
defects associated with molecular heterogeneity. A large-scale
epidemiological survey conducted in Guangxi reported that the carrier
rate of the IVS-II-5 G>C mutation was 0.02%.[7] No IVS-II-5 G>C mutation was detected in the screening of 47,500 people in Baise city[8] or 130,318 in Yulin city in Guangxi.[9] Only one case with the IVS-II-5 G>C mutation was identified among 189,414 individuals screened in Fujian, China.[10]
These studies confirmed that the IVS-II-5 G>C mutation is rare in
the Chinese population. However, in the present study, 13 (54.2%) of
the 24 undiagnosed patients with TDT or NTDT had co-inherited the
IVS-II-5G>C mutation, which included 7 (53.8%) from Hechi City. In
addition, 13 (68.4%) of the 19 heterozygous were from Hechi City, which
was not included in the large-scale epidemiological survey previously
conducted in Guangxi. Hence, it seemed that the mutation was
concentrated in Hechi city. However, a large-scale epidemiological
investigation is needed to confirm this suspicion.
There have been
relatively few reported cases of co-inheritance of the IVS-II-5 G>C
mutation compounded with other β-thalassemia mutations. Thus, the
impact on phenotype remains unclear.[4,5,11]
Three cases previously reported with the IVS-II-5 G>C mutation
compounded with -TCTT at codons 41–42 had transfusion-dependent
thalassemia. Only one (14.3%) of seven patients in the present study
had the TDT phenotype, while 6 (85.7%) had the NTDT phenotype.
Interestingly, one patient required 3–4 blood transfusions per year but
no longer after splenectomy. In addition, blood transfusions were no
longer needed after splenectomy in three patients, as Hb levels were
maintained at >6.8 g/dL.[11] The efficacy of
splenectomy for the treatment of thalassemia is dependent on the
destruction site of red blood cells. However, the efficacy of
splenectomy is poor in some patients with ineffective hematopoiesis,[12]
suggesting a greater extent of erythrocyte destruction in the spleen of
patients with the IVS-II-5 G>C mutation as compared to those with
other types of β-thalassemia. Hence, splenectomy is relatively
effective in these patients. Clinical manifestations were more severe
with the IVS-II-5 G>C/codon 17 A>T genotype, as most patients had
the TDT phenotype. In patients with the IVS-II-5 G>C and +A at
codons 71–72, our data were insufficient to predict the phenotype as
only one was diagnosed with TDT. Among the patients with severe disease
in the present study, none carried both IVS-II-5 and β+-thalassemia
mutations, which indirectly confirmed that the clinical symptoms might
not be severe for patients with IVS II-5 G>C and β+-thalassemia mutations. Zhao et al.[5]
reported a case of compounded heterozygosity for IVS II-5 G>C and
IVS-II-672 A>C with normal Hb and MCV levels, although there is no
evidence that the IVS-II-672 A>C sequence variant is pathogenic. In
addition, although the influence of co-inheritance of α-thalassemia on
the phenotype was ruled out, the influence of other globin gene
modifications on the severity of clinical presentation remains unclear.
Analysis
of heterozygotes with the IVS-II-5 G>C mutation showed that in 12
(63.2%) of 19 individuals, MCV was > 80 (reference range, 80–100) fL
and in 11 (57.9%) of 19, MCH was >27 (reference range, 27–34) pg.
When screening β-thalassemia carriers, it is not uncommon to identify
individuals with normal or mildly reduced red cell indices. Subjects
with this phenotype may be carriers of very mild or silent β-gene
mutations associated with high residual β-globin chain output. Unlike
the IVS-II-1 G>T mutation, which causes β0-thalassemia, the IVS-II-5 G>C mutation did not completely abolish normal splicing.[11] Jiang et al.[11]
found that normally spliced RNA was the dominant form of the IVS-II-5
G>C mutation. Therefore, co-inheritance of α-thalassemia has a
significant effect on red cell indices, particularly the MCV and MCH,
which may be normalized.[13] Furthermore, mutations to the KLF1 gene have been associated with normal MCV and MCH.[14]
Although co-inheritance of α-thalassemia was excluded from our cohort,
and other factors may contribute to differences in RBC parameters
between individuals. The HbA2
levels of all heterozygotes were <4.0%, of which 13 (68.4%) were
below the diagnostic cutoff of 3.5%. Of the 19 heterozygotes for the
IVS-II-5 G>C mutation, 8 (42.1%) were silent with completely normal
HbA2
and MCV levels. Because whole blood cell and Hb composition analysis
are required for a diagnosis of anemia and screening of thalassemia,
the key RBC parameters and the constitution of Hb were compared among
patients with other β+-thalassemia mutations or IVS-II-5 G>C
carriers (Figure 1). Significant differences in HbA2,
HbF, MCV, MCH, and MCHC values were evident between patients with other
β+-thalassemia mutations (IVS-II-654 C>T or -28 A>G) and IVS-II-5
G>C carriers (P<0.001). IVS-II-5 G>C carriers have significantly higher Hb levels than IVS-II-654 C>T carriers (P=0.002).
Thus, the IVS-II-5 G>C mutation is responsible for milder anemia
than other mutations described in China, although the variant was
described as the β+ hematological phenotype in a prior publication.[11]
Based solely on the screening results, there was no indication of
β-thalassemia, which is prone to a missed diagnosis. In addition, the
IVS-II-5 G>C defect is undetectable by traditional methods. DNA
sequencing is usually required, which prevents the detection of more
patients and missed diagnoses, and may eventually have adverse
consequences in genetic counseling. Thus, it is recommended that if a
partner is diagnosed as a carrier of thalassemia and indices and HbA2 are borderline, then molecular screening for the IVS-II-5 G>C mutation by NGS should be performed.
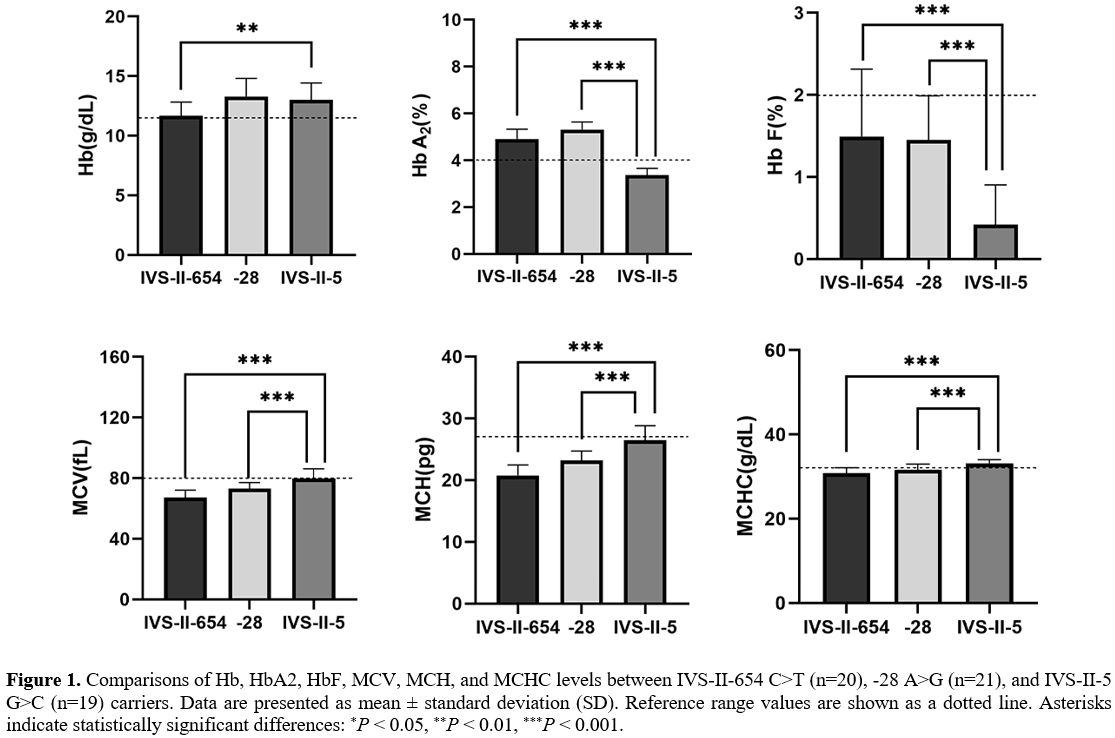 |
Figure 1. Comparisons of
Hb, HbA2, HbF, MCV, MCH, and MCHC levels between IVS-II-654 C>T
(n=20), -28 A>G (n=21), and IVS-II-5 G>C (n=19) carriers. Data
are presented as mean ± standard deviation (SD). Reference range values
are shown as a dotted line. Asterisks indicate statistically
significant differences: *P < 0.05, **P < 0.01, ***P < 0.001. |
Conclusions
In
conclusion, the present study results showed that co-inheritance of the
IVS-II-5 G>C defect with other mutations to globin genes may account
for a considerable portion of the incidence of undiagnosed moderate and
severe β-thalassemia in Guangxi. On the other hand, MCV and HbA2 levels
in most patients heterozygous for IVS-II-5 G>C were normal and,
thus, often misdiagnosed. Hence, routine screening of this nucleotide
substitution and NGS are recommended for genetic counseling of
suspected cases, especially in populations with a significantly high
frequency.
Acknowledgements
We are grateful to our patients for participating in this study.
Ethic statement
The study protocol was approved by the Medical Ethics Committee of the 923rd
Hospital of the Joint Logistics Support Force of the People's
Liberation Army and the First People's Hospital of Zigong. All patients
provided written informed consent.
Funding
This
study was supported by the Scientific Research Project of Guangxi
Zhuang Autonomous Region Health Committee (Z20201266) and Key Science
and Technology Project of Zigong (2020YXY04).
References
- Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med. 2021;384(8):727-43. https://doi.org/10.1056/NEJMra2021838 PMid:33626255
- Weatherall
DJ. Phenotype-genotype relationships in monogenic disease: lessons from
the thalassaemias. Nat Rev Genet. 2001;2(4):245-55. https://doi.org/10.1038/35066048 PMid:11283697
- Higgs DR. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med. 2013;3(1):a011718. https://doi.org/10.1101/cshperspect.a011718 PMid:23284078 PMCid:PMC3530043
- Jiang
NH, Liang S, Su C, Nechtman JF, Stoming TA. A novel beta-thalassemia
mutation [IVS-II-5 (G-->C)] in a Chinese family from Guangxi
Province, P.R. China. Hemoglobin. 1993;17(6):563-7. https://doi.org/10.3109/03630269309043498 PMid:8144358
- Zhao
L, Qing J, Liang Y, Chen Z. A novel compound heterozygosity in Southern
China: IVS-II-5 (G > C) and IVS-II-672 (A > C). Hemoglobin.
2016;40(6):428-30. https://doi.org/10.1080/03630269.2016.1252387 PMid:27829298
- Viprakasit
V, Ekwattanakit S. Clinical Classification, Screening and Diagnosis for
Thalassemia. Hematol Oncol Clin North Am. 2018;32(2):193-211. https://doi.org/10.1016/j.hoc.2017.11.006 PMid:29458726
- Xiong
F, Sun M, Zhang X, Cai R, Zhou Y, Lou J, et al. Molecular
epidemiological survey of haemoglobinopathies in the Guangxi Zhuang
Autonomous Region of southern China. Clin Genet. 2010;78(2):139-48. https://doi.org/10.1111/j.1399-0004.2010.01430.x PMid:20412082
- He
S, Qin Q, Yi S, Wei Y, Lin L, Chen S, et al. Prevalence and genetic
analysis of α- and β-thalassemia in Baise region, a multi-ethnic region
in southern China. Gene. 2017;619:71-5. https://doi.org/10.1016/j.gene.2016.02.014 PMid:26877226
- He
S, Li J, Li DM, Yi S, Lu X, Luo Y, et al. Molecular characterization of
α- and β-thalassemia in the Yulin region of Southern China. Gene.
2018;655:61-4. https://doi.org/10.1016/j.gene.2018.02.058 PMid:29477874
- Huang
H, Xu L, Chen M, Lin N, Xue H, Chen L, et al. Molecular
characterization of thalassemia and hemoglobinopathy in Southeastern
China. Sci Rep. 2019;9(1):3493. https://doi.org/10.1038/s41598-019-40089-5 PMid:30837609 PMCid:PMC6400947
- Jiang
NH, Liang S. The beta+-thalassemia mutation [IVS-II-5 (G-->C]
creates an alternative splicing site in the second intervening
sequence. Hemoglobin. 1999;23(2):171-6. https://doi.org/10.3109/03630269908996161 PMid:10335984
- Al-Salem AH, Nasserulla Z. Splenectomy for children with thalassemia. Int Surg. 2002;87(4):269-73.
- Galanello
R, Paglietti E, Melis MA, Crobu MG, Addis M, Moi P, et al. Interaction
of heterozygous beta zero-thalassemia with single functional
alpha-globin gene. Am J Hematol. 1988;29(2):63-6. https://doi.org/10.1002/ajh.2830290202 PMid:3189303
- Perseu
L, Satta S, Moi P, Demartis FR, Manunza L, Sollaino MC, et al. KLF1
gene mutations cause borderline HbA(2). Blood. 2011;118(16):4454-8. https://doi.org/10.1182/blood-2011-04-345736 PMid:21821711
[TOP]