Diego A Vargas-Hernández1, Adriana Catalina Uscategui-Ruiz1, Andrés Jesus Prada-Rueda1 and Consuelo Romero-Sánchez2,3.
1 Hospital Militar Central, Department of Internal Medicine, Universidad Militar Nueva Granada, Medical School, Bogotá, Colombia.
2 Hospital Militar Central, Rheumatology and Immunology Department/Clinical Immunology Group, Bogotá, Colombia.
3 Universidad El Bosque, Cellular and Molecular Immunology Group/INMUBO, Bogotá, Colombia.
Correspondence to:
Diego A. Vargas-Hernández. Hospital Militar Central, Department of
Internal Medicine, Tv 3 No 49-00 Bogotá, CO 110231 3598888, Universidad
Militar Nueva Granada, Medical School Bogota, CO, 6500000. ORCID:
https://orcid.org/0000-0002-2584-5195. E-mail:
dvargashdez@gmail.com u0401551@unimilitar.edu.co
Published: March 1, 2023
Received: September 05, 2022
Accepted: February 15, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023015 DOI
10.4084/MJHID.2023.015
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background: Sickle
cell trait (SCT) is an autosomal recessive blood disorder in which
patients are heterozygous carriers for hemoglobin S (HbAS) and are
usually asymptomatic. We performed a descriptive analysis of clinical
manifestations and outcomes associated with SCT.
Methods: This
was a descriptive, cross-sectional study that included patients with
SCT from 2014 to 2020 at Hospital Militar Central, the reference center
of the Military forces in Bogota, Colombia.
Results: Of
647 hemoglobin electrophoresis analyzed, we identified 51 patients with
SCT, including 43 males (84.3%) and eight females (15.7%), with a
median age of 22 years (IQR 15–36 years). Of these, 28 (54.8%) were
Afro-Colombian, 23 (45.1%) were Colombian mestizos, and 31/51 (60.8%)
of patients were active military members. Twenty-four patients (47.1%)
were asymptomatic, and Twenty-seven patients (52.9%) were symptomatic
(systemic complications); Most of the patients who presented symptoms
were active military members of the Colombian military forces. Splenic
complications were the most important (85.2%), p=0.0005, and there was
a wide spectrum of splenic complications. In addition, we found
significant elevations in leukocytes, bilirubin, LDH, and CRP. Eighteen
patients (66.7%) received medical management, five (18.5%) required
splenectomy, and only 5.9% of patients were sent for genetic counseling.
Conclusions: Military Personnel is a population with a high risk
of developing symptoms, and splenic complications were the most
relevant in symptomatic patients. Most patients received medical
treatment, and 18.5% of patients required splenectomy. Our results
reflect the absence of redirection of these patients to genetic
counseling.
|
Introduction
Sickle
cell trait (SCT) is an autosomal recessive inherited blood disorder
caused by the substitution of glutamic acid for valine due to an
abnormal allele mutation within the β globin gene, with the other
allele of the gene without alteration. These patients are heterozygous
or carriers for hemoglobin S (HbAS); in contrast, if the other allele
of the β globin gene also contains the mutation, patients are
considered homozygous for the mutation (HbSS), or if patients have
another mutation (e.g., HbC or β-thalassemia), the individual will
develop sickle cell disease (SCD) among which hemoglobin variants SS,
SC, Sβ-thalassemia are included.[1,2]
The HbS gene is distributed worldwide and affects millions of people
with an especially high frequency in sub-Saharan Africa, the countries
of the Middle East, and India.[2] The overall estimate
of incidence in the United States for SCT was 15.5 cases per 1,000
births and is more prevalent in the black population (73.1 per 1,000)
and less prevalent in Hispanic newborns (6.9 per 1,000 newborns);[3] it is estimated that the number of newborns with SCD globally will increase to more than 400,000 by 2050.[4]
Compared to SCD, which is associated with serious conditions and
complications, most patients with SCT are asymptomatic, usually living
their entire lives without a diagnosis. Consequently, in countries with
no adequate screening programs, it is uncommon to diagnose this
pathology, and descriptions of symptomatic patients with SCT are rare.
Furthermore, most literature is limited to case reports or series and a
systematic review of cases.[1,5–9] We did not find any reports or studies conducted in Colombia on patients with SCT.
Patients with SCT present symptoms only in extreme conditions of high
altitude, severe dehydration, or high- intensity physical activity,[10–12] which is explained by red blood cell sickling and increased mechanical fragility due to the polymerization of hemoglobin HbS[13,14] with consequent microvascular occlusion resulting in complications or clinical conditions that can be life-threatening.[7,15,16]
Symptoms are common with exposure to low oxygen tension or altitude,
including nonpressurized aircraft cabins, or in people exercising in
mountains or the military in high-altitude locations.[7,16]
When observing complications or manifestations, patients with SCT may present with splenic infarction,[6,9] venous thromboembolism (PE and DVT),[1,17] renal involvement (including proteinuria, hematuria, and chronic kidney disease),[1,18] rhabdomyolysis,[19,20] sudden death,[21]
and others; in paraclinical data, increased levels of bilirubin,
lactate dehydrogenase (LDH), and increased reticulocyte count have been
described, and on some occasions, anemia has been observed.[18,22,23]
Diagnostic confirmation of SCT is performed by hemoglobin
electrophoresis in gel or capillary electrophoresis or high-performance
liquid chromatography (HPLC).[24,25] The presence of
SCT is established by finding both hemoglobin A (HbA) and hemoglobin S
(HbS), with an amount of HbA greater than that of HbS (i.e., HbAS);
typically, hemoglobin S levels are between 20% and 45%.[26,27]
Based on this, we conducted a descriptive observational,
cross-sectional study in a quaternary care hospital in Bogota,
Colombia. We included patients with an SCT diagnosis and described
clinical manifestations, paraclinical and imaging findings, and the
treatment to which they were subjected. In addition, we compared the
clinical and paraclinical variables of patients with SCT who had
clinical manifestations and patients who were asymptomatic.
Material and Methods
Type of Study.
This report is a descriptive, cross-sectional study with an analytical
component that includes patients from the Hospital Militar Central, a
reference center for military forces in Bogota, Colombia, with a
diagnosis of SCT confirmed by hemoglobin electrophoresis in the
institutional laboratory between January 2014 and December 2020.
Inclusion criteria.
We included all patients who underwent hemoglobin electrophoresis at
the Hospital Militar Central, including-active military members of the
Colombian military forces and nonmilitary members (family, wife,
children, and parents) who attend the Hospital Militar Central. In
Colombia and its military forces, there are no screening programs for
hemoglobinopathies, so the reasons for which hemoglobin electrophoresis
was performed were variable; it was performed on symptomatic patients,
patients with a family history, and patients with abnormalities in
blood count, among other reasons.
Exclusion criteria. We exclude patients with a diagnosis of SCD defined as patients with HbSS, HbSC, HbSβ+ thalassemia or HbSβ0 thalassemia, repeat patients, or those without data in the clinical history.
Statistical analysis.
We built a database of important variables, and statistical analyses
were carried out using IBM SPSS Statistics; quantitative variables are
expressed as the mean, median, and interquartile ranges, as
appropriate; qualitative variables are expressed as absolute values and
percentages. The qualitative variables were compared using the
chi-square or Fisher's exact test. The quantitative variables between
the clinical presentation groups were analyzed using the nonparametric
Mann–Whitney U test. A p-value <0.05 was considered statistically
significant. The project was endorsed by the Research Ethics Committee
of the Hospital Militar Central, code-2021-030.
Results
A
total of 647 hemoglobin electrophoresis results were analyzed, from
which institutional medical history information was obtained; 123
(19.01%) abnormal results were found among patients with some
hemoglobinopathy, and a total of 90 patients (13.91%) with HbS were
found, of which 39 met exclusion criteria, resulting in a total of 51
patients (7.88%) diagnosed with SCT (Figure 1).
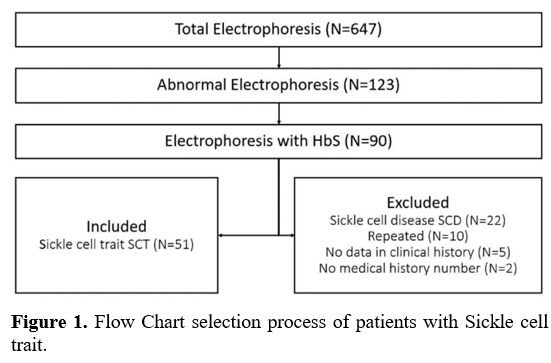 |
- Figure 1. Flow Chart selection process of patients with Sickle cell trait.
|
In
the descriptive analysis, we found a total of 51 patients, of which 43
were male (84.3%) and 8 were female (15.7%), and the median age was 22
years old (IQR 15–36 years). Of the Colombian population analyzed, 28
(54.9%) were Afro-Colombian and 23 (45.1%) Colombian mestizos;
additionally, 31/51 (60.8%) of patients were active military members of
the Colombian military forces. These 51 patients in the analysis
presented a percentage of HbS with a median of 38 (IQR 37-39),
predominating between 35–40% (Table 1).
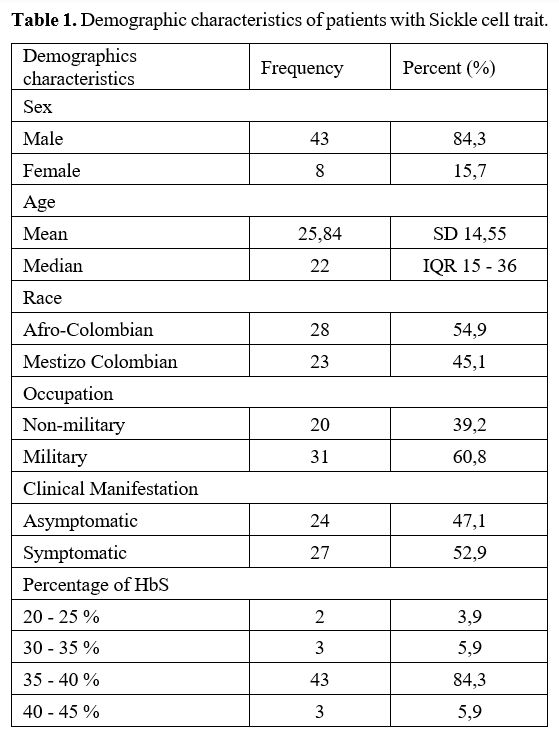 |
- Table 1. Demographic characteristics of patients with Sickle cell trait.
|
We
found 24/51 patients (47.1%) with a diagnosis of SCT who had never
presented with any symptoms or clinical manifestations associated with
the diagnosis (classified as "asymptomatic" patients) and 27/51
patients (52.9%) who presented with systemic complications (splenic,
hepatic, renal, urological, or other), which led to the diagnosis of
the disease (classified as "symptomatic" patients). Most of the
patients who presented symptoms were active military members of the
Colombian military forces 24/27 (88,8%).
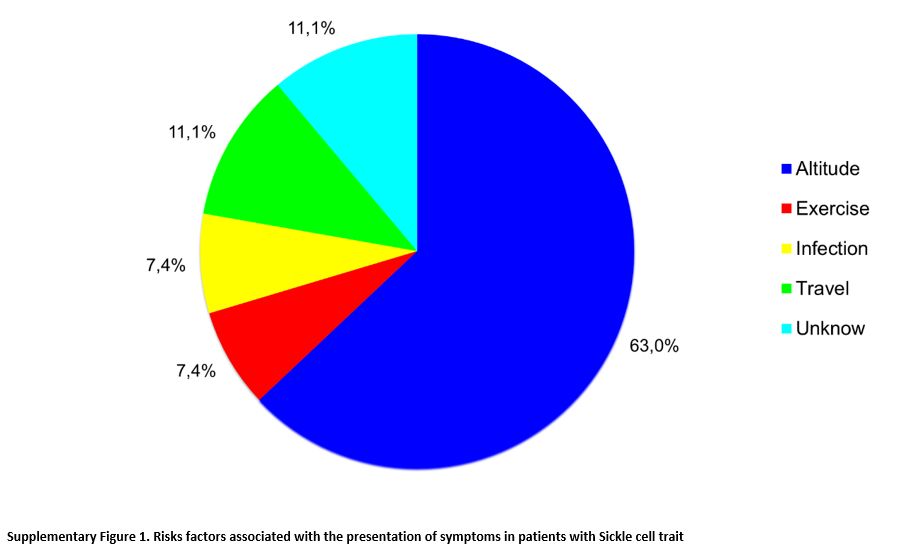
In patients classified as symptomatic (n=27), clinical manifestations
were associated mainly with a change in altitude in 17 (63.0%) and,
less frequently, with physical activity in two (7.4%), infection in two
(7.4%), travel in three (11.1%), or no known factor in three (11.1%)
(p= 0.0005) (Supplementary Figure 1).
High altitude was the risk factor most commonly associated with
clinical complications, with a likelihood ratio (LR) of 29.3. The three
patients who presented clinical manifestations associated with travel
were active military patients with symptoms associated with long trips
by land, in which there were no significant changes at the altitude of
the cities.
There was no association of any comorbidity (smoking, hypertension,
diabetes mellitus, dyslipidemia, cancer or autoimmune disease.), or any
known family history of hemoglobinopathy S with the development of
symptoms (p = 0.054 and p = 0.139, respectively).
Symptomatic patients presented some characteristic symptoms and signs,
the most important of which were abdominal pain and fever. The pain was
increased by palpation and splenomegaly (p = 0.0001) (Table 2).
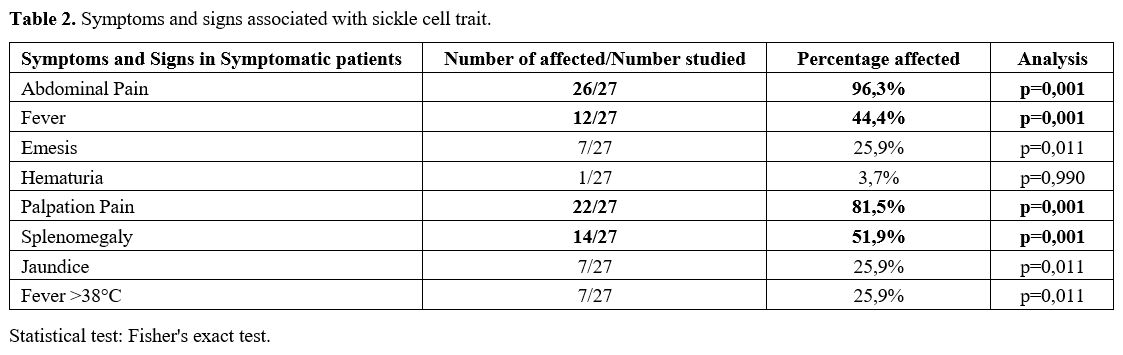 |
- Table 2. Symptoms and signs associated with sickle cell trait.
|
In
symptomatic patients, splenic complications were the most frequent
systemic manifestations in 23/27 patients (85.2%) vs. not having
splenic complications in four (14.8%)] (p=0.0005); splenic infarction
occurred in 10/23 of these patients (43.5%) and was the most common
manifestation; however, other splenic complications were also found,
including splenomegaly 5/23 (21.7%), subcapsular hematoma 4/23 (17.4%),
splenic abscess 2/23 (8.7%), and splenic rupture 2/23 (8.7%) (Figure 2).
The other systemic complications were present in 4/27 patients (14.8%);
these were distributed among three patients with hepatic manifestation
(11.1%) (i.e., cholelithiasis, jaundice, or liver abscess) and in one
patient with a urological manifestation (3.7%) (i.e., priapism).
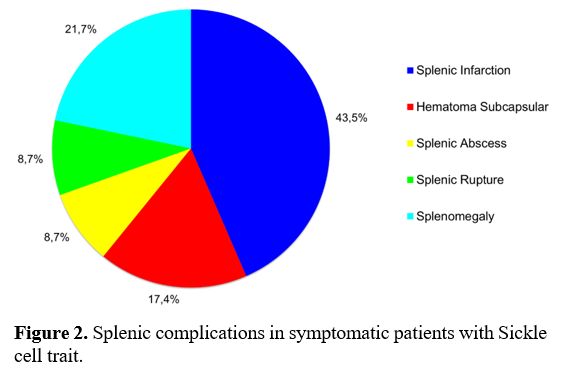 |
- Figure 2. Splenic complications in symptomatic patients with Sickle cell trait.
|
Among
the relevant paraclinical data taken in the first medical contact (in
the emergency room in symptomatic patients, and the medical
consultation in asymptomatic), statistically significant differences
were found in the leukocyte count and in the levels of bilirubin, LDH,
and C-reactive protein (CRP) in patients who were symptomatic vs.
asymptomatic (Table 3, Figure 3).
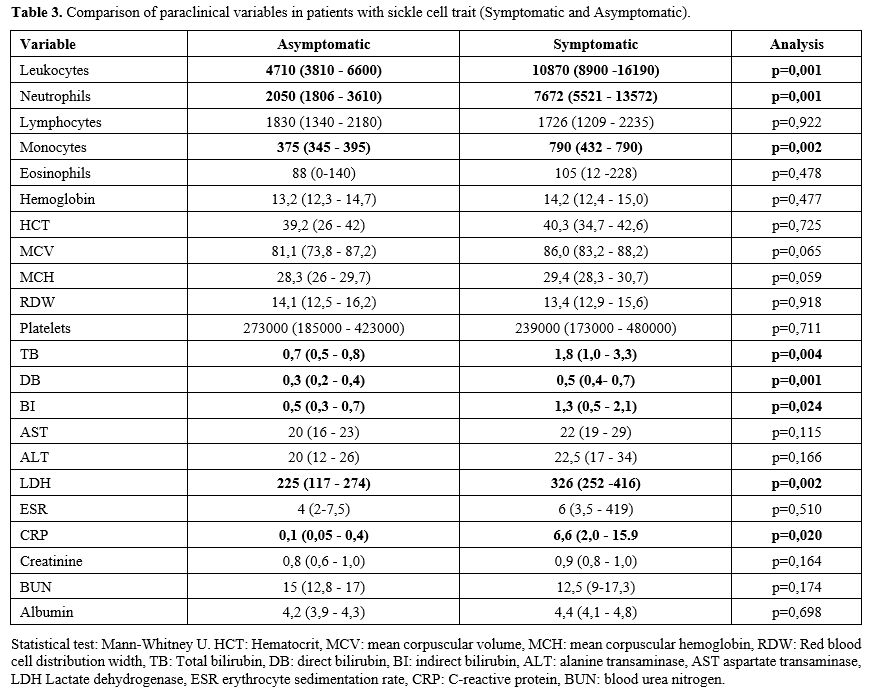 |
Table 3. Comparison of paraclinical variables in patients with sickle cell trait (Symptomatic and Asymptomatic). |
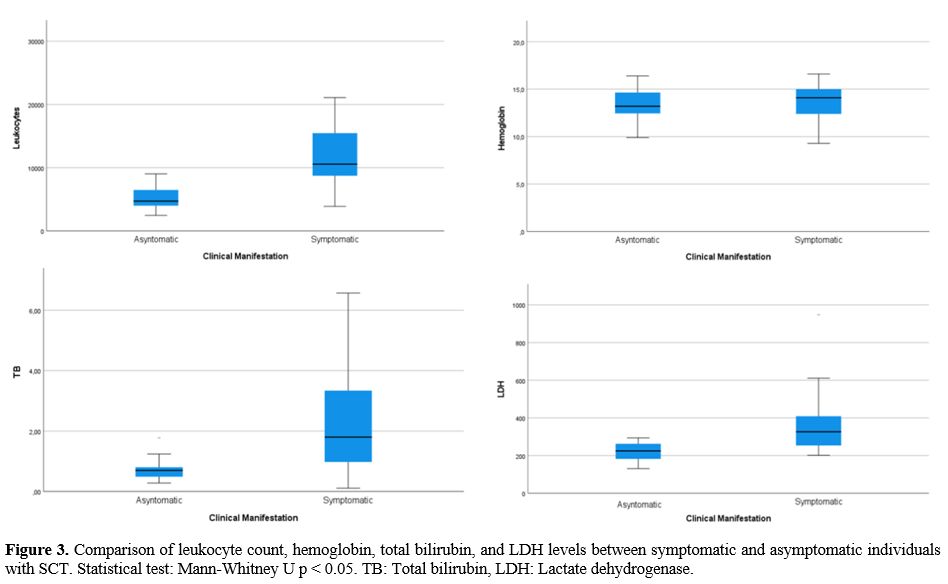 |
Figure
3 Comparison of leukocyte count, hemoglobin, total bilirubin, and LDH
levels between symptomatic and asymptomatic individuals with SCT.
Statistical test: Mann-Whitney U p < 0.05. TB: Total bilirubin, LDH:
Lactate dehydrogenase.
|
We
found differences among the treatments performed in symptomatic
patients: medical management was performed in 18 patients (66.7%),
interventional radiology in three patients (11.1%), and surgery in six
(22.2%) (p= 0.002). Among the surgical procedures, five (18.5%)
required splenectomy for different causes, and one patient required
cholecystectomy (3.7%) for cholelithiasis. However, there was no
statistically significant association between the need for splenectomy
and the percentage of HbS; no patients died in our study.
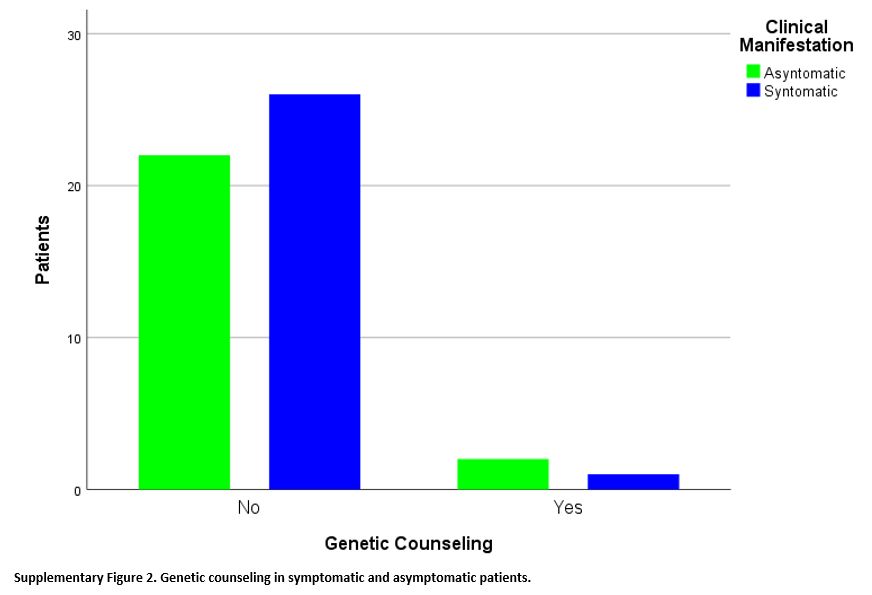
Finally, of the 51 patients with SCT, only three (5.9%) received
genetic counseling, vs. 48 patients (94.1%) did not. These three
patients were distributed between the two groups of patients in similar
proportions: 1/27 in symptomatic patients and 2/24 in asymptomatic
patients, without a statistically significant difference between the
two groups (p=0.595) with Fisher's exact test (Supplementary Figure 2).
Discussion
The HbS gene is distributed worldwide, and its prevalence varies according to geographical region and country.[2]
In our study, we found the presence of HbS in 13.91% of the studied
population, with the presence of SCT in 7.88% of patients statistically
more frequent in males than in females. Classically, it has been
described that hemoglobinopathy S is associated with the black race or
people of African descent; however, in our study, we found that 45.1%
of the patients were Colombian Mestizos and 54.9% of patients with SCT
were Afro- Colombians (African descent). Due to the wide distribution
and ethnic mixtures of Colombia, SCT should be suspected in patients
with compatible clinical manifestations, independent of race or
ethnicity. The ancestry of the Colombian population has proceeded
essentially from three racial groups: European, African, and Native
American origin,[28] which is common for most South American countries.[29]
Although SCT is largely considered a benign carrier state, there are
clear reports of clinical complications. Most of these associations
have been described in epidemiological studies,[9,30] which found a clear association with chronic diseases, such as chronic kidney disease.[1]
However, acute complications, such as splenic infarction, are
considered rare, and the literature is limited to a few descriptions.[8,9] To the best of our knowledge, this is one of a few studies with the most SCT patients with acute symptoms.
In symptomatic patients, the most frequent systemic complication was
splenic in 85.2% of cases. In addition, it has been described that the
spleen is an organ highly prone to injury in SCD due to the unique
characteristics of its microvasculature, along with the fact that
sickle red globules decrease deformability and increase adhesion,[31,32]
both of which mechanisms could also play an important role in splenic
injury in SCT; however, there is no complete clarity on the mechanism
in these patients.
High altitude was the main risk factor for presenting clinical
manifestations in 63% of cases, along with a lower percentage of
physical activity and infections. The places where clinical
manifestations occurred most frequently in patients with symptomatic
SCT were Bogotá and Pasto-Nariño, Colombia, where there are cities
located at an altitude of 2,630 meters above sea level and 2,527 meters
above sea level, respectively.
Compared with the largest reported study,[9] a
systematic review of case studies between 1970 and 2020, consisting of
54 articles with 85 cases of splenic infarction in individuals with
SCT, found that 29% of cases occurred at an altitude between 2000 and
3000 m (similar to that of our patients), 46% occurred between 3000 and
4000 m, and 3% occurred at more than 4000 m. A low percentage of
patients presented symptoms at less than 2000 meters. Physical activity
was also an important risk factor for these patients;[9]
physical activity or exercise was another trigger factor found in our
patients. Military personnel are potentially at increased risk of
triggering symptoms due to displacement to high- altitude regions and
strenuous training;[16] therefore, it is important to
evaluate for hemoglobinopathies in patients in these types of
professions, who present with splenic manifestations when exposed to
high altitude, despite not having a clear risk factor or family history
known of SCT or SCD. In our study, most of the patients who presented
symptoms were active military members of the Colombian military forces
(88,8%); this explains the detection of a high number of SCT patients
with acute symptoms.
The amount of circulating HbS can influence the prevalence of clinical
complications in SCT. The co-inheritance of α-thalassemia (which
reduces HbS levels) decreases the probability of clinical presentation.[30,33] In the same previously reported study,[9]
the percentage of HbS varied from 29.8 to 46.5%, and only four cases
(5%) had HbS less than 35%. In our study, no symptomatic patient had
HbS less than 35%. Some reports find that co-inheritance of
α-thalassemia and SCA, or SCD, has been associated with a lower MCV and
a milder phenotype in SCA patients, e.g., lower stroke rates. Despite
this, data are scarce on the co-inheritance of α-thalassemia and Sickle
cell trait SCT,[33] and the effects remain unclear even in the African setting.[34]
In our study, we perform hematological phenotypes, not hematological
genotypes, or molecular diagnostic testing for the detection of 3.7 kb
α-globin to evaluate the co- existing sickle cell trait/alpha
thalassemia double heterozygosity.
We found a systematic review that described the spectrum of splenic
complications in patients with SCD (defined by the genotypes HbSS,
HbSC, HbSβ+ thalassemia, or HbSβ0 thalassemia) in Africa;[34]
they did not include patients with SCT. The spectrum of splenic
complications reported includes splenomegaly in 12% to 73.2%,
hypersplenism in 0.1% to 5%, splenic sequestration crisis in 2% to 3%,
or as splenic rupture, splenic abscess, or splenic infarction, which
were reported only in isolated studies.[34] Despite the important
difference between the two pathologies (SCD and SCT), in our study, we
also found a wide spectrum of splenic complications in SCT (splenic
infarction, splenomegaly, splenic abscess, among others), being the
most important systemic complications in these patients. In conclusion,
although high-altitude splenic infarction is not uncommon and it is the
main splenic complication that is reported in the literature, our
findings emphasize that it is not the only splenic complication that
SCT patients can have, but rather, there is a wide spectrum of splenic
complications that must be included as classical manifestations of SCT.
In paraclinical data, increased bilirubin levels, LDH, reticulocyte count, and sometimes anemia have been described.[18,22,23]
By contrast, we found no difference in hemoglobin levels or platelets
among the symptomatic or asymptomatic groups; All complications are
associated with different pathophysiological conditions in which the
leukocyte count can increase,[14] which explains why
a clear difference was found in WBC counts, with higher counts in
symptomatic patients. However, although hemoglobin levels were similar,
an interesting finding was statistically significant higher levels of
bilirubin and LDH, possibly reflecting a low grade of hemolysis among
"sickling" red blood cells, as described in these patients, which we
can associate with the clinical presentation.[13–15]
Therefore finding patients with splenic complications, increased LDH,
and increased bilirubin should lead us to suspect a diagnosis of SCT,
even in the absence of anemia.
Most cases of splenic complication can be successfully treated with
hydration, analgesia, rest, oxygen, and other complementary measures.
Splenectomy is indicated mainly in cases of splenic rupture with
intraperitoneal hemorrhage, splenic abscess, or symptomatic massive
splenomegaly and spleen sequestration crisis.[1,18]
In our study; most patients were treated medically with symptomatic and
supportive management, and 18.5% of patients required splenectomy for
associated complications of those previously mentioned; patients who
required splenectomy received prophylactic vaccination, which is
essential in preventing secondary complications in the future.[35]
One patient with SCT during hospitalization was taken for
cholecystectomy for symptomatic cholelithiasis; although it has not yet
been described that SCT increases the risk of cholelithiasis, chronic
hemolysis could have contributed to its presentation in this patient.
Finally, of the 51 patients, only three (5.9%) were sent for genetic
counseling; there was no difference between symptomatic and
asymptomatic patients who were sent for genetic counseling (p=0.595);
it is very important to appropriately refer to genetic counseling
because of the type of inheritance of this pathology. There is
misinformation among patients about the meaning of being a carrier and
its implications for health and reproduction.[24]
That is why strategies are necessary to increase the use of genetic
counseling and improve the information of doctors and specialists in
hematology and internal medicine, and pre-and postconception counseling
is of great importance with high public health impact.[36-38]
There are no screening programs for hemoglobinopathies in Colombia and
its military population. In our study, we found an ascertainment bias.
The percentage of patients with SCT who either had or did not have a
complication may not represent a true percentage, given that hemoglobin
electrophoresis was performed on symptomatic patients, patients with a
family history, patients with abnormalities in the blood count, or
patients with an incidental diagnosis. Further studies are required to
assess the prevalence of SCT in the population and the percentage of
symptomatic patients. However, the fact that we found similar
percentages of patients with or without symptoms allows us to compare
them and describe the percentage of the different splenic complications.
Conclusions
SCT
is a rare disease that is usually asymptomatic; the main risk factor
for presenting with any symptoms is high altitude. Military Personnel
is a population with a high risk of developing symptoms; in symptomatic
patients, splenic complications were the most important, and splenic
infarction was the most common; however, splenic infarction was not the
only complication. As in SCD, there is also a wide spectrum of splenic
complications in patients with SCT. Diagnostic search should be
emphasized in patients with elevated bilirubin and LDH. Most patients
received medical treatment, and only 18,5% required splenectomy. Our
results reflect the absence of redirection of these patients to genetic
counseling, which can impact public health.
Strengths
In
our study, we obtained data from different geographical regions of
Colombia, highlighting that the study population is enriched with
active military members, a population already at risk of developing
symptoms, which explains the detection of many SCT patients with acute
symptoms.
Acknowledgments
To
the Immunology Laboratory of the Hospital Militar Central and the
Instituto de Referencia Andino and to the Asociación Colombiana de
Inmunología ACOI.
References
- Naik RP, Smith-Whitley K, Hassell KL, Umeh NI, de
Montalembert M, Sahota P, et al. Clinical outcomes associated with
sickle cell trait: A systematic review. Annals of Internal Medicine
2018;169:619-27. https://doi.org/10.7326/M18-1161 PMid:30383109 PMCid:PMC6487193
- Piel
FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, Temperley WH,
Williams TN, Weatherall DJ, Hay SI. Global epidemiology of sickle
haemoglobin in neonates: a contemporary geostatistical model-based map
and population estimates. Lancet. 2013 Jan 12;381(9861):142-51. doi:
10.1016/S0140-6736(12)61229-X. https://doi.org/10.1016/S0140-6736(12)61229-X PMid:23103089
- Ojodu
J, Hulihan MM, Pope SN, Grant AM, Centers for Disease Control and
Prevention (CDC). Incidence of Sickle Cell Trait - United States, 2010.
Morbidity and Mortality Weekly Report 2014;63:1155.
- Piel
FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of
sickle cell anaemia in children under five, 2010-2050: modelling based
on demographics, excess mortality, and interventions. PLoS Medicine
2013;10. https://doi.org/10.1371/journal.pmed.1001484 PMid:23874164 PMCid:PMC3712914
- Asfaw
SH, Falk GA, Morris-Stiff G, Tuthill RJ, Moorman ML, Samotowka MA. A
Unique Cause of Intestinal and Splenic Infarction in a Sickle Cell
Trait Patient. Case Reports in Surgery 2013;2013:1-3. https://doi.org/10.1155/2013/580453 PMid:23738181 PMCid:PMC3662113
- Gonzalez
L, Shapiro AF, Tafur A, Plaza-Meneses C, Sabando B. Splenic Infarct
Secondary to High Altitude Exposure in Sickle Cell Trait Patients: A
Case Series. Cureus 2020;12:e9815. https://doi.org/10.7759/cureus.9815
- Cook
AL. Splenic infarction in a high-altitude traveler with undiagnosed
sickle cell trait. Wilderness and Environmental Medicine
2008;19:318-20. https://doi.org/10.1580/08-WEME-LE-189.1 PMid:19099334
- Goodman
J, Hassell K, Irwin D, Witkowski EH, Nuss R. The Splenic Syndrome in
Individuals with Sickle Cell Trait. High Altitude Medicine &
Biology 2014;15:468-71. https://doi.org/10.1089/ham.2014.1034. PMid:25361178 PMCid:PMC4273194
- Jefferson
JM, Sims WM, Umeh N, Byeon YJJ, Abdallah KE, Bonham VL, et al. Splenic
infarction in sickle cell trait: A comprehensive systematic review of
case studies. EJHaem 2021;2:585-600. https://doi.org/10.1002/jha2.248. PMid:34870278 PMCid:PMC8635393
- Lane PA. Splenic Syndrome at Mountain Altitudes in Sickle Cell Trait. JAMA 2011;253:2251. https://doi.org/10.1001/jama.1985.03350390093033 PMid:3974118
- Murano
T, Fox AD, Anjaria D. Acute splenic syndrome in an African-American
male with sickle cell trait on a commercial airplane flight. Journal of
Emergency Medicine 2013;45. https://doi.org/10.1016/j.jemermed.2013.05.009. PMid:23810115
- Longo
T, Shaines M. Case report: Exertional rhabdomyolysis in a spin class
participant with sickle cell trait. F1000Research 2019;7. https://doi.org/10.12688/f1000research.16326.2 PMid:31372209 PMCid:PMC6659762
- Presley
TD, Perlegas AS, Bain LE, Ballas SK, Nichols JS, Sabio H, et al.
Effects of a single sickling event on the mechanical fragility of
sickle cell trait erythrocytes. Hemoglobin 2010;34:24-36. https://doi.org/10.3109/03630260903546999. PMid:20113285 PMCid:PMC3226741
- Blinder MA, Russel S. Exertional sickling: questions and controversy. Hematology Reports 2014;6:5502. https://doi.org/10.4081/hr.2014.5502 PMid:25568759 PMCid:PMC4274478
- Seegars MB, Brett AS. Splenic infarction associated with sickle cell trait at low altitude. Hematology 2015;20:607-9. https://doi.org/10.1179/1607845415Y.0000000024 PMid:26133225
- Yanamandra
U, Das R, Malhotra P, Varma S. A Case of Autosplenectomy in Sickle Cell
Trait Following an Exposure to High Altitude. Wilderness and
Environmental Medicine 2018;29:85-9. https://doi.org/10.1016/j.wem.2017.08.021. PMid:29331296
- Bucknor
MD, Goo JS, Coppolino ML. The risk of potential thromboembolic, renal
and cardiac complications of sickle cell trait. Hemoglobin
2014;38:28-32. https://doi.org/10.3109/03630269.2013.832689. PMid:24099594
- Tsaras
G, Owusu-Ansah A, Boateng FO, Amoateng-Adjepong Y. Complications
Associated with Sickle Cell Trait: A Brief Narrative Review. American
Journal of Medicine 2009;122:507-12. https://doi.org/10.1016/j.amjmed.2008.12.020. PMid:19393983
- Nelson
DA, Deuster PA, Carter R, Hill OT, Wolcott VL, Kurina LM. Sickle Cell
Trait, Rhabdomyolysis, and Mortality among U.S. Army Soldiers. New
England Journal of Medicine 2016;375:435-42. https://doi.org/10.1056/nejmoa1516257. PMid:27518662 PMCid:PMC5026312
- Webber
BJ, Nye NS, Harmon KG, O'Connor FG. Exertional Rhabdomyolysis, Sickle
Cell Trait, and "Military Misdirection." Current Sports Medicine
Reports 2021;20:562-3. https://doi.org/10.1249/JSR.0000000000000897 PMid:34622822
- Buchanan
BK, Siebert DM, Zigman Suchsland ML, Drezner JA, Asif IM, O'Connor FG,
et al. Sudden Death Associated With Sickle Cell Trait Before and After
Mandatory Screening. Sports Health 2020;12:241-5. https://doi.org/10.1177/1941738120915690 PMid:32271134 PMCid:PMC7222668
- Fernando
C, Mendis S, Upasena A, Costa Y, Williams H, Moratuwagama D. Splenic
Syndrome in a Young Man at High Altitude with Undetected Sickle Cell
Trait. Journal of Patient Experience 2018;5:153-5. https://doi.org/10.1177/2374373517747905 PMid:29978033 PMCid:PMC6022946
- Yeral M, Boğa C. Is sickle cell trait really innocent? Turkish Journal of Hematology 2021;38:159-60. https://doi.org/10.4274/tjh.galenos.2020.2020.0344 PMid:33053967 PMCid:PMC8171209
- Busse B, Tepedino M-F, Rupprecht W, Klein H-G. Stepwise diagnostics of hemoglobinopathies. LaboratoriumsMedizin 2016;39. https://doi.org/10.1515/labmed-2016-0009
- Amer
Wahed, Andres Quesada, Amitava Dasgupta, Chapter 4 - Hemoglobinopathies
and thalassemias, Editor(s): Amer Wahed, Andres Quesada, Amitava
Dasgupta, Hematology and Coagulation (Second Edition), Academic Press,
2020, Pages 51-75, ISBN 9780128149645, https://doi.org/10.1016/B978-0-12-814964-5.00004-8
- Giordano
PC. Strategies for basic laboratory diagnostics of the
hemoglobinopathies in multi-ethnic societies: interpretation of results
and pitfalls. International Journal of Laboratory Hematology
2013;35:465-79. https://doi.org/10.1111/IJLH.12037. PMid:23217050
- Serjeant GR. The natural history of sickle cell disease. Cold Spring Harbor Perspectives in Medicine 2013;3. https://doi.org/10.1101/cshperspect.a011783 PMid:23813607 PMCid:PMC3784812
- Ossa
H, Aquino J, Pereira R, Ibarra A, Ossa RH, Pérez LA, et al. Outlining
the Ancestry Landscape of Colombian Admixed Populations. PLoS ONE
2016;11:e0164414. https://doi.org/10.1371/journal.pone.0164414 PMid:27736937 PMCid:PMC5063461
- Homburger
JR, Moreno-Estrada A, Gignoux CR, Nelson D, Sanchez E, Ortiz-Tello P,
et al. Genomic Insights into the Ancestry and Demographic History of
South America. PLOS Genetics 2015;11:e1005602. https://doi.org/10.1371/journal.pgen.1005602 PMid:26636962 PMCid:PMC4670080
- Xu JZ, Thein SL. The carrier state for sickle cell disease is not completely harmless. Haematologica 2019;104:1106-11. https://doi.org/10.3324/haematol.2018.206060 PMid:31097635 PMCid:PMC6545856
- el
Hoss S, Cochet S, Marin M, Lapouméroulie C, Dussiot M, Bouazza N, et
al. Insights into determinants of spleen injury in sickle cell anemia.
Blood Advances 2019;3:2328-36. https://doi.org/10.1182/bloodadvances.2019000106 PMid:31391165 PMCid:PMC6693014
- Brousse
V, Buffet P, Rees D. The spleen and sickle cell disease: The sickled
spleen. British Journal of Haematology 2014;166:165-76. https://doi.org/10.1111/bjh.12950 PMid:24862308
- Rumaney
MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Kengne AP, Ngogang J, et
al. The Co-Inheritance of Alpha-Thalassemia and Sickle Cell Anemia Is
Associated with Better Hematological Indices and Lower Consultations
Rate in Cameroonian Patients and Could Improve Their Survival. PLoS ONE
2014;9. https://doi.org/10.1371/journal.pone.0100516 PMid:24978191 PMCid:PMC4076272
- Ladu
AI, Aiyenigba AO, Adekile A, Bates I. The spectrum of splenic
complications in patients with sickle cell disease in Africa: a
systematic review. British Journal of Haematology 2021;193:26-42. https://doi.org/10.1111/bjh.17179 PMid:33161568
- Luu
S, Spelman D, Woolley IJ. Post-splenectomy sepsis: preventative
strategies, challenges, and solutions. Infection and Drug Resistance
2019;12:2839. https://doi.org/10.2147/IDR.S179902 PMid:31571940 PMCid:PMC6748314
- Acharya
K, Lang CW, Ross LF. A pilot study to explore knowledge, attitudes, and
beliefs about sickle cell trait and disease. Journal of the National
Medical Association 2009;101:1163-72. https://doi.org/10.1016/S0027-9684(15)31113-5 PMid:19998646
- Pecker
LH, Naik RP. The current state of sickle cell trait: Implications for
reproductive and genetic counseling. Blood 2018;132:2331-8. https://doi.org/10.1182/blood-2018-06-848705 PMid:30487130 PMCid:PMC6265653
- Ali EH, Alkindi S, Mohamed AO, Awadalla KE,
Abdlgadir O, Adam G, Magdi M, Ibrahim AK, Ghebremeskel K. Adverse
Pregnancy Outcomes in Sickle Cell Trait: a Prospective Cohort Study
Evaluating Clinical and Haematological Parameters in
Postpartum Mothers and Newborns. Mediterr J Hematol Infect Dis. 2023
Jan 1;15(1):e2023002. https://doi.org/10.4084/MJHID.2023.002 PMid:36660349 PMCid:PMC9833303
SUPPLEMENTARY MATERIAL
 |
Supplementary Figure 1. Risks factors associated with the presentation of symptoms in patients with Sickle cell trait |
 |
Supplementary Figure 2. Genetic counseling in symptomatic and asymptomatic patients.
|
[TOP]