Sohini Chattopadhyay1, Sharon
Lionel1, Sushil Selvarajan1, Anup J Devasia1, Anu Korula1, Uday Kulkarni1,
Fouzia NA1, Eunice Sindhuvi1, Kavitha M Lakshmi1, Alok Srivastava1, Aby
Abraham1, Vikram Mathews1 and Biju George1.
1 Department of Hematology, Christian Medical College, Vellore, India
Correspondence to: Dr.
Biju George (ORCID ID:
0000-0002-9847-9501), Professor. Department of
Hematology, Christian Medical College, Vellore, India. Ph:
+91-416-2282352 Fax: +91-416-2226449. Email:
biju@cmcvellore.ac.in
Published: July 1, 2023
Received: December 22, 2022
Accepted: May 31, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023039 DOI
10.4084/MJHID.2023.039
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Hematopoietic stem cell transplantation (HSCT) is the only curative
option for patients with Fanconi Anemia (FA) with hematological
abnormalities.
Materials and Methods: This is a retrospective analysis of patients with FA who underwent a matched-related donor HSCT.
Results:
Sixty patients underwent 65 transplants between 1999-2021 using a
fludarabine-based low-intensity conditioning regimen. The median age at
transplant was 11 years (range: 3-37). Aplastic anemia (AA) was the
underlying diagnosis in 55 (84.6%), while 8 (12.4%) had myelodysplastic
syndrome (MDS) and 2 (3%) had acute myeloid leukemia (AML). The
conditioning regimen used was Fludarabine with low-dose
Cyclophosphamide for aplastic anemia and Fludarabine with low-dose
Busulfan for MDS/AML. Graft versus host disease (GVHD) prophylaxis
consisted of Cyclosporine and methotrexate. Peripheral blood was the
predominant stem cell graft source (86.2%). Engraftment occurred in all
but one patient. The median time to neutrophil and platelet engraftment
was 13 days (range: 9-29) & 13 days (range: 5-31), respectively.
Day 28 chimerism analysis showed complete chimerism in 75.4 % and mixed
chimerism in 18.5%. Secondary graft failure was encountered in 7.7%.
Grade II–IV acute GVHD occurred in 29.2%, while Grade III-IV acute GVHD
occurred in 9.2%. Chronic GVHD was seen in 58.5% and was limited in
most patients. The median follow-up is 55 months (range: 2-144) &
the 5-year estimated overall survival (OS) is 80.2 ± 5.1%. Secondary
malignancies were noted in 4 patients. The 5-year OS was significantly
higher in patients undergoing HSCT for AA (86.6 + 4.7%) as compared to
MDS/AML (45.7+16.6%) (p= 0.001).
Conclusion:
SCT using a fully matched donor provides good outcomes with
low-intensity conditioning regimens in patients with FA who have
aplastic marrow.
|
Introduction
Fanconi
Anemia (FA) is the most common inherited bone marrow failure syndrome
classically characterized by somatic malformations, progressive bone
marrow failure, and predilection to both hematological and solid organ
malignancies.[1,2] The incidence of the latter increases with exposure
to alkylating and DNA cross-linking agents.[3,4] Despite advances in
the therapeutic strategies to manage FA and its myriad manifestations,
allogeneic hematopoietic stem cell transplantation (HSCT) remains the
only therapy that corrects the hematological manifestation of FA.[5]
HSCT for FA was attempted as early as the 1980s, with the initial
transplants employing a preparatory regimen combining high doses of
Cyclophosphamide (200mg/kg) and Total Body Irradiation (TBI) similar to
the regimens used for acquired aplastic anemia.[4,6] Outcomes were
poor, with high morbidity and mortality - mainly related to
regimen-related toxicity (RRT) and graft versus host disease, coupled
with an increased predisposition to late post-transplant
malignancies.[6] This outcome was attributed to the hypersensitivity of
FA cells to high doses of radiation and Cyclophosphamide.[7,8] In the
next decade, a reduction in the doses of Cyclophosphamide and Total
Body Irradiation (TBI) was realized to reduce treatment-related
mortality, but that resulted in poor engraftment and graft
function.[9-11] Fludarabine, an antimetabolite with potent
immunosuppressive action, was incorporated into the conditioning
regimens in the mid-’90s. Fludarabine, not associated with DNA
cross-linking, substantially reduced the incidence of GVHD/RRT in
patients with FA.[12] We have previously described a small series of
patients who underwent transplants using a combination of Fludarabine
and low-dose Cyclophosphamide with promising outcomes.[12] We describe
a larger experience using a fludarabine-based preparatory regimen for
HSCT in patients with FA.
Materials and Methods
This
study is a retrospective analysis of patients with FA who underwent a
matched-related donor HSCT in the Department of Haematology, Christian
Medical College Vellore, between 1990 and 2021. It was approved by the
local institutional Ethics committee.
Patients and donors. The
diagnosis of FA was confirmed using chromosomal breakage analysis (CBA)
studies with mitomycin C. In patients with equivocal results on CBA,
the diagnosis was confirmed either by analysis of the ubiquitination
status of FANC- D2 on peripheral blood or skin fibroblasts or by
mutation analysis. Western Blot has been available for use since 2015
at our centre. This study included only patients who received stem
cells from a matched related (sibling/ non-sibling) donor; alternative
stem-cell donor transplants were excluded. All donors were screened and
confirmed to be negative by chromosomal breakage analysis studies.
Conditioning Regimen and GVHD prophylaxis.
The choice of conditioning regimen was based on the type of
hematopoietic failure - aplastic anemia (AA) or myelodysplastic
syndrome (MDS)/ acute myeloid leukemia (AML) at HSCT.
Patients with AA received a combination of Fludarabine (30 mg/m2/day
x 6 days) and a low dose of Cyclophosphamide (10 mg/kg/day x 2 days).
Four patients initially received low-dose ATG (ATGAM 10 mg/kg/day x 4
days), but since 2016, ATG was omitted from the preparative regimen.
Patients with MDS/AML received Fludarabine (30 mg/m2/day
x 6 days) and intravenous low-dose Busulfan (2.4 mg/kg/day IV x 2
days). The dose of Busulfan was adjusted after the first dose to target
a total AUC of 5000 – 6000 ng/ml.
Graft versus Host Disease (GVHD) prophylaxis consisted of Cyclosporine and a short course of methotrexate for all patients.
Transplant outcomes. The
primary endpoint of the analysis was overall survival at 5 years.
Neutrophil and platelet engraftment was defined as per standard CIBMTR
criteria. Whole blood chimerism using short tandem repeats was assessed
on Day 28, day 60, day 100, and at one year. Primary graft failure was
defined as failure to achieve an (ANC >500/mm3) by day +28, while secondary graft failure was defined as evidence of initial engraftment (ANC >500/mm3), followed by subsequent fall in counts (ANC<500/mm3)
for 7 continuous days. Secondary endpoints included engraftment,
regimen-related toxicity, and cumulative incidence of acute and chronic
GVHD, which were defined and graded by standard Glucksberg
criteria.[11] Patients were screened for the development of secondary
malignancies at each follow-up visit.
Statistical Analysis.
Statistical analyses were performed using IBM SPSS software - version
24. Continuous variables were summarized as medians, range, and
categorical variables as percentages. Overall survival was estimated
using the Kaplan-Meier estimators and Cox regression analysis, and
comparisons between groups were conducted with a chi-square test or a
Fisher's exact test (2-sided) wherever appropriate. P < 0 .05 was
considered significant.
Results
Baseline characteristics.
Between 1990 and December 2021, 60 patients with FA underwent 65
transplants using a fully matched related donor. Baseline
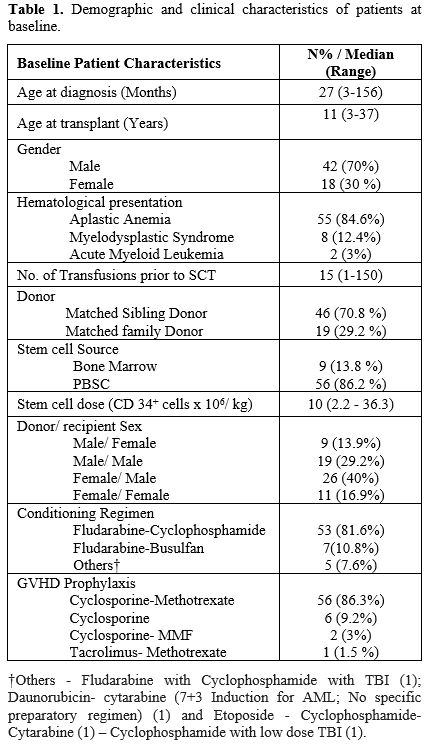
characteristics are described in Table 1.
The median age was 27 months (3-156) at diagnosis and 11 years (3-37)
at HSCT. Indications for HSCT included: AA in 55 transplants
(84.6%), MDS in 8 (12.4%), and AML in 2 (3%). Prior to HSCT, most
(89.2%) had failed treatment with androgens
(oxymetholone/stanozolol/danazol). The median number of transfusions
received prior to HSCT was 15 (range 1-150).
 |
- Table 1. Demographic and clinical characteristics of patients at baseline
|
Transplant. Sixty
transplants were done upfront, 3 patients underwent a second transplant
for rejection, and 1 required two transplants because of disease
relapse. The stem cell source included G-CSF-stimulated peripheral
blood stem cells (PBSC) in 56 and unmanipulated bone marrow (BM) in 9.
The median cell dose infused was 10 × 106 CD 34 cells/kg (range: 2.2–36.3). For all second transplants, PBSC was the graft source used.
Engraftment and Chimerism.
Engraftment occurred in all (98.4%) except one who expired less than
two weeks after SCT due to gram-negative septicemia. The median time
for neutrophil engraftment was 13 days (9–29), and also for platelet
engraftment with a range of 5-31. Day 28 chimerism was complete donor
chimerism in 50 (76.9%) transplants, of which 43 (66.1%) maintained
complete donor chimerism on follow-up. Mixed donor-recipient chimerism
was noted in 12 (18.5%) on day 28. All patients with mixed chimerism
had aplastic anemia at the time of HSCT. Seven patients with complete
donor chimerism on day 28 were noted to have mixed chimerism on
follow-up. As per institutional protocol, a reduction of
immunosuppression was attempted in 19 transplants with mixed chimerism,
and 13 (68.4%) attained complete donor chimerism during subsequent
follow-up. One patient required donor lymphocyte infusion to attain
complete donor chimerism. Secondary graft failure was noted in 5 (7.7%)
patients, of which 3 had initially achieved complete donor chimerism on
day 28. A second stem cell transplant with reduced-intensity
conditioning was offered to the patients with SGF, of which 3 opted for
the same. Only one patient attained engraftment and complete chimerism
on Day 28 following the second transplant.
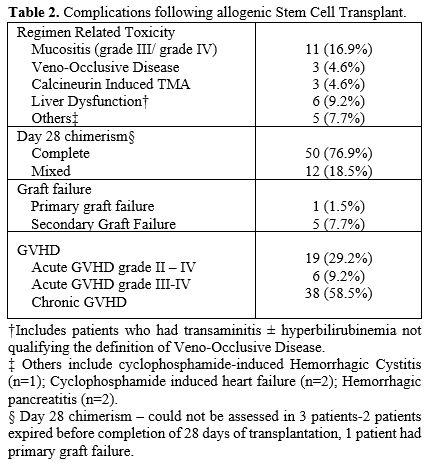
Regimen Related toxicity (RRT) and GVHD. Grade 3-4 mucositis was seen in 11 (16.9%) transplants (Table 2).
Liver dysfunction [transient elevation of liver enzymes and
hyperbilirubinemia] was noted in 6 (9.2%), while veno-occlusive disease
was diagnosed in 3 (4.5%), and hemorrhagic pancreatitis was noted in 2
(3.4%). There were no deaths related to RRT. There was no significant
difference in the incidence of mucositis (p = 0.35) or veno-occlusive
disease (p = 1.0) between patients having AA or AML/MDS at the time of
HSCT.
 |
- Table 2. Complications following allogenic Stem Cell Transplant
|
The
Day 100 cumulative incidence of acute graft versus host disease (GvHD)
was 29.2%, while grade III-IV GVHD was 9.2%. Chronic GVHD was noted in
38 patients (58.5%) on follow-up; this was limited in 23 (35.9%) and
extensive in 15 transplants (22.6%).
Infections.
Febrile neutropenia occurred in all transplants though bacteremia was
documented in only 13 (20%), and it was mainly gram-negative infections
more than gram-positive (69.2% vs. 30.7%). Viral reactivation
(Cytomegalovirus) necessitating therapy was seen in 19 (29.2%)
patients, of which ten patients had underlying grade III/IV GVHD and
were on systemic corticosteroids. Six patients (9.2%) developed
possible invasive fungal disease (IFD) based on imaging.
Secondary Malignancies.
Of the 60 patients who underwent HSCT, 4 (6.7%) patients developed
second malignancies – mainly squamous cell carcinoma of the head and
neck on follow-up at a median of 8 years post HSCT (range: 6-13 years).
They were treated with surgery ± radiotherapy. Two patients attained
remission and are on follow-up, while the other two succumbed to the
malignancy. One patient with aplastic anemia transformed into acute
myeloid leukemia post-SCT. Amongst the cohort of patients undergoing
SCT for MDS/AML, four patients had a relapse/ progression to AML on
follow-up.
Survival Outcomes. Forty-six
patients are alive at a median follow-up of 55 months (2-144 months).
The 5-year overall survival in our cohort is 80.2% ± 5.1% (Figure 1).
Six patients died due to infective complications, five expired due to
secondary malignancies/ relapse of acute myeloid leukemia, two due to
graft failure, and one due to extensive chronic GVHD.
 |
- Figure
1 shows the overall survival at 5 years.
|
The presence of MDS/AML at the time of HSCT was the only factor noted to have independently influenced survival (Table 3).
The 5-year OS was significantly lower in patients who underwent
transplants for MDS/AML (45.7 + 16.6%) compared to aplastic anemia
(86.6 + 4.7%) (p= 0.001) (Figure 2).
The choice of conditioning regimen did not impact survival, though the
univariate analysis demonstrated better survival in patients who
received Fludarabine - Cyclophosphamide. This improvement was
attributed to the choice of conditioning regimen being closely linked
to the underlying hematological disorder. Age at the time of HSCT also
did not influence the 5-year OS in our study (p=0.35) (Figure 3).
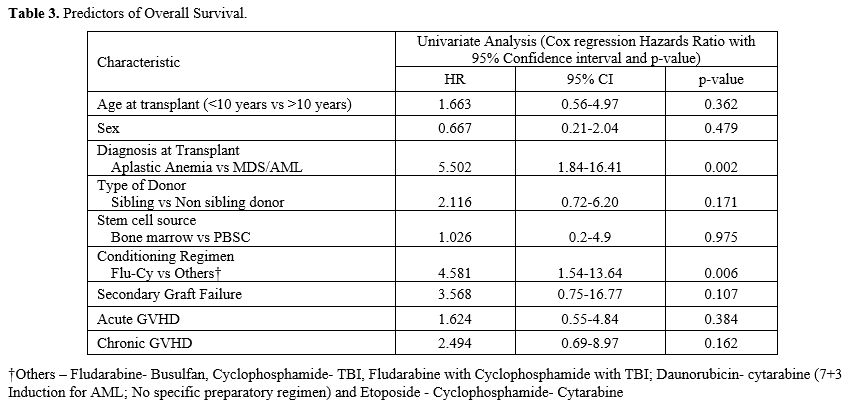 |
Table 3. Predictors of overall survival |
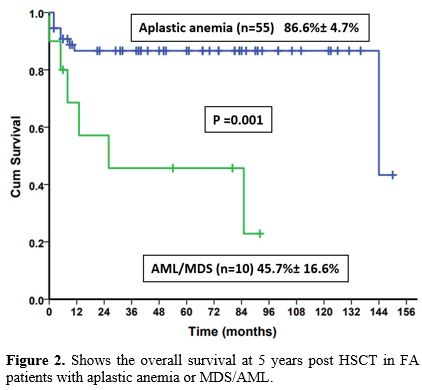 |
Figure 2 shows the overall survival at 5 years post HSCT in FA patients with aplastic anemia or MDS/AML.
|
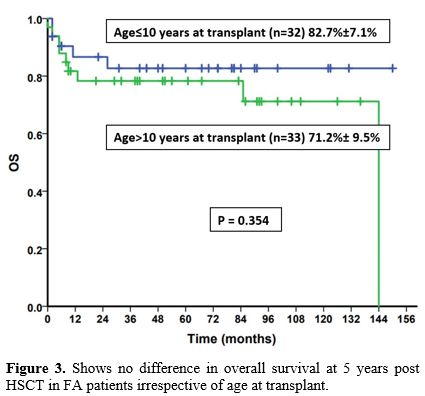 |
Figure 3 shows no difference in overall survival at 5 years post HSCT in FA patients irrespective of age at transplant.
|
Discussion
The
advent of Fludarabine-based reduced-intensity conditioning regimens has
led to a massive reduction in treatment-related mortality and long-term
complications of HSCT, i.e., the incidence of secondary malignancies
and chronic GVHD, thus leading to better long-term survival. There is
limited data available on the outcomes of allogeneic SCT from
resource-limited settings, and we report our experience in allogeneic
stem cell transplants for patients with FA.
We observed a
5-year-overall survival of 80% in our study population, which was
comparable to the multicenter study conducted by Latour et al. (76%),
Ayas et al. (85%), Smetsers et al. (76%) and Farzin et al.
(89%).[10,13-15]
All patients received fludarabine-based preparatory
regimens, with no mortality related to regimen-related toxicity or
primary graft failure. Data from the Chinese Bone Marrow Registry
(CBMTR) suggested that OS and EFS were both 100% with the use of
Fludarabine and Cyclophosphamide in patients with FA.[16]
Although
the overall survival of SCT for aplastic anemia in FA shows promising
outcomes, the results were not similar for FA patients with MDS/AML.
The cohort of patients with MDS/AML had significantly lower overall
survival (46%), corroborating previous data suggesting that clonal
evolution at HSCT was a major predictor of the outcome.[13,15,17,18]
This datum is similar to that from the CIBMTR, where in a study of 113
patients with FA, the outcome of patients with MDS/acute leukemia was
43%.[19]
Though the rates of acute GVHD (grade III-IV) were low
(9.2%), we experienced a higher incidence of chronic GVHD (58.5%) when
compared to the EBMT group (acute GVHD 19%; chronic GVHD 20%).[13] This
may be related to the higher use of peripheral blood stem cells (86.2%)
in our cohort in comparison to other studies that used predominantly
bone marrow harvested stem cells (Latour et al. (66%) and Farzin et al.
(91%)).[13,14] Although peripherally derived stem cells are known to be
associated with a greater risk of chronic GVHD, we have used them in
our patients, per our experience with PBSC grafts in acquired aplastic
anemia. Engraftment and immune reconstitution are hastened with the use
of PBSC grafts, which reduce the incidence of severe sepsis and,
thereby, mortality in our setting. However, we are considering using
Bone Marrow as the graft source, given the higher incidence of chronic
GVHD.
Secondary graft failure was documented in 9.1% of the
transplants, which again was similar to that published by the EBMT
group (6%).[13] The primary cause of mortality in our study was
infectious complications.
Age at transplant has been identified as
one of the main variables influencing an SCT's overall outcome. When
transplanted in the first decade of life, FA patients have been shown
to have consistently better outcomes in various studies.[6,13] In our
cohort, we did not find such a difference with age, which may reflect
better tolerance of patients to reduced intensity fludarabine-based
conditioning regimens.
The distinct genomic instability of FA
leads to an increased propensity towards secondary
malignancies.[8,20,21] The hazard is 2%/y at age 24, 4%/y at age 30,
and close to 8%/y at age 40.[7,20] In addition, SCT has been postulated
to increase further the risk of secondary malignancies in patients with
FA.[7,22,23] The German Fanconi Anemia Registry demonstrated that
patients undergoing SCT had a 3.8-fold higher risk of developing solid
organ malignancies than those who did not receive an SCT.[22] However,
the incidence of secondary malignancies in our cohort was 6.7%,
corresponding to the risk portended by the disease per se. This value
was per the findings of Rosenberg et al. but contradicted what was
observed in the EBMT group (15%).[7,13] Using non-irradiation-based
conditioning regimens and better modalities for limiting and treating
chronic GVHD has probably influenced this moderation in the incidence
of solid organ malignancies.
This study describes a large
cohort of patients with Fanconi anemia that have undergone HSCT in
India. The major drawbacks of this study are the retrospective nature
of data and the heterogeneity of the patients enrolled in it. Genetic
studies defining the underlying FA mutations were unavailable for all
the enrolled patients. Hence, identifying subsets of patients with a
propensity toward high-risk disease and the probability of higher
regimen-related toxicity was not feasible. However, the rarity of the
disease limits more extensive prospective trials for FA.
To
summarize, HSCT remains the principal treatment option to correct the
hematological manifestations of FA. Given the high incidence of GVHD,
it may be preferable to use bone marrow grafts, especially in children.
Though the overall survival rates are on the rise, long-term morbidity
in the form of chronic GVHD and secondary malignancies remains a
formidable setback to a successful HSCT, requiring further
modifications in the approach to SCT in FA.
Author Contributions
George
B and Chattopadhyay S conceptualized the study, provided data, analyzed
the data, and wrote the manuscript. Lionel S, Selvarajan S, Devasia AJ,
Korula A, Kulkarni U, Aboobacker FN, Sindhuvi E, Srivastava A, Abraham
A, and Mathews V contributed patient data to the study. Lakshmi KM
analyzed the data and provided statistical support. All authors were
responsible for the critical review and revision of the manuscript.
References
- Butturini A, Gale RP, Verlander PC, Adler-Brecher B, Gillio AP,
Auerbach AD. Hematologic abnormalities in Fanconi anemia: an
International Fanconi Anemia Registry study. Blood. 1994 Sep
1;84(5):1650-5. https://doi.org/10.1182/blood.V84.5.1650.1650
PMid:8068955
- Alter BP. Bone Marrow Failure
Syndromes. Clin Lab Med. 1999 Mar 1;19(1):113-34.
https://doi.org/10.1016/S0272-2712(18)30131-8 PMid:10403077
- Gluckman
E, Devergie A, Dutreix J. Radiosensitivity in Fanconi anaemia:
application to the conditioning regimen for bone marrow
transplantation. Br J Haematol. 1983;54(3):431-40.
https://doi.org/10.1111/j.1365-2141.1983.tb02117.x PMid:6344915
- Gluckman
E, Auerbach AD, Horowitz MM, Sobocinski KA, Ash RC, Bortin MM, et al.
Bone marrow transplantation for Fanconi anemia. Blood. 1995 Oct
1;86(7):2856-62.
https://doi.org/10.1182/blood.V86.7.2856.bloodjournal8672856
PMid:7670120
- Dufour C. How I manage patients with
Fanconi anaemia. Br J Haematol. 2017;178(1):32-47.
https://doi.org/10.1111/bjh.14615 PMid:28474441
- MacMillan
ML, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia
- when and how? Br J Haematol. 2010;149(1):14-21.
https://doi.org/10.1111/j.1365-2141.2010.08078.x PMid:20136826
- Rosenberg
PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi
anemia. Blood. 2003 Feb 1;101(3):822-6.
https://doi.org/10.1182/blood-2002-05-1498 PMid:12393424
- Auerbach
AD, Wolman SR. Susceptibility of Fanconi's anaemia fibroblasts to
chromosome damage by carcinogens. Nature. 1976 Jun;261(5560):494-6.
https://doi.org/10.1038/261494a0 PMid:934283
- Tan
PL, Wagner JE, Auerbach AD, Defor TE, Slungaard A, Macmillan ML.
Successful engraftment without radiation after fludarabine-based
regimen in Fanconi anemia patients undergoing genotypically identical
donor hematopoietic cell transplantation. Pediatr Blood Cancer. 2006
May 1;46(5):630-6. https://doi.org/10.1002/pbc.20538
PMid:16078221
- Ayas M, Al-Jefri A, Al-Seraihi A,
Elkum N, Al-Mahr M, El-Solh H. Matched-related allogeneic stem cell
transplantation in Saudi patients with Fanconi anemia: 10 year's
experience. Bone Marrow Transplant. 2008 Aug;42 Suppl 1:S45-8.
https://doi.org/10.1038/bmt.2008.114 PMid:18724300
- Glucksberg
H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical
manifestations of graft-versus-host disease in human recipients of
marrow from HL-A-matched sibling donors. Transplantation. 1974
Oct;18(4):295-304. https://doi.org/10.1097/00007890-197410000-00001
PMid:4153799
- George B, Mathews V, Shaji RV,
Srivastava V, Srivastava A, Chandy M. Fludarabine-based conditioning
for allogeneic stem cell transplantation for multiply transfused
patients with Fanconi's anemia. Bone Marrow Transplant. 2005
Feb;35(4):341-3. https://doi.org/10.1038/sj.bmt.1704785
PMid:15640819
- Peffault de Latour R, Porcher R,
Dalle JH, Aljurf M, Korthof ET, Svahn J, et al. Allogeneic
hematopoietic stem cell transplantation in Fanconi anemia: the European
Group for Blood and Marrow Transplantation experience. Blood. 2013 Dec
19;122(26):4279-86. https://doi.org/10.1182/blood-2013-01-479733
PMid:24144640
- Farzin A, Davies SM, Smith FO,
Filipovich A, Hansen M, Auerbach AD, et al. Matched sibling donor
haematopoietic stem cell transplantation in Fanconi anaemia: an update
of the Cincinnati Children's experience. Br J Haematol.
2007;136(4):633-40. https://doi.org/10.1111/j.1365-2141.2006.06460.x
PMid:17367413
- Smetsers SE, Smiers FJ, Bresters D,
Sonnevelt MC, Bierings MB. Four decades of stem cell transplantation
for Fanconi anaemia in the Netherlands. Br J Haematol. 2016
Sep;174(6):952-61. https://doi.org/10.1111/bjh.14165
PMid:27470218
- Xu L, Lu Y, Chen J, Sun S, Hu S,
Wang S, et al. Fludarabine- and low-dose cyclophosphamide-based
conditioning regimens provided favorable survival and engraftment for
unmanipulated hematopoietic cell transplantation from unrelated donors
and matched siblings in patients with Fanconi anemia: results from the
CBMTR. Bone Marrow Transplant. 2023 Jan;58(1):106-8.
https://doi.org/10.1038/s41409-022-01838-9 PMid:36257981
- Giardino
S, de Latour RP, Aljurf M, Eikema DJ, Bosman P, Bertrand Y, et al.
Outcome of patients with Fanconi anemia developing myelodysplasia and
acute leukemia who received allogeneic hematopoietic stem cell
transplantation: A retrospective analysis on behalf of EBMT group. Am J
Hematol. 2020 Jul;95(7):809-16. https://doi.org/10.1002/ajh.25810
PMid:32267023
- Peffault de Latour R, Soulier J.
How I treat MDS and AML in Fanconi anemia. Blood. 2016 Jun
16;127(24):2971-9. https://doi.org/10.1182/blood-2016-01-583625
PMid:27020090
- Ayas M, Saber W, Davies SM, Harris
RE, Hale GA, Socie G, et al. Allogeneic Hematopoietic Cell
Transplantation for Fanconi Anemia in Patients With Pretransplantation
Cytogenetic Abnormalities, Myelodysplastic Syndrome, or Acute Leukemia.
J Clin Oncol. 2013 May 1;31(13):1669-76.
https://doi.org/10.1200/JCO.2012.45.9719 PMid:23547077
PMCid:PMC3635221
- Kutler DI, Singh B, Satagopan J,
Batish SD, Berwick M, Giampietro PF, et al. A 20-year perspective on
the International Fanconi Anemia Registry (IFAR). Blood. 2003 Feb
15;101(4):1249-56. https://doi.org/10.1182/blood-2002-07-2170
PMid:12393516
- Alter BP. Fanconi anemia and the
development of leukemia. Best Pract Res Clin Haematol.
2014;27(0):214-21. https://doi.org/10.1016/j.beha.2014.10.002
PMid:25455269 PMCid:PMC4254647
- Rosenberg PS,
Alter BP, Ebell W. Cancer risks in Fanconi anemia: findings from the
German Fanconi Anemia Registry. Haematologica. 2008 Apr;93(4):511-7.
https://doi.org/10.3324/haematol.12234 PMid:18322251
- Rosenberg
PS, Socié G, Alter BP, Gluckman E. Risk of head and neck squamous cell
cancer and death in patients with Fanconi anemia who did and did not
receive transplants. Blood. 2005 Jan 1;105(1):67-73.
https://doi.org/10.1182/blood-2004-04-1652 PMid:15331448