Nasir Arefinia1,2, Ramin Yaghobi3, Amin Ramezani4,5 and Jamal Sarvari1,6.
1 Department of Bacteriology and Virology, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran.
2 School of Medicine, Jiroft University of Medical Sciences, Jiroft, Iran.
3 Shiraz Transplant Research Center, Shiraz University of Medical Sciences, Shiraz, Iran.
4
Department of Medical Biotechnology, School of Advanced Medical
Sciences and Technologies, Shiraz University of Medical Sciences,
Shiraz, Iran.
5 Shiraz Institute for Cancer Research, School of Medicine, Shiraz University of Medical Sciences, Shiraz, Iran.
6 Gastroenterohepatology Research Center, Shiraz University of Medical Sciences, Shiraz, Iran.
Correspondence to: Dr Jamal Sarvari. Tel: +9871-32307953(4543), Fax: +9871-12304069. Email:
sarvarij@sums.ac.ir
Published: July 1, 2023
Received: March 26, 2023
Accepted: June 18, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023042 DOI
10.4084/MJHID.2023.042
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Background:
Mutations in the SARS-CoV-2 genome might influence pathogenicity,
transmission rate, and evasion of the host immune system. Therefore,
the purpose of the present study was to investigate the genetic
alteration as well as assess their effects on the receptor binding
domain (RBD) of the spike and the putative RNA binding site of the RdRp
genes of SARS-CoV-2 using bioinformatics tools.
Materials and Method:
In this cross-sectional study, 45 confirmed COVID-19 patients using
qRT-PCR were included and divided into mild, severe, and critical
groups based on the severity of the disease. RNA was extracted from
nasopharyngeal swab samples using a commercial kit. RT-PCR was
performed to amplify the target sequences of the spike and RdRp genes
and sequence them by the Sanger method. Clustal OMEGA, MEGA 11
software, I-mutant tools, SWISS-MODEL, and HDOCK web servers were used
for bioinformatics analyses.
Results:
The mean age of the patients was 50.68±2.73. The results showed that
four of six mutations (L452R, T478K, N501Y, and D614G) in RBD and three
of eight in the putative RNA binding site (P314L, E1084D, V1883T) were
missense. In the putative RNA binding site, another deletion was
discovered. Among missense mutations, N501Y and V1883T were responsible
for increasing structural stability, while others were responsible for
decreasing it. The various homology models designed showed that these
homologies were like the Wuhan model. The molecular docking analysis
revealed that the T478K mutation in RBD had the highest binding
affinity. In addition, 35 RBD samples (89.7%) and 33 putative RNA
binding site samples (84.6%) were similar to the Delta variant.
Conclusion:
Our results indicated that double mutations (T478K and N501Y) in the S
protein might increase the binding affinity of SARS-CoV-2 to human ACE2
compared to the wild-type (WT) strain. Moreover, variations in the
spike and RdRp genes might influence the stability of encoded
proteins..
|
Introduction
The
SARS-CoV-2 outbreak occurred in Wuhan, China, in December 2019 and
quickly became a pandemic by spreading to all countries worldwide,
resulting in significant public health problems and even economic
distress for nations.[1,2] As of the end of the year,
more than 608 million confirmed COVID-19 cases were reported worldwide,
with 6.5 million confirmed deaths.[3]
Coronaviruses
are divided into three genera, including Alpha, Beta, and Gamma
coronaviruses; SARS, MERS, and SARS-CoV-2 are members of the Beta
coronavirus. All coronaviruses have a genome consisting of a
positive-sense single-strand RNA molecule. SARS-CoV2 encoded four
structural proteins, including spike, 16 nonstructural proteins (NSP),
and nine accessory proteins, including the RNA-dependent RNA polymerase
(RdRp) (also named nsp12).[4]
The spike glycoprotein mediates receptor binding and fusion of the viral and cellular membranes.[4]
In addition, it is subject to proteolytic cleavage by host proteases
(i.e., trypsin and furin) at two sites at the polybasic furin cleavage
site (PCS), located at the boundary between the S1 and S2 subunits
(S1/S2 site). The binding of ACE2 with Spike S1 (Amino Acids (AA):
15–685) protein allows the virus to adhere to the lung epithelial
cells. A small, independently folded subdomain of S1, described as the
receptor-binding domain (RBD), directly binds ACE2 (AA: 320–540) when
the virus engages the target cells.[5] The virus is
predicted to be zoonotic in origin, and mutations in the surface
glycoprotein structure enable its transmission to human hosts.[6]
There has been evidence of SARS-CoV-2 mutations that have a substantial functional effect on the virus.[7]
A mutation resulting in an AA substitution in the spike protein (D614G)
emerged early in the epidemic and spread rapidly through Europe and
North America, particularly.[8] During a joint study
in the United States and the Netherlands in September 2020, researchers
discovered that the D614G mutation of the spike protein of SARS-CoV-2
was predominant and affected disease presentation.[9]
Two important mutations in the RBD at positions R407I and A930V have
been identified in India, increasing the molecule's dependence on its
receptor.[10] Another mutation in the RBD position
was N501Y, which American researchers showed to neutralize human
convalescent or post-vaccination sera in vitro.[11,12]
The emergence and spread of a SARS-CoV-2 lineage that contains several
nonsynonymous spike mutations, including mutations that affect key
sites in the RBD (resulting in K417N, E484K, and N501Y substitutions)
in South Africa may have functional importance.[4]
Specifically, residues G431 and S514 in SARS-CoV-2 RBD are important
for S protein stability. An experimental study indicated that an S
missense mutation such as D614G contributed to the dominant pandemic
form and could stabilize the entire S protein.[13]
SARS-CoV-2 RdRp is the prime constituent of the replication/transcription machinery prone to mutation.[14] RdRp is a primary target for antiviral inhibitors like Remdesivir,[15]
Favipiravir, Galidesivir, and Ribavirin,[16] as well as some other
potential drugs such as Filibuvir, Cepharanthine, Simeprevir, and
Tegobuvir.[16] The RdRp mutation may alter the
rigidity of the RdRp protein structure, which can exert its effects
through altered interaction with the RNA pattern or with other
components of the transcription/replication machine, thereby altering
the mutation rate.[17] A comparative analysis shows
that some mutations are specific to a certain region; for example,
T265I, P5828L, and Y5865C are unique to the United States and have not
been reported from Europe or Asia, while P4715L is predominant in
Europe. Moreover, four significant mutations T265I (nsp 2), P4715L (nsp
12), P5828L, and Y5865C (both at nsp 13), were identified in important
nonstructural proteins, which function either as replicas or helicase.[18]
As
the virus spreads to new places, it alters its protein sequence by
introducing mutations in its genome that help it survive better in the
host.[19] As an efficient unique pathogen, this virus
often mutates its proteins so that it can still infect the host cells,
change its pathogenicity and transmission rate, and evade the host
immune system.[20] Even when profitable strategies
are discovered and engaged, viruses' high rate of genetic change
frequently leads to drug resistance or vaccine escape.[21]
With cautious optimism at the recent decline in cases, there is still
anxiety about subsequent waves of infections after the relaxation of
containment procedures in various cities and countries.[22]
Although
the mutation rate in SARS-CoV-2 is much lower than that of other RNA
viruses, including seasonal flu viruses, knowing these mutations can
help us create targeted treatments through more effective vaccines and
antiviral therapy and reveal its pathogenesis, transmission rate, and
spread. Therefore, our studies focused on the mutation rate and
assessed their effects on the spike's receptor binding domain (RBD) and
the putative RNA binding site of the RdRp genes of SARS-CoV-2 using
bioinformatics tools.
Materials and methods
Study population.
From July 28 to September 1, 2021, 45 COVID-19 patients were recruited
consecutively from Afzalipour Hospital, affiliated with Kerman
University of Medical Science. The selected patients were divided into
three groups, mild, severe, and critical, based on the disease severity
according to the World Health Organization (WHO) criteria.[24]
The inclusion criteria were as follows: 1) patients that met the
diagnostic standard for SARS CoV-2 virus based on the Center for
Disease Control and Prevention (CDC) definition as an acute respiratory
disease with laboratory-confirmed SARS CoV-2 infection by qRT-PCR;[25]
2) positive results from throat swabs; 3) patients with an intense
cough with bloody or purulent sputum, chest pain, severe vomiting,
diarrhea, dehydration, and dyspnea; 4) patients with complete clinical
information; 5) patients who did not complain about other infectious
diseases, history of hematological diseases, or immune system
disorders; and 6) patients who were inoculated with any of the Corona
vaccines. A structured questionnaire was used to collect demographic
characteristics, temperature, number of breaths per minute, weekly
exercise, smoking, and a history of underlying disease. Written
informed consent was obtained from each participant.
Sampling.
Nasopharyngeal samples were taken from all patients using a Dacron swab
and then transferred to Viral Transporter Media (VTM) in a cool box at
the research laboratory in the School of Medicine, Kerman University of
Medical Sciences.
RNA extraction.
Viral RNA was extracted from throat samples (one sample, one patient)
using a highly pure Viral RNA Kit (Roche, Product no: 11858882001)
based on manufacturer instructions.
Primer design and reverse transcriptase polymerase chain reaction (RT-PCR).
The RBD domain sequence, the spike-glycoprotein gene-cleavage site, and
the putative RNA binding site of RdRp of the ORF1ab gene were extracted
from the NCBI database. The AlleleID program also designed primers for
the desired domains.
The region of the RBD-cleavage site of the S
glycoprotein and the putative RNA binding domain of the RdRp was
amplified by PCR methods. The primer sequences, annealing temperatures,
and product size are shown in Table 1.
 |
- Table
1. The sequences of the primers used for amplification of RBD–cleavage
site of the spike and Putative RNA binding site of the RdRP using the
PCR method.
|
This
reaction was performed in two steps. In the first step, the reverse
transcriptase (RT) PCR method was performed in one step, and the
components were combined in a single tube. The RT-PCR was performed in
a standard protocol in a 25 μl volume containing the primer, RNA
template, and 2X one-step RT-PCR master mix (Qiagen, Germany). In the
second step, the conventional PCR technique with the specific internal
primers on previous products and Taq DNA Polymerase Master Mix RED 2X
(Ampliqon) was performed. 12.5 µl of a 2×master mix, 10 pmol of each
primer, and 150 ng of extracted RNA were used. The reaction was
performed in an Eppendorf thermal cycler under the following
conditions: First denaturation for 5 min at 95°C followed by 40 cycles:
40 s at 95°C, 35 s at Ta °C, and 40 s at 72°C. A final extension at
72°C for 5 min was performed at the end of the cycles.
The PCR
products were electrophoresed on a 1.5% agarose gel. A 100-bp molecular
weight marker was used for band detection. The target bands were sliced
for gel purification, which was performed by the MinElute PCR
Purification Kit (Qiagen GmbH) according to the manufacturer's
protocol.
Sanger Sequencing. Purified amplified products were sent to the 1st
BASE Company (Gemini) for Sanger sequencing. Of 45 samples, 39 results
for the RBD–cleavage site and the putative RNA binding site were clear
and perfect and used for sequence analysis.
Mutational analysis.
The sequences obtained from the studied region were aligned and
complemented with CLC 6 and Clustal OMEGA. To confirm the results, we
compared the sequences with the existing database using the online
BLAST search tool (www.ncbi.nlm.nih.gov).
To find the related mutations of the RBD–cleavage site and putative RNA
binding site, we compared the sequences with the reference strain Wuhan-Hu sequence using the Clustal OMEGA algorithm.
To identify the
nucleotide variations, we performed multiple sequence alignments using
Clustal OMEGA,[26] and the sequence of the strain Whuhan-Hu-1 (GenBank
accession number: 045512) was used as a reference genome. The alignment
file was analyzed using the MVIEW program of Clustal OMEGA.[27]
Detection of mutation Spectrum.
Clustal OMEGA was used again for the multiple sequence alignment of
each protein, which was further analyzed by MVIEW. The amino acid
variations were identified in each protein by comparing it to the
protein of the reference strain. Further, both nucleotide and amino
acid variations were compared to study the types of mutations.
Prediction of mutational effects.
The structural and functional effects of the missense variants and the
stability change were analyzed using different prediction tools.
I-mutant was employed to analyze the stability change with all the
parameters set to default.[28] Additionally, Mutpred2 was adopted to predict the molecular consequences and functional effects of these mutations.[29]
Homology modeling of spike proteins and model validation. The BLASTp program at the NCBI interface (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins)
was used to find the most suitable template for homology modeling.
Getting the protein databank reservoir (PDB), we identified spike
protein with PDB ID: 6M0J as a suitable template, as it has 99.59%
sequence similarity and 94% coverage with the target sequence. The
homology modeling of all mutant spike proteins, along with the spike
protein of the reference, was done using SWISS-MODEL.[30] The predicted model was validated by adopting Rampage and ERRAT.[31]
Molecular docking of Spike Protein with ACE2 receptor.
The molecular docking approach was employed to investigate the
interaction of mutant spike protein with the human ACE2 receptor.
First, the crystal structure of human ACE2 (PDB ID: 6D0G) was obtained
from the Protein Data Bank (PDB), and PyMOL was used to clean the
structure to remove all the complex molecules and water.[32]
The HDOCK webserver was used to predict the interaction between Spike
protein and the human ACE2 receptor through protein-protein molecular
docking.[33]
Phylogenic analysis.
Phylogenetic and sequence analysis based on RBDs and RdRp was performed
from all 39 clear and perfect isolates. In the phylogenic analysis, we
considered 6 variants of concern (VOCs), including B.1.1.529
(UZI26581.1), P.1 (QXU68443.1), B.1.617.2 (UAL04647.1), B.1.1.7
(UFQ05186.1), B.1.351 (UFQ05198.1), and whuhan strain (YP 009724390.1)
as reference sequences. All sequences were clustered using MEGA 11
software. Finally, the evolutionary history was inferred using the
Maximum Likelihood method as a statically model, the bootstrap method
as a type of phylogenic (~1000 bootstrap), and the Tamura-Nei model as
a substitution method.
Results
Demographic characteristics of the subjects.
In this cross-sectional study, 45 COVID-19 patients were included. Of
these, 39 samples had clear perfect sequence results with an average
age of 50.38 ± 2.13, including 21 males and 18 females (Table 2).
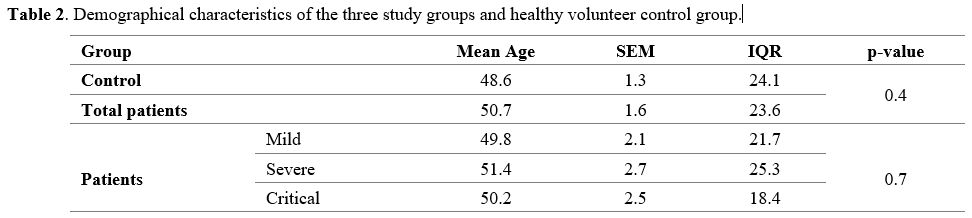 |
- Table
2. Demographical characteristics of the three study groups and healthy volunteer control group
|
Mutation Spectrum of SARS-CoV-2 isolates.
Of 45 PCR products sent for sequencing, 39 samples had clear perfect
sequencing results and were used for analysis. Analysis of all 39
Kerman isolates revealed a total of 8 single nucleotide variants in the
putative RNA binding site and 6 in the RB-cleavage site. Besides, one
deletion was also found in those isolates, which was responsible for
the deletion of putative RNA binding site isolates (Table 3).
Three of the eight putative RNA binding site mutations were missense
mutations, and five were synonymous. In addition, four of the six
mutations in the RBD domain were missense, and the other two were
synonymous (Table 3).
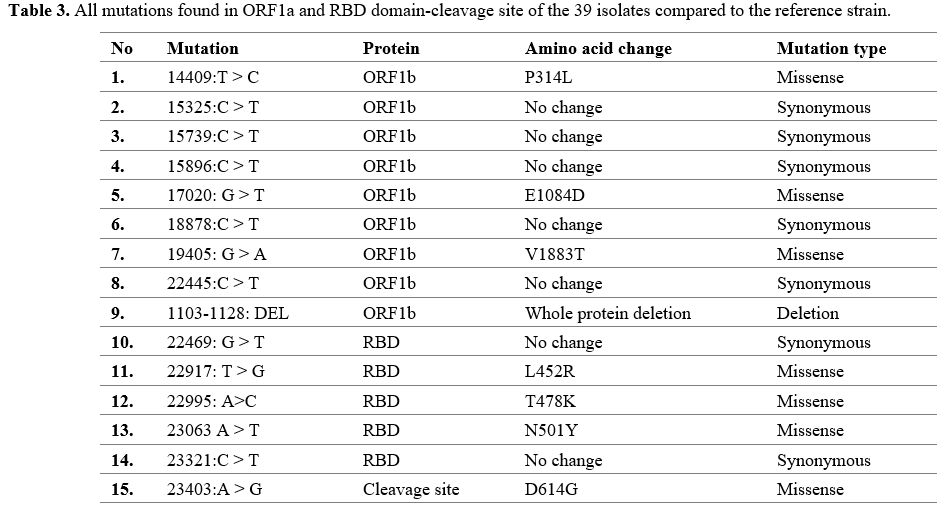 |
- Table 3. All mutations found in ORF1a and RBD domain-cleavage site of the 39 isolates compared to the reference strain.
|
Mutational effects.
Mutational effects analysis of the three missense mutations located in
the putative RNA binding site revealed that two of them were
responsible for decreasing structural stability and another for
increasing it. Furthermore, four missense mutations occurred in the RBD
and cleavage site domains; three of them were responsible for decreased
structural stability, and one for increased structural stability (Table 4).
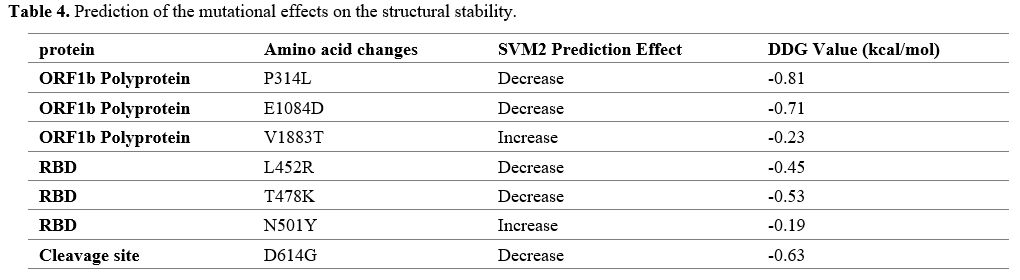 |
- Table 4. Prediction of the mutational effects on the structural stability.
|
Prediction and validation of the homology models.
Five models were generated using the template PDB ID: 6M0J: one for the
spike protein of the reference strain and the other four for three
different mutant isolates from Kerman. Mutant models 1, 2, 3, and 4 are
designed for L452R, T478K, N501Y, and D614G mutations, respectively.
The validation assessment scores of these four models were mostly
similar to the template, which proved the reliability of these models (Table 5).
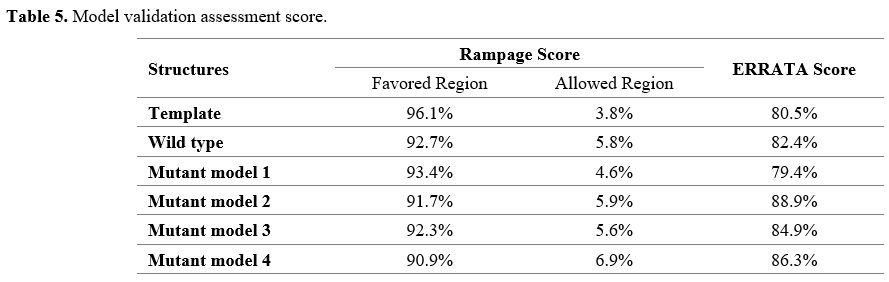 |
- Table 5. Model validation assessment score.
|
Analysis of the interaction between spike proteins and human ACE2 receptor.
The HDOCK server was used to predict the interaction between the 3D
models of reference spike proteins, mutant models, and the human ACE2
receptor. This molecular docking analysis confirmed that the T478K
mutation had the highest binding affinity (-143.1), followed by N501Y
(-142.3) and L452R (139.9). For three spike protein models, this study
showed that the spike protein domain, rather than the complete protein,
amino acids 345 to 527, was involved in the interactions (Table 6).
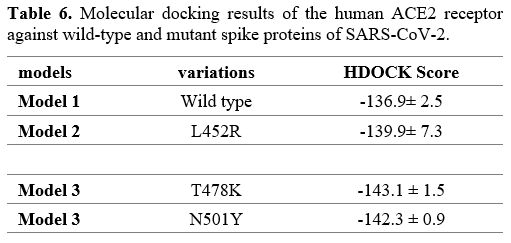 |
- Table 6. Molecular docking results of the human ACE2 receptor against wild-type and mutant spike proteins of SARS-CoV-2.
|
Phylogenetic analysis.
The RBD and putative RNA binding site sequencing results of the current
study were compared to the world's registered prevalent concern
variants, and detailed specifications are shown in Figures 1 and 2.
According to the comparison results and different variants, the
sequences confirmed the introduction of species by overcoming Delta
species. It was found that most SARS-CoV-2 viruses circulating in
Kerman, Iran, from July to September 2021 were similar to the Delta
variant (B.1.617.2), with 90% and 98% genetic homology, respectively (Figures 1 and 2).
The prevalence of RBD VOCs with Pangolin Lineages B.1.617.2/Delta was 89.74%, and B.1.1.529/Omicron (5.10%) (Figure 1).
In two samples (5.1%), in addition to the Delta variant, some RBD
mutations of the Omicron variant (N501Y and T478K) were added to this
type in patients in this area that might gradually dominate the
previous species. Also, in a similar pattern, the prevalence of
putative RNA binding sites of VOCs was 81.39% for B.1.617.2/Delta (Figure 2)...
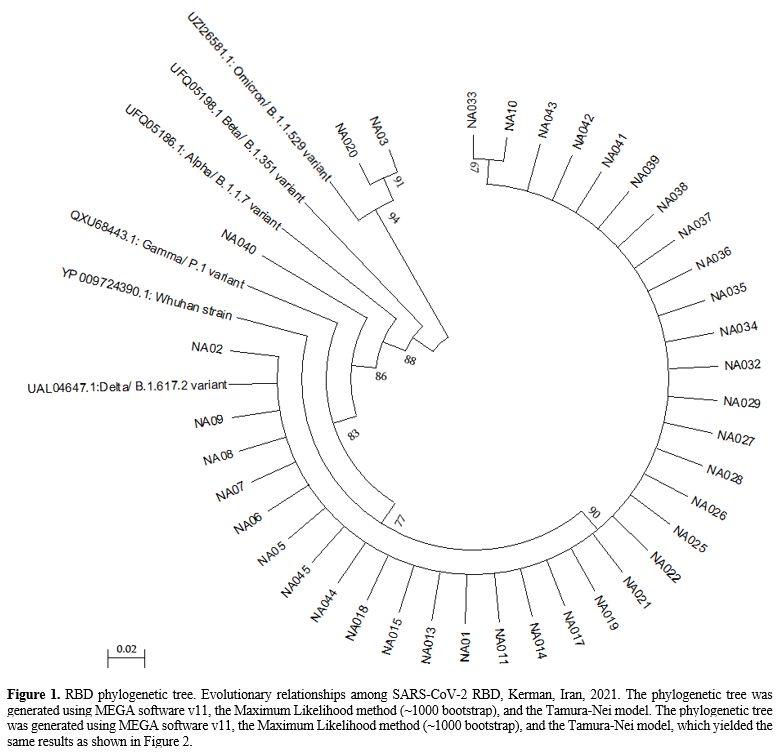 |
Figure 1. RBD phylogenetic
tree. Evolutionary relationships among SARS-CoV-2 RBD, Kerman, Iran,
2021. The phylogenetic tree was generated using MEGA software v11, the
Maximum Likelihood method (~1000 bootstrap), and the Tamura-Nei model.
The phylogenetic tree was generated using MEGA software v11, the
Maximum Likelihood method (~1000 bootstrap), and the Tamura-Nei model,
which yielded the same results as shown in Figure 2.
|
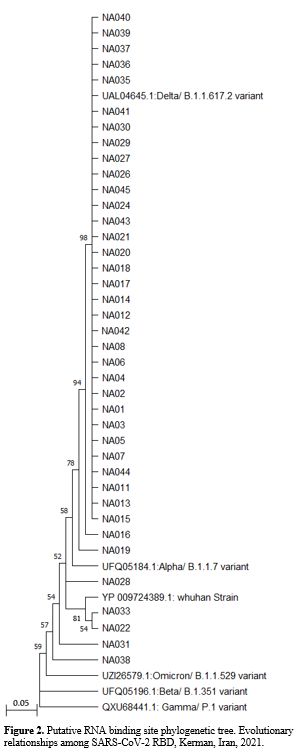 |
Figure 2.
Putative RNA binding site phylogenetic tree. Evolutionary relationships
among SARS-CoV-2 RBD, Kerman, Iran, 2021.
|
Discussion
SARS-CoV-2
has become a global challenge, affecting millions and taking thousands
of lives daily. Along with other studies, viral and host genetic
studies can give a significant clue to understanding the pathogenesis
of COVID-19 and combating SARS-CoV-2.[34-36] In
addition to the critical therapeutic target, the genomic sequence data
may provide insights into the pattern of global spread, dynamics of
evolutions, transmission rate, escape of the host defense,
and importance in unraveling the molecular pathogenesis mechanism of
SARS-CoV2.[37]
In this study, we found some
mutations in the SARS-CoV-2 isolated in Kerman, Iran, which may give
insights into the transmission of SARS-CoV-2, the genetic diversity of
the S gene (RBD and cleavage site domain), and ORF1ab (putative RNA
binding) and predict the impacts of mutations. A total of 15 mutations
were identified with a deletion, of which less than half were
synonymous (7 mutations). Among the six mutations found in the RBD
domain and cleavage site of the S glycoprotein, L452R, T478K, N501Y,
and D614G were identified as missense mutations. This variant, which
has evolved to become the predominant genotype worldwide, can evade
cellular immunity, have high infectivity, and increase host glycolysis
and fusogenicity, increasing the number of hospitalized patients with
high morbidity and mortality.[38,39] These mutations
also had the potential to evade the protective antibodies derived from
the convalescent sera or vaccine origin, as well as those used in
therapy, which could favor virus expansion and compromise infection
control.[40] The results of a study have revealed a
set of mutations, such as D614G and P314L, that influence the outcomes
of SARS-CoV-2 infection and guide the development of SARS-CoV-2
vaccines.[41]
Moreover, our molecular docking
analysis revealed that these mutations in the RBD domain affected the
interaction with the ACE2 receptor, suggesting that this mutation is
more responsive to hACE2, indicating the potential for transmission.
T478K (-143.1) has the highest binding affinity, followed by N501Y
(-142.3) and L452R (-139.9). In this regard, a study indicated that
some mutations such as Q493R, N501Y, S371L, S373P, S375F, Q498R, and
T478K significantly contributed to high binding affinity with human
ACE2, which is consistent with our results.[42]
Another study showed that mutations T478K and N501Y, particularly, had
the highest binding affinity to Glucose Regulated Protein 78 (GRP-78)
as a co-receptor for virus attachment to the host cell.[39]
Large
deletion mutations can cause a defect in the production and function of
the desired protein; some scientists have suggested that large
deletions in OFR1ab may not encode the target protein.[43]
Large deletion mutations have also been found in other scientists'
studies; for instance, an 80-nucleotide deletion in ORF7a was also
reported in a study conducted in Arizona.[44]
Previous studies have shown that the lack of these side proteins
(ORF8b) has adverse effects on viral replication, pathogenesis, and
structural expression of the protein.[45] In another
study, researchers indicated that loss of some of the SARS-CoV-2
proteins, such as ORF1a and ORF7, could cause a much more significant
restriction of the spread of the virus into the host and may also lead
to less pathogenicity of the virus, resulting in a meager infection
rate and mortality compared to the other countries, which are
consistent with the study conducted by Keng and his colleagues.[45]
Furthermore,
compared to the reference, variations in the putative RNA binding site
of the RdRp caused substitutions of one or more amino acids in the
isolates from Kerman, Iran. Among the eight mutations found in the
Putative RNA binding site, mutations P314L, E1084D, and V1883T were
identified as missense ones. Some of the mutations were found to affect
the structural stability of the proteins rather than alter their
molecular functions, and some of those altered their molecular
functions. The putative RNA binding site in ORF1ab is essential for
replicating the viral genome into a helical ribonucleocapsid (RNP).[46]
These functions may not be affected much by those
mutations; Mutpred2 predicted that these mutations did not alter
any molecular consequences of the proteins, which is consistent with
the study conducted by Wrapp.[47] Parvez and
colleagues' molecular docking analysis revealed no significant
interactions between the putative RNA binding site and the genome.[48]
Doga Eskier et al. reported that the third most common RdRp,
15324C>T, decreased the mutation rate created by RdRp, while the
14408C>T mutation had the opposite effect. They also suggested that
the 14408C>T mutation contributed to co-mutations' dominance in
Europe and elsewhere.[49]
The S gene displays
higher tolerance for positive selection in mutant isolates early during
the appearance of the double mutant genotype. It undergoes increasing
negative selection over time, whereas the RdRp region in the mutant
isolates shows strong negative selection throughout the pandemic.[50]
Conclusions
Our
findings suggest that mutations may reduce the stability of spike and
RdRp, whereas variations such as N501Y and V1883T may increase
stability. Moreover, according to various homology models, these
homologies were similar to those in the WT strain. Furthermore, the
mutated structures (T478K and L452R) that were seen in the RBD domain
might increase the binding affinity of SARS-CoV-2 to human ACE2
compared to WT[51] because of the significant changes
in the electrostatic and van der Waals (vdW) interactions. Further
studies are recommended to clarify the results.
Acknowledgements
The
present study was extracted from a thesis written by Nasir Arefinia,
which was financially supported by a grant from Shiraz University of
Medical Sciences (No: 22168).
Ethics statement
The study was approved by the local Ethics Committee of Shiraz University of Medical Sciences (IR.SUMS.REC.1400.737).
References
- Arefinia N, Ghoreshi Z-S, Alipour AH, Reza Molaei
H, Samie M, Sarvari J. Gastrointestinal Manifestations in Patients
Infected with SARS-CoV-2. Iran J Med Microbiol. 2022;16(4):271-81. https://doi.org/10.30699/ijmm.16.4.271
- Sharif-zak
M, Abbasi-jorjandi M, Asadikaram G. Immunobiology CCR2 and DPP9
expression in the peripheral blood of COVID-19 patients : Influences of
the disease severity and gender. Immunobiology [Internet].
2022;227(2):152184. https://doi.org/10.1016/j.imbio.2022.152184 PMid:35131543 PMCid:PMC8806394
- No Title [Internet]. [cited 2022 August 31]. https://www.worldometers.info/coronavirus/
- Wu
F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, et al. A new coronavirus
associated with human respiratory disease in China. Nature.
2020;579(7798):265-9. https://doi.org/10.1038/s41586-020-2008-3 PMid:32015508 PMCid:PMC7094943
- Shang
J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of
receptor recognition by SARS-CoV-2. Nature. 2020 May;581(7807):221-4. https://doi.org/10.1038/s41586-020-2179-y PMid:32225175 PMCid:PMC7328981
- Andersen
KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of
SARS-CoV-2. Vol. 26, Nature medicine. 2020. p. 450-2. https://doi.org/10.1038/s41591-020-0820-9 PMid:32284615 PMCid:PMC7095063
- Tegally
H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, et
al. Emergence and rapid spread of a new severe acute respiratory
syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike
mutations in South Africa. MedRxiv. 2020; https://doi.org/10.1101/2020.12.21.20248640
- Plante
JA, Liu Y, Liu J, Xia H, Johnson BA, Lokugamage KG, et al. Spike
mutation D614G alters SARS-CoV-2 fitness. Nature [Internet]. Nature
Publishing Group; 2020. https://doi.org/10.1101/2020.09.01.278689
- Butowt
R, Bilinska K, Von Bartheld CS. Chemosensory dysfunction in COVID-19:
integration of genetic and epidemiological data points to D614G spike
protein variant as a contributing factor. ACS Chem Neurosci.
2020;11(20):3180-4. https://doi.org/10.1021/acschemneuro.0c00596 PMid:32997488 PMCid:PMC7581292
- Saha
P, Banerjee AK, Tripathi PP, Srivastava AK, Ray U. A virus that has
gone viral: amino acid mutation in S protein of Indian isolate of
Coronavirus COVID-19 might impact receptor binding, and thus,
infectivity. Biosci Rep. 2020 May;40(5). https://doi.org/10.1042/BSR20201312 PMid:32378705 PMCid:PMC7225408
- Jangra
S, Ye C, Rathnasinghe R, Stadlbauer D, Alshammary H, Amoako AA, et al.
SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. The
Lancet Microbe [Internet]. 2021 Jul 1;2(7):e283-4. https://doi.org/10.1016/S2666-5247(21)00068-9 PMid:33846703
- Teng
S, Sobitan A, Rhoades R, Liu D, Tang Q. Systemic effects of missense
mutations on SARS-CoV-2 spike glycoprotein stability and
receptor-binding affinity. Brief Bioinform. 2021 Mar;22(2):1239-53. https://doi.org/10.1093/bib/bbaa233 PMid:33006605 PMCid:PMC7665319
- Teng
S, Sobitan A, Rhoades R, Liu D, Tang Q. Systemic effects of missense
mutations on SARS-CoV-2 spike glycoprotein stability and
receptor-binding affinity. Brief Bioinform. 2021;22(2):1239-53. https://doi.org/10.1093/bib/bbaa233 PMid:33006605 PMCid:PMC7665319
- Singh D, Yi S V. On the origin and evolution of SARS-CoV-2. Exp Mol Med. 2021;53(4):537-47. https://doi.org/10.1038/s12276-021-00604-z PMid:33864026 PMCid:PMC8050477
- Gordon
DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O'Meara MJ, et al. A
SARS-CoV-2-Human Protein-Protein Interaction Map Reveals Drug Targets
and Potential Drug-Repurposing. bioRxiv [Internet]. 2020 Jan
1;2020.03.22.002386. http://biorxiv.org/content/early/2020/03/27/2020.03.22.002386.abstract
- Pachetti
M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al.
Emerging SARS-CoV-2 mutation hot spots include a novel
RNA-dependent-RNA polymerase variant. J Transl Med. 2020;18(1):1-9. https://doi.org/10.1186/s12967-020-02344-6 PMid:32321524 PMCid:PMC7174922
- Pachetti
M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, et al.
Emerging SARS-CoV-2 mutation hot spots include a novel
RNA-dependent-RNA polymerase variant. J Transl Med [Internet].
2020;18(1):179. https://doi.org/10.1186/s12967-020-02344-6 PMid:32321524 PMCid:PMC7174922
- Banerjee
S, Seal S, Dey R, Mondal KK, Bhattacharjee P. Mutational spectra of
SARS-CoV-2 orf1ab polyprotein and signature mutations in the United
States of America. J Med Virol. 2021;93(3):1428-35. https://doi.org/10.1002/jmv.26417 PMid:32779784 PMCid:PMC7436414
- Sackman
AM, McGee LW, Morrison AJ, Pierce J, Anisman J, Hamilton H, et al.
Mutation-Driven Parallel Evolution during Viral Adaptation. Mol Biol
Evol [Internet]. 2017 Dec 1;34(12):3243-53. https://doi.org/10.1093/molbev/msx257 PMid:29029274 PMCid:PMC5850295
- Srivastava
M, Hall D, Omoru OB, Gill HM, Smith S, Janga SC. Mutational landscape
and interaction of sars-cov-2 with host cellular components.
Microorganisms. 2021;9(9):1794. https://doi.org/10.3390/microorganisms9091794 PMid:34576690 PMCid:PMC8464733
- McKeegan
KS, Borges-Walmsley MI, Walmsley AR. Microbial and viral drug
resistance mechanisms. Trends Microbiol [Internet]. 2002 Oct
1;10(10):s8-14. https://doi.org/10.1016/S0966-842X(02)02429-0 PMid:12377562
- Agrawal
L, Poullikkas T, Eisenhower S, Monsanto C, Bakku RK, Chen M-H, et al.
Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for
Potential Therapeutic Targets. Antibodies. 2021;10(1):3. https://doi.org/10.3390/antib10010003 PMid:33440681 PMCid:PMC7839017
- Malik
JA, Ahmed S, Mir A, Shinde M, Bender O, Alshammari F, et al. The
SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to
new challenges. J Infect Public Health [Internet]. 2022;15(2):228-40. https://doi.org/10.1016/j.jiph.2021.12.014 PMid:35042059 PMCid:PMC8730674
- Organization
WH. COVID-19 clinical management: living guidance, January 25 2021
[Internet]. Geneva PP - Geneva: World Health Organization https://apps.who.int/iris/handle/10665/338882
- Emergency F, Only U, Only R. Real-Time RT-PCR Diagnostic Panel For Emergency Use Only. 2021.
- Madeira
F, Park Y mi, Lee J, Buso N, Gur T, Madhusoodanan N, et al. The
EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids
Res [Internet]. 2019 Jul 2;47(W1):W636-41. https://doi.org/10.1093/nar/gkz268 PMid:30976793 PMCid:PMC6602479
- Brown
NP, Leroy C, Sander C. MView: a web-compatible database search or
multiple alignment viewer. Bioinformatics. 1998;14(4):380-1. https://doi.org/10.1093/bioinformatics/14.4.380 PMid:9632837
- Capriotti
E, Fariselli P, Casadio R. I-Mutant2.0: predicting stability changes
upon mutation from the protein sequence or structure. Nucleic Acids Res
[Internet]. 2005 Jul 1;33(suppl_2):W306-10. https://doi.org/10.1093/nar/gki375 PMid:15980478 PMCid:PMC1160136
- Pejaver
V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, et al.
Inferring the molecular and phenotypic impact of amino acid variants
with MutPred2. Nat Commun. 2020;11(1):1-13.
https://doi.org/10.1038/s41467-020-19669-x PMid:33219223
PMCid:PMC7680112
- Schwede
T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein
homology-modeling server. Nucleic Acids Res. 2003;31(13):3381-5. https://doi.org/10.1093/nar/gkg520 PMid:12824332 PMCid:PMC168927
- Lovell
SC, Davis IW, Arendall III WB, De Bakker PIW, Word JM, Prisant MG, et
al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation.
Proteins Struct Funct Bioinforma. 2003;50(3):437-50. https://doi.org/10.1002/prot.10286 PMid:12557186
- DeLano WL. Pymol: An open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr. 2002;40(1):82-92.
- Yan
Y, Zhang D, Zhou P, Li B, Huang S-Y. HDOCK: a web server for
protein-protein, and protein-DNA/RNA docking based on a hybrid
strategy. Nucleic Acids Res. 2017;45(W1):W365-73. https://doi.org/10.1093/nar/gkx407 PMid:28521030 PMCid:PMC5793843
- Hashemi
SMA, Thijssen M, Hosseini SY, Tabarraei A, Pourkarim MR, Sarvari J.
Human gene polymorphisms and their possible impact on the clinical
outcome of SARS-CoV-2 infection. Arch Virol [Internet]. 2021/05/02.
2021 Aug;166(8):2089-108. https://doi.org/10.1007/s00705-021-05070-6 PMid:33934196 PMCid:PMC8088757
- Arefinia
N, Ramezani A, Farokhnia M, Arab Zadeh AM, Yaghobi R, Sarvari J.
Association between expression of ZBP1, AIM2, and MDA5 genes and
severity of COVID-19. EXCLI J. 2022 Sep 1;21:1171-1183. https://doi.org/10.17179/excli2022-5141 PMID: 36320810; PMCID: PMC9618740.
- Nasir
Arefinia, Ramin Yaghoubi, Amin Ramezani, Mehrdad Farokhnia, Ali
Mohammad Arab Zadeh JS. Association of IFITM1 Promoter Methylation with
the severity of SARS CoV-2 infection. Clin Lab. 2023; https://doi.org/10.7754/Clin.Lab.2022.220622 PMid:37057950
- Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Reports [Internet]. 2020;19:100682. https://doi.org/10.1016/j.genrep.2020.100682 PMid:32300673 PMCid:PMC7161481
- Wahid
M, Jawed A, Mandal RK, Dailah HG, Janahi EM. Variants of SARS-CoV-2 ,
their effects on infection , transmission and neutralization by
vaccine-induced antibodies. 2021;5857-64.
- Elfiky
AA, Ibrahim IM, Elgohary AM. SARS-CoV-2 Delta Variant is Recognized
Through GRP78 Host-Cell Surface Receptor, In Silico Perspective. Int J
Pept Res Ther [Internet]. 2022;28(5):1-11. https://doi.org/10.1007/s10989-022-10450-w PMid:36034049 PMCid:PMC9395890
- Di
Giacomo S, Mercatelli D, Rakhimov A, Giorgi FM. Preliminary report on
severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike
mutation T478K. J Med Virol. 2021 Sep;93(9):5638-43. https://doi.org/10.1002/jmv.27062 PMid:33951211 PMCid:PMC8242375
- Huang
F, Chen L, Guo W, Zhou X, Feng K, Huang T, et al. Identifying COVID-19
Severity-Related SARS-CoV-2 Mutation Using a Machine Learning Method.
Life (Basel, Switzerland). 2022 May;12(6). https://doi.org/10.3390/life12060806 PMid:35743837 PMCid:PMC9225528
- Bhattacharya
M, Sharma AR, Dhama K, Agoramoorthy G, Chakraborty C. Omicron variant
(B.1.1.529) of SARS-CoV-2: understanding mutations in the genome,
S-glycoprotein, and antibody-binding regions. GeroScience [Internet].
2022;44(2):619-37. https://doi.org/10.1007/s11357-022-00532-4 PMid:35258772 PMCid:PMC8902853
- Alam
S, Mahfujur M, Morshed N. Since January 2020 Elsevier has created a
COVID-19 resource center with free information in English and Mandarin
on the novel coronavirus COVID- 19. The COVID-19 resource centre is
hosted on Elsevier Connect, the company's public news and information .
2020;(January).
- Mercatelli D, Giorgi FM. Geographic and genomic distribution of SARS-CoV-2 mutations. Front Microbiol. 2020;11:1800. https://doi.org/10.3389/fmicb.2020.01800 PMid:32793182 PMCid:PMC7387429
- Keng
C-T, Choi Y-W, Welkers MRA, Chan DZL, Shen S, Gee Lim S, et al. The
human severe acute respiratory syndrome coronavirus (SARS-CoV) 8b
protein is distinct from its counterpart in animal SARS-CoV and
down-regulates the expression of the envelope protein in infected
cells. Virology [Internet]. 2006;354(1):132-42. https://doi.org/10.1016/j.virol.2006.06.026 PMid:16876844 PMCid:PMC7111915
- Hoffmann
M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al.
SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a
clinically proven protease inhibitor. Cell. 2020;181(2):271-80. https://doi.org/10.1016/j.cell.2020.02.052 PMid:32142651 PMCid:PMC7102627
- Wrapp
D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, et al.
Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation.
Science (80- ) [Internet]. 2020 Mar 13;367(6483):1260 LP - 1263. https://doi.org/10.1126/science.abb2507 PMid:32075877 PMCid:PMC7164637
- Parvez
MSA, Rahman MM, Morshed MN, Rahman D, Anwar S, Hosen MJ. Genetic
analysis of SARS-CoV-2 isolates collected from Bangladesh: insights
into the origin, mutation spectrum, and possible pathomechanism.
bioRxiv [Internet]. 2020 Jan 1;2020.06.07.138800. http://biorxiv.org/content/early/2020/06/07/2020.06.07.138800.abstract
- Eskier D, Karakülah G, Suner A, Oktay Y. RdRp mutations are associated with SARS-CoV-2 genome evolution. PeerJ. 2020;8:e9587. https://doi.org/10.7717/peerj.9587 PMid:32742818 PMCid:PMC7380272
- Berrio
A, Gartner V, Wray GA. Positive selection within the genomes of
SARS-CoV-2 and other Coronaviruses independent of impact on protein
function. PeerJ. 2020;8:1-24. https://doi.org/10.7717/peerj.10234 PMid:33088633 PMCid:PMC7571416
- Starr
TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, et al.
Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals
Constraints on Folding and ACE2 Binding. Cell [Internet]. 2020 Sep
3;182(5):1295-1310.e20. https://doi.org/10.1016/j.cell.2020.08.012 PMid:32841599 PMCid:PMC7418704
[TOP]