Ugo Testa1, Germana Castelli1 and Elvira Pelosi1,*.
1 Department of Oncology, Istituto Superiore di Sanità, Rome, Italy.
Correspondence to: Dr
Ugo Testa. Department of Oncology, Istituto Superiore di Sanità, Viale
Regina Elena 299, 00161, Rome, Italy.
Published: July 1, 2023
Received: May 8, 2023
Accepted: June 1, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023038 DOI
10.4084/MJHID.2023.038
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
TP53-mutated
myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) form a
distinct and heterogeneous group of myeloid malignancies associated
with poor outcomes. Studies carried out in the last years have in part
elucidated the complex role played by TP53 mutations in the
pathogenesis of these myeloid disorders and in the mechanisms of drug
resistance. A consistent number of studies has shown that some
molecular parameters, such as the presence of a single or multiple TP53
mutations, the presence of concomitant TP53 deletions, the association
with co-occurring mutations, the clonal size of TP53 mutations, the
involvement of a single (monoallelic) or of both TP53 alleles
(biallelic) and the cytogenetic architecture of concomitant chromosome
abnormalities are major determinants of outcomes of patients. The
limited response of these patients to standard treatments, including
induction chemotherapy, hypomethylating agents and venetoclax-based
therapies and the discovery of an immune dysregulation have induced a
shift to new emerging therapies, some of which being associated with
promising efficacy. The main aim of these novel immune and nonimmune
strategies consists in improving survival and in increasing the number
of TP53-mutated MDS/AML patients in remission amenable to allogeneic
stem cell transplantation.
|
Introduction
Genetic classification of AML.
The myeloid malignancies form a group of related cancers generated by
the malignant transformation of hematopoietic stem/progenitor cells,
including acute myeloid leukemia (AML) and myelodysplastic syndromes
(MDS). AMLs form a heterogeneous group of hematological malignancies
characterized by a considerable complexity of molecular alterations,
clonal development, and consistent defects in cell
differentiation/maturation, associated with expansion of immature
leukemic elements.
Acute myeloid leukemia (AML) is a
heterogeneous and complex disease, characterized by the uncontrolled
proliferation of progenitor leukemic cells that progressively
accumulate and display variable degrees of differentiation blockade.
The incidence of AML is age-dependent, rising markedly at an age of ≥60
years, with a median age at diagnosis of about 68-70 years.[1,2]
The incidence of AML in Europe increased from 3.48 in 1976 to 5.06
cases per 100,000 people in 2013, a phenomenon at least in part related
to the ageing of the population.[3]
The
identification and classification of cellular and molecular
abnormalities occurring in AML was of fundamental importance for the
understanding of the pathogenesis of these leukemias and for the
development of a more rational approach for their treatment. Thus, the
initial classification of AML, the French-American-British (FAB)
classification was based on the evaluation of the hematopoietic cell
lineage of leukemic cells and of their differentiation stage, based on
cytological and cytochemical techniques. The development of techniques
in the study of cytogenetic abnormalities introduced new fundamental
criteria in the classification of AMLs, reflected in the World Health
Classifications of AML proposed in 2001 and 2008.[4,5]
AMLs
are a heterogeneous group of hematological malignancies, characterized
by a complexity of molecular alterations and clonal development. In the
last years, considerable progresses have been made in the
characterization of the molecular abnormalities underlying AMLs, with
the identification of recurrent chromosomal alterations and of gene
mutations, allowing the classification of these leukemias in various
subgroups, characterized by different genetic alterations and response
to current treatments.[6-9] This molecular
classification identified some major molecular subtypes: (i) AMLs
characterized by peculiar translocation events (balanced
rearrangements) leading to the formation of fusion genes and
correspondent fusion proteins, including inv(6), t(15;17), t(8;21),
inv(3), MLL fusions and t(6;9); (ii) AMLs exhibiting
chromatin-spliceosome gene abnormalities, including mutations of genes
involved in RNA splicing (SRSF2, SF3B1, U2AF1, ZRSR2),
chromatin and transcription; (iii) AMLs characterized by TP53
mutations, complex karyotype alterations and copy-number chromosome
alterations; (iv) AMLs displaying mutations of the nucleophosmin 1
(NPM1) gene; (v) AMLs characterized by double CEBPA mutation; (vi) AMLs
with IDH2R172 mutation, defined as a distinct subgroup for the mutual exclusivity with NPM1 mutation and other class-defining lesions.[8,9] AMLs with mutated RUNX1
have been included in the WHO classification as a provisional entity in
the category of AMLs with recurrent genetic abnormality.[10]
AMLs
were characterized in the context of other tumors, solid and
hematological tumors, by a relatively low number of mutations in coding
genes, but a high number of driver genes, of whom a part is related to
leukemia-specific driver genes and driver genes observed also in other
tumors.[11]
The genes most frequently mutated
in AMLs are represented by: mutations of the tyrosine kinase membrane
receptor Flt3, more frequently (about 30% of adult AMLs) with
Flt3-Internal Tandem Duplication (FLT3-ITD) and less frequently (about 10%) with FLT3-Tyrosine Kinase Domain (FLT3-TKD) mutations; mutations of the NPM1 gene observed in 30-35% of cases; mutations of the methyltransferase DNMT3A (DNA methyltransferase 3A) gene (20-30% of AMLs); NRAS (15-20% of cases); mutations of the transcription factor RUNX1 (15% of AMLs); the methylcytosine dioxygenase 2 ten-eleven-translocation (TET) TET2 gene (15-20% of AMLs); the isocitrate dehydrogenase 2 (IDH2) gene (10-15% of AMLs) and IDH1 gene (5-10%); mutations of the additional sex coombs-like 1 (ASXL1), a transcriptional regulator (10-20%); mutations of the transcription factor runt-related transcription factor 1 (RUNX1)
gene occurring in 5-15% of cases; mutations of the tumor suppressor
gene TP53, occurring in about 10% of cases; mutations of the
transcription factor CCAAT/enhancer-binding protein α (CEBPA) (10%); mutations of the zinger finger transcription factor Wilm’s tumor 1 (WT1) observed in <10% of cases; mutations of the enhancer of zeste homolog 2 (EZH2), a histone methyltransferase (5-10%); somatic mutations of the transcription factor GATA2 (<5%); mutations of the transcription factors BCL6 corepressor (BCOR) and BCL6 corepressor like 1 (BCORL1) (4%); mutations of the cohesion complex genes (SMC1A, SMC3, RAD21, STAG1, STAG2) occurring in 6-12% of cases; mutations of splicing factor genes (SRSF2, ZRZF2, ZF3B1, U2AF1) observed in about 18% of cases.[12]
The identification of genetic abnormalities in AMLs was of fundamental
importance for the understanding of leukemia pathogenesis, for the
identification of new therapeutic targets and for the identification of
biomarkers suitable to monitor the response to anti-leukemia therapy.[12]
Metzler
et al. explored the association of driver gene mutations with clinical
characteristics and cytogenetic alterations. The major findings of this
analysis showed that: DNMT3A and NPM1 mutations were more common in women than in men; RUNX1, SRSF2, ASXL1, STAG2 and BCOR were less common in women than in men; FLT3-ITD mutations were associated with high blast cell counts; mutations in SRSF2, ASXL1, STAG2, U2AF1, RUNX1 and PTPN11
are more frequent in secondary AMLs (sAMLs, AMLs developing from a
pre-existing myelodysplastic syndrome or a myeloproliferative disorder)
than in de novo-occurring AMLs; TP53 mutations were more frequent in therapy-related AMLs (tAMLs); mutations at the level of DNMT3A, FLT3, NPM1, IDH1, IDH2 and CEBPA are present predominantly at the level of patients with normal karyotype.[13]
According
to various molecular criteria, the European Leukemia Net stratified
AMLs into three risk subgroups, with favorable prognosis (comprising
t(15;17), t(8;21), inv(6), biallelic mutated CEBPA and NPM1 mutant (without FLT3-ITD), intermediate prognosis (encompassing NPM1 mutant with FLT3-ITDlow,
t(9;21) and various cytogenetic abnormalities not classified as
favorable or adverse) and adverse prognosis (comprising monosomy 7 and
5, deletion of long arm (q) chromosome 7, abnormalities of 3q, 17p and
11q, multiple cytogenetic abnormalities, NPM1 wt and FLT3-ITDhigh, TP53 mutations associated with complex karyotype, ASXL1 mutations, t(6;9) and t(3;3) groups.[14]
Importantly, a recent study by Herold and coworkers on 1116 adult AML
patients not selected by genetics validated the ELN-2017 classification
and showed that: (i) in 599 patients <60 years, the OS was 64% for
ELN-2017 favorable, 42% for intermediate-risk and 20% for adverse-risk
AMLs; (i) in 517 patients >60 years, corresponding 5-year OS was
37%, 16% and 6%.[15] Patients with biallelic CEBPA mutations or inv(16) displayed a good prognosis; in contrast, patients with TP53 mutations displayed a particularly poor outcome.[15]
Recently,
Fleming and coworkers proposed a machine-learning (ML) approach to
develop a hierarchical prognostic risk model that hierarchically
categorizes cytogenetic and molecular factors into groupings that
accurately predict survival.[16] This approach was
used to explore two large cohorts of AML patients: this ML approach
allowed to classify the analyzed AMLs into four prognostic groups: good
(30%), intermediate (26%), poor (26%) and very poor (18%); the ELN2017
classification evaluated these AML as: good (39%), intermediate (31%)
and poor (30%).[16] It is important to note that in
this system of AML prognostication a large number of molecular
parameters were taken in account: complex karyotype, inv(16), CEBPAdmut, inv(3)/t(3;3), FLT3-ITD, spliceosome mutations (U2AF1, SRSF2 or SF3B1), NPM1mut (in the absence of FLT3-ITD), t(8;21), MLL translocations, NRASmut, TP53mut, ASXL1mut.[16]
This evaluation system allowed the prognostication of many AML
subgroups: (i) in the group characterized by complex karyotype, the
presence of high-risk monosomies or chromosomal abnormalities or TP53 mutations have a very poor prognosis, whereas complex karyotype without these alterations have a better prognosis; (ii) CEBPAdmut AMLs have a good prognosis, particularly when associated with NRAS mutations; (iii) co-occurrence of FLT3-ITD and spliceosome mutations was associated with very negative outcome; (iv) FLT3-ITD high allelic ratio (>0.5) have a very poor prognosis when present in the absence of concomitant NPM1 mutations; (v) triple mutant NPM1/DNMT3A/FLT3-ITD display a poor prognosis: (vi) AMLs with spliceosome mutations display a poor prognosis when associated with ASXL1 mutations or ASXL1 heterozygous deletion; (vii) among NPM1-mutant AMLs, NRAS co-mutations identified a subgroup associated with good prognosis, whereas those associated with IDH1 mutations display an intermediate prognosis; (vii) the presence of KIT mutations in t(8;21) AMLs was associated with an intermediate prognosis.[16]
Recently,
a functional genomic analysis was performed on a large cohort of 562
AML patients based on whole exome sequencing, RNA-sequencing and ex
vivo drug sensitivity analyses.[17] This approach showed several relevant findings: (i) a sensitivity of FLT3-ITD mutant AMLs to FLT3 inhibitors; (ii) NRAS-mutant AMLs resistant to most of drugs, but sensitive to MAPK inhibitors; (iii) IDH2-mutant AMLs are sensitive to several drugs, whereas the contrary is true for IDH1-mutant AMLs; (iv) RUNX1-mutant
AMLs are sensitive to PIK3C/MTOR inhibitors; (v) AMLs with mutations of
spliceosome genes display a peculiar pattern of drug sensitivity; (vi)
triple mutant NPM1/FLT3/DNMT3A AMLs are sensitive to ibrutinib.[17]
This study was further extended through an integration of functional
genomic resources represented by molecular, clinical and drug response
data; this approach allowed to identify genetic and cell
differentiation state features that predict drug response.[18] Interestingly, modeling of clinical outcome revealed a single gene, PEAR1, among the best predictors of patient survival, particularly for young AML patients.[18]
Tazi
and coworkers, through the analysis of the genomic profile of 223 AML
patients, proposed a classification and risk-stratification. Clustering
analysis based on cytogenetic alterations and gene mutations allowed to
identify 16 non-overlapping clusters classifying 100% of patients. Some
cytogenetic subgroups were identified based on cytogenetic alterations.
One cytogenetic subgroup was defined by complex karyotype (≥3
unbalanced cytogenetic abnormalities), corresponding to about 10% of
all patients and characterized by frequent TP53
alterations (about 65%), paucity of other mutations, older age and poor
outcomes; another cytogenetic subgroup was characterized by the
presence of ≥13 trisomies (most frequently involving +8, +11, +13, +21
and +22), corresponding to about 2% of all AMLs and associated with
infrequent TP53 mutations (4%) and with a prognosis more favorable
compared to the complex karyotype subgroup, even when ≥3 aneuploidies
were present; patients with ≤2 aneuploidies (11% of all AML patients),
enriched for MDS-related total or partial monosomies, -7(7q-) or
-5(5q-) were clustered with sAML subgroups; other cytogenetic subgroups
are those characterized by the presence of translocation events, such
as t(15;17), t(8;21), inv(16), t(11;x), t(6;9).[19]
The sAML cluster is the second largest cluster (28.4% of all patients)
and is characterized by the presence of classifying mutational events,
including SRSF2, U2AF1, SF3B1, ZRSR2, ASXL1, EZH2, BCOR , STAG2 as well as RUNX1, SERTBP1 and MLLPTD;
the patients comprised in this cluster were characterized by an older
age, lower blast counts and higher incidence of antecedent
hematological disease (AHD) and displayed a different prognosis
according to the number of class-defining gene mutations. The sAML
cluster is subdivided into two subgroups: sAML like-1 with single
mutations (4.7% of all AMLs); sAML like-2 with ≥2 mutations (23.7% of
all AMLs) is enriched in AHD and is associated with worse outcomes; RUNX1 mutations were observed at similar frequencies in sAML1 and sAML2 subgroups.[19] WT1 mutations, when observed in the absence of concomitant CEBPAbi
and t(8;21), defined a distinct subgroup and represented about 2% of
all AMLs and involved patients of younger age and englobed two
prognostic subgroups, following the absence (intermediate risk) or the
presence (adverse risk) of concomitant FLT3-ITD mutations.[19] DNMT3A/IDH1 or IDH2
mutant AMLs represent a rare subgroup (1%) of AMLs and are associated
with adverse outcomes. 6% of patients, not clustering with any
class-defining molecular event, are classified as not otherwise
specified (mNOS). NPM1-mutant
AMLs represent the largest subgroup (31.8% of all AMLs) and display an
intermediate or adverse risk following their co-mutational status.
About 2% of AMLs displayed apparently not relevant mutational events.[19] FLT3 and NRAS
mutations are distributed in the various subgroups and are not
class-defining mutations. This genetic classification, together with
clinical criteria, allowed to define the probability of response and of
disease relapse for the various molecular AML subgroups. Thus, this
analysis supported a risk stratification of AML subgroups implying a
classification of (i) NPM1, inv(16), t(8;21), t(15;17), biCAEBPA and no events subgroups as a favorable-risk AML cluster; (ii) sAML1, t(6;9), mNOS, t(11;x), DNMT3A/IDH1-2 and trisomies is an intermediate-risk AML cluster; (iii) TP53-complex karyotype, sAML2 and inv(3) is an adverse-risk AML group.[18] The concomitant presence of FLT3-ITD in NPM1 subgroup induced the shift of a part of these AMLs from the favorable to the intermediate risk cluster; the presence of FLT3-ITD mutations in AMLs pertaining to the intermediate-risk group induced their shift to an adverse risk condition.[19]
Other
recent studies have provided a detailed molecular characterization of
AMLs with myelodysplasia-related changes (AML-MRC). Gao et al. reported
the results of the genomic profiling of 293 newly diagnosed AML
patients and observed that 28.5% of these patients displayed AML-MRC;
particularly, several notable differences in rate of mutation of genes
recurrently mutated were observed: the mutation rates of ASXL1 (25% vs 8.7%) NRAS (17.9% vs 8.1%), PTPN11 (11.9% vs 5%), SETBP1 (6% vs 0.6%), SRSF2 (11.9% vs 5.5%), TP53 (16.7% vs 1.2%) and U2AF1 (17.9% vs 7.5%) were higher in AML-MRC than in those without MRC, while the rates of FLT3-ITD (3.6% vs 15.5%), KIT (0% vs 6.2%), WT1 (3.6% vs 9.9%), NPM1 (1.2% vs 21.7%) and CEBPA (4.8% vs 24.2%) were lower in AML-MRC compared to those without MRC.[20] At clinical level, AML-MRC were characterized by older age, low WBC counts and inferior outcomes.[20]
Kang
et al. have evaluated 45 AML-MRC patients; genetic aberrations in these
patients were analyzed using an RNA-based NGS pane assay; using this
approach, 4 gene fusions of KMT2A-SEPT9, KMT2A-ELL, NUP98-NSD1 and RUNX1-USP42 were observed.[21]
AML-MRC patients have been classified into one of these three
subgroups: (i) patients with history of prior MDS or MDS/MPN
(AML-MRC-H); (ii) patients with MDS-defining cytogenetic abnormalities
(AML-MRC-C); (iii) patients with >50% dysplasia in at least two
hematopoietic lineages (AML-MRC-M).[20] 33% of AML-MRC-H, 56% of
AML-MRC-M and 96% of AML-MRC-C patients have complex karyotype
abnormalities. TP53 gene was the most frequently mutated gene in these patients and all these patients are included in the AML-MRC-C subgroup; ASXL1 and SRSF2 mutations were preferentially associated with the AML-MRC-M subgroup and were frequently co-mutated; IDH1-2 genes were also frequently mutated and their mutations are distributed in all three AML-MRC subgroups.[21]
The
evaluation of genomic profile of AMLs had a clinical value at
prognostic level. The presence of some genetic mutations had a clearly
negative prognostic impact: (i) a systematic analysis of the literature
data showed that in adult AML patients, the presence of TP53 mutations predicted inferior overall survival compared to patients TP53-WT [22]; (ii) a meta-analysis of literature data showed that AML patients with ASXL1 mutations have a significantly poor prognosis compared to those without mutations;[23] in intermediate risk AML patients, the presence of WT1 mutations was associated with a significantly increased risk of relapse after transplantation.[24] Secondary AML-like gene mutations other than ASXL1 (SRSF2, STAG2, BCOR, U2AF1, EZH2, SF3B1, ZRSR2) identify a subset of intermediate-risk AML patients (about one-third) with a worse outcome (shorter OS and EFS).[25]
The main aim of induction chemotherapy consists in achieving clinical
remission and a condition of negativity of measurable residual disease
(MRD), a key prognostic factor in AML. The analysis of a cohort of 211
AML patients molecularly characterized by NGS and studies for MRD by
immunophenotyping assay after induction chemotherapy and allogeneic
stem cell transplantation (allo-SCT).[26] 35% of patients achieved MRD-, 27% MRD+ and 38% persistent disease; after subsequent therapies 34% of patients with MRD+ and 26% of those with persistent disease achieved a condition of MRD-.[26] Mutations in CEBPA, NRAS, KRAS and NPM1 predicted high frequencies of MRD-, while mutations in TP53, SF3B1, ASXL1 and RUNX1 and karyotypic abnormalities (inv(3), monosomy 5 or 7) predicted low rates of MRD-.[26] Furthermore, patients with fewer individual clones have a higher probability of achieving MRD-.[26]
For patients who underwent allo-SCT, outcomes were favorable for those
who achieved a condition of MRD negativity early after induction
chemotherapy or after subsequent therapy.[26]
In
addition to studies of characterization of genomic alterations, the
gene expression studies have also contributed to capture and to define
the heterogeneity of AML disease, showing gene expression changes in
large part related to underlying genomic alterations. Particularly,
transcriptomic information helped to improve the ENL system of
prognostic evaluation of ELN system.[27] The whole
transcriptomic RNA sequencing HAMLET (Human AML Expedited
Transcriptomics) was established as a single, comprehensive, and
flexible platform for AML diagnostics; this platform allows the
simultaneous detection of fusion genes, small variants, tandem
duplications, and gene expression.[28] HAMLET showed the potential to provide accurate comprehensive diagnostic information relevant for AML classification.[28]
Using a base pairing approach, eliminating batch effects across
heterogeneous patient cohorts and transcriptomic data, Kong and
coworkers developed and immunity and pyroptosis-related
prognostic signature, consisting of 15 genes, that predicts
consistently and accurately AML patients’ survival, with a better
performance compared to other 10 existing signatures.[29]
Several
studies exploring gene expression profile of AMLs identified
transcriptomic signatures whose scoring may complement the European
Leukemia Net classification. Thus, through the analysis of genes
differentially expressed in different types of cytogenetically defined
AML subtypes, Nehme et al. identified 22 CODEG (commonly deregulated
genes) that provided a robust prognostic signature that was predictive
of outcomes of AML patients.[30] An artificial neural
network -based machine learning approach to a publicly available data
set for a large cohort of AML patients led to the identification of a
3-gene signature comprising CALCRL, CD109 and LSP1,
which was predictive of outcomes; this 3-gene signature separated the
AML patients classified following ELN 2017 into subgroups with
different risk probabilities and allowed the identification of AML
patients with high-risk features.[31] Docking et al.
used expression data derived from 145 AML patients to develop a novel
prognostic score strongly associated with patient outcomes; this risk
score combined with standard molecular guidelines, allowed the
re-stratification of more than 20% of AML patients into correct risk
groups.[32] Furthermore, this transcriptomic analysis
allowed to identify a subset of high-risk AML patients characterized by
dysregulated integrin signaling and TP53 or RUNX1 mutations, potentially treatable with inhibitors of focal adhesion kinase.[32]
Another
approach was based on the characterization of genes whose expression
was deregulated in leukemic stem cells (LSCs), the cells that initiate
and maintain the leukemic process and that, for their quiescent state,
are resistant to therapy and are responsible for relapse. Thus, Ng et
al. identified 17 genes that are differentially expressed in LSC+ cells fractions compared to LSC- cell fractions.[33]
The investigation of this LSC17 score in five independent cohorts of
AML patients showed its capacity to accurately predict initial therapy
resistance; furthermore, patients with high LSC17 scores showed poor
outcomes with current treatments, including allo-SCT.[33]
Bill and coworkers have evaluated the association between the LSC17
score status and the mutational profile in AML patients and showed that
some mutations are significantly less frequent in LSC17-genehigh compared to LSC17-genelow (biallelic CEBPA, GAT2, KIT), while other mutations were significantly more frequent in LSC17-genehigh patients that in LSC17-genelow patients (ASXL1, DNMT3A, FLT3-ITD, KMT2A, RUNX1, SRSF2, STAG2, TET2 and TP53).[34]
Furthermore, AMLs with complex karyotype or with inv(3) have much more
frequently a high LSC17-gene score; however, a part of patients with an
adverse risk following ELN2017 display a LSC17-gene score low.[34]
Importantly, two large cohorts of AML patients, one of younger (<60
years) and another one of older (>60 years) patients, showed that a
high LSC17 gene score was associated with a significantly shorter PFS
and OS compared to those with a low LSC17 gene score.[34]
Given the results of these studies, Ng and coworkers have developed the
LSC17 test in the context of a certified diagnostic laboratory, thus
generating a clinical grade test.[35] Values from the
LSC17 test to clinical outcome were established in a large cohort of
AML patients, thus determining a median assay value that can be used
for clinical risk evaluation of individual patients with de novo
diagnosed AML.[35] A recent study explored the
predictivity of the risk by LSC17 signature in a large cohort (1503
primary AMLs) of pediatric AML patients and provided evidence that
while LSC17 scores were prognostic for EFS and OS in every age whole
AML category (0-10 years, 10-18 years, 18-30 years), they were no
longer predictive of survival within established cytomolecular risk
groups.[36] Thus, it was identified a distinct
molecular signature, LSC4, englobing all the genes initially found to
be upregulated in adult LSCs [33], that was more predictive than LSC17 in pediatric AML cytomolecular subtypes.[36] The LSC47 signature contributed to build a robust relapse prediction model in pediatric AML patients.[36]
A
recent study reported the results of a transcriptome-based
classification of 655 Chinese AML patients and allowed the
identification through enhanced consensus clustering of 8 gene
expression subgroups (G1 to G8) with unique features. The first four
subgroups corresponded the well-known t(15;17) (G1), CBFB-MYH11 (G2), RUNX1-RUNXT1 (G3), biallelic CEBPA (G4);
The G5 subgroup (myelodysplasia-related/-like) included clinical,
cytogenetic and genetic features resembling secondary AML; most NPM1 mutations and KMT2A and NUP98 fusions clustered into G6-G8, displaying high expression of HOXA/B genes and various differentiation stages: HOX-committed (G6), HOX-primitive (G7) and HOX-mixed (G8).[37]
Importantly, each subgroup was associated with distinct prognosis and
response to therapy, thus supporting the clinical applicability of this
gene expression-based AML classification.[37]
Single
cell RNA sequencing studies carried out in the last years have
consistently contributed to defining the complex and heterogeneous
cellular hierarchies of AMLs. A fundamental study by van Galen and
coworkers, through a combination of transcriptomics and mutational
analyses in single cells from AML patients allowed to define the
existence of multiple functional cellular subsets and their associated
genetic drivers.[38]
The use of a machine learning classifier allowed to distinguish a
spectrum of leukemic cells corresponding at various stages of
differentiation, whose abundances greatly varied between patients and
between subclones in the same tumor. According to their transcriptional
profile six types of leukemic cells have been identified, including
HSC-like, Progenitor-like, GMP-like, Promonocyte-like, Monocyte-like,
DC-like. seven clusters (A to G) of AMLs have been identified: the
cluster A contained mainly t(15;17) AMLs and some FLT3-ITD
mutated AMLs and have a GMP-like transcriptomic profile; the cluster B
consisted exclusively of t(8;21) AMLs and shows a GMP-like
transcriptomic profile; the cluster F almost exclusively implies CBFB-MYH11 AMLs and displays high monocyte-like and DC-like scores; the cluster C involves TP53 and RUNX1
mutated AMLs and AMLs with complex cytogenetics and other cytogenetic
abnormalities and some AMLs with normal karyotype and shows high
HSC-like and Progenitor-like scores; the cluster G involves the same
AML types described for cluster C and also CEBPA-mutated
AMLs and displays a wide spectrum of differentiation types; clusters D
and E comprise a large number of AMLs and mainly involve AMLs with
normal karyotype, largely represented by NPM1-mutant
AMLs, but largely different in their cell type compositions, the
cluster D being enriched in undifferentiated HSC/Progenitor-like cell
signatures and englobes multiple FLT3-ITD mutant leukemias, while the cluster E was enriched for monocyte-like and DC-like cell signatures and harbored FLT3-TKD leukemias.[38]
The analysis of primitive AML cells at single-cell level showed that
these cells exhibit a dysregulated transcriptional program, involving
co-expression of stemness-related genes and of myeloid priming genes
and their number is associated with a negative prognosis.[38]
A
second study, in part based on single-cell studies, was performed by
Zeng and coworkers who provided an analysis of the cellular and
molecular heterogeneity of AMLs through the study of the complex
cellular hierarchies present in these leukemias.[39]
This study was based on a peculiar strategy through which the cellular
hierarchies of more than 1,000 AML patients were characterized by gene
expression deconvolution on bulk AML transcriptomes using single-cell
reference profiles of distinct AML stem, progenitor, and mature cell
types.[39]
Using this approach, 864 AML patient samples were analyzed, providing
evidence that clustering based on the composition of leukemia
hierarchies revealed four distinct subtypes; primitive (shallow
hierarchy, LSPC-enriched), mature (step hierarchy, enriched for
monocyte-like and cDC-like blasts), GMP (enriched by granulo-monocytic
progenitor-like blasts) and intermediate (balanced distribution). The
hierarchies of different AMLs were separated according to two principal
components (PC1 and PC2): PC1, spanning a continuum from primitive to
GMP and PC2, spanning from primitive to mature.[39]
Hierarchies generated by cytogenetic alterations are dispersed along
the primitive versus GMP axis, with adverse cytogenetic alterations
generating primitive hierarchies, while favorable cytogenetic
alterations generating GMP-enriched hierarchies.[39]
Cellular hierarchies generated by driver mutations and their
combinations were distributed along the primitive versus mature axis
(PC2), reflecting their effect on cell differentiation.[39]
The PC1 axis well captures patient prognosis with GMP-like enriched
class being predictive of favorable outcomes, while the primitive-like
enriched group being associated with poor outcomes.[39]
In contrast to PC1, the PC2 axis was not predictive of prognosis.
Hierarchy composition of AMLs consistently changes between diagnosis
and relapse with a clear increase of total LSPC populations at relapse.[39] The primitive to mature axis (PC2) correlates with ex vivo drug sensitivity.[39]
At the level of gene expression, the PC1 axis was well captured by the
LSC17 gene expression scoring assay; from the LSC17, through regression
on PC2, it was derived a LSC7 that captures the primitive>mature
axis and predicted drug sensitivity: a high LSC7 score predicted
sensitivity to drugs such as venetoclax and azacitidine active on
primitive AMLs, while a low LSC7 score predicted sensitivity to drugs
such everolimus or selumetinib preferentially active on mature AMLs.[39]
The identification of cellular hierarchies present in the different
AMLs represents an important tool to better understand leukemia
development and to predict and define drug sensitivity.[39]
De novo, secondary and therapy-related AMLs. AMLs can be classified into three different groups following their origin as de novo,
secondary (sAML) and therapy-related AML (tAML). sAML and tAML are
recognized as AML clinical subtypes. Following the WHO classification
of myeloid neoplasms, sAMLs are defined as AMLs occurring after an
antecedent myeloid neoplasia, such as a myelodysplastic syndrome (MDS)
or a myeloproliferative neoplasm (MPN), independently of the therapy
used for the treatment of these disorders. tAMLs are defined as AMLs
occurring as a late complication related to the mutagenic potential of
cytotoxic chemotherapy and/or radiotherapy for a neoplastic or
non-neoplastic disease.[40]
A Danish
population-based study carried on 3055 AML patients diagnosed in the
lapse of 13 years from 2000 to 2013 showed that 73.6% of cases
correspond to de novo AMLs. 19.8% to sAMLs and 8.3% to tAMLs.[41] tAMLs were mostly related to solid tumors or to lymphoproliferative disorders.[41]
An antecedent myeloid disorder (sAML) or prior cytotoxic exposure
(tAML) was associated with a reduced rate of complete remission and
decreased overall survival compared to de novo AMLs.[41]
Molecular
profiling studies of a large set of AML samples have identified four
different groups of mutations: secondary type mutations specific to
sAML (SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR and STAG2); de novo mutations (NPM1, CBF); TP53 mutations; pan-AML mutations (FLT3, NRAS, KRAS, RUNX1, CEBPA, GATA2).[42] Some remarkable differences been shown in the frequency of several molecular abnormalities between sAMLs, tAMLs and de novo AMLs, as well as between sAMLs and tAML: (i) the presence of mutations in SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR or STAG2 was specific for sAMLs; tAMLs frequently displayed TP53 mutations (23% of cases) and in a 33% of cases harbored secondary-type mutations in SRSF2, SF3B1, U2AF1, ZRS2, ASXL1, AZH2, BCOER or STAG2.[42]
Finally, the group of sAML showed a consistent degree of heterogeneity
with a first subset characterized by the presence of secondary type
mutations, a second subset characterized by the presence of de novo or pan-AML mutations and a third set characterized by the presence of TP53 mutations.
Nazha et al. confirmed through the analysis of a large set of primary and secondary AMLs that mutations of the genes DHX29, ASXL1, SF3B1, BCOR, PRPF8, CBL, BCORL1, EZH2, STAGF2, JAK2, U2AF1, TET2 are more specific for sAML, whereas CEBPA, IDH2, DNMT3A, NPM1 and FLT3 mutations are more specific for primary de novo AMLs.[43]
The cytogenetic profile showed that sAMLs were more frequently than
pAMLs classified as pertaining to an unfavorable risk category.[43]
Patients with tAML are older and display more frequently than patients
with pAML cytogenetic abnormalities including monosomal (-7, -5 or 5q-,
7q-) and complex karyotypes, events associated with a poor outcome.[44]
More recent studies on a large set of tAML patients confirmed the
decrease of the frequency of normal karyotype (30% vs 46%) and the
increase of complex karyotype (29% vs 16% in sAML, compared to pAMLs.[45]
tAMLs represent the most aggressive and chemo-resistant malignancies with a 5-year survival of <10%.[46]
The 2016 WHO classification of myeloid neoplasms classified the myeloid
neoplasms occurring after therapy, including tMDS, tMDS/MPN and tAML as
a unique clinical entity, called tMN (therapy-related myeloid
neoplasm).[10] Therefore, several studies have
considered tMDS and tAML together. As for tAMLs, tMDSs are observed in
patients treated for solid tumors (54%) or hematological disease (43%);
tMDSs are observed in patients treated with chemotherapy alone or
combined chemo-radiotherapy.[47] tMDSs compared to
pMDSs display a higher proportion of cases pertaining to high/very
high-risk scoring, a higher proportion of cases with multiple
cytogenetic aberrations, and shorter overall survival.[47]
Al mutational level, tMDSs show some remarkable quantitative
differences compared to pMDSs. Thus, Ok et al. reported a frequency of TP53 mutations higher in tMDS than in pMDS (35.7% vs 17.7%, respectively).[48] Lindsley and coworkers confirmed that tMDSs have a clearly higher frequency of TP53 mutations compared to pMDSs (38% vs 14%, respectively); they observed also that tMDSs display a lower mutational rate of SF3B1, ASXL1, U2AF1 and JAK2 mutations compare to pMDSs; finally, DNMT3A mutations were more frequent in tMDS compared to pMDS.[49]
Thus, although there are some remarkable quantitative differences
between tMDS and pMDS in cytogenetics, gene mutations and epigenetics,
there are no specific markers to distinguish between these two MDS
forms.[50]
The ELN2022 guidelines for myeloid
neoplasms introduced important changes to the AML classification
through the removal of the categories of AMLs with
myelodysplasia-related changes (AML-MRC) and therapy-related myeloid
neoplasms. These changes were based on two different criteria: (i) a
prior history of MDS or prior exposure to therapy are now considered as
only diagnostic qualifiers; (ii) genetic characteristics, rather than
clinical history, are mostly relevant in classifying different AML
subgroups.[51] According to the new proposed
classification, three different hierarchical groups are defined: (i)
mutated TP53 with VAF >10% (MDS/AML if blasts 10-19% and AML if
blasts >20%); (ii) AMLs with myelodysplasia-related gene mutation (ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1 and ZRSR2); (iii) AMLs with myelodysplasia-related cytogenetic abnormality.
The
current pathogenetic interpretation of tAML development implies the
origin from the expansion of clonal hematopoiesis clones due to the
mutagenic activity of cytotoxic chemotherapy or radiotherapy;
alternatively, new mutations occur in the normal HSC compartment and
progressively drive the leukemic process. The first mechanism seems to
play a major role in the development of tAMLs. Clonal hematopoiesis of
undetermined potential (CHIP) is a biological event associated with age
observed in healthy individuals and corresponding to the presence in
their blood/bone marrow of clonal mutations at the level of DNMT3A, TET2 and ASXL1 genes; a fraction of the individuals with CHIP develop an hematological neoplasm later.[52] In addition to the three genes mentioned above, mutations of the epigenetic modifiers IDH1 and IDH2 and of the splicing factor genes SF3B1. SRSF2 and U2AF1, of TP53 and JAK2
genes are also observed at the level of CHIP. Pre-AML cases of clonal
hematopoiesis are characterized by more mutations per sample, higher
mutant allele frequencies and enrichment of mutations in specific genes
(such as TP53, IDH1, IDH2, DNMT3A, TET2 and spliceosome genes).[53,54] Detection of clonal mutations ≥0.01 VAF identifies subjects at increased risk for developing AML.[55]
The cumulative analysis on CHIP mutations and on the risk of developing
AML suggests that the considerable variation observed in variant allele
frequencies among individuals is mainly driven by chance differences in
the timing of mutation acquisition combined with differences in the
cell-intrinsic fitness of variants: thus, CHIP development reflects a
stochastic process of acquisition of mutations by hematopoietic stem
cells and possible clonal expansion driven by some mutations with
increased fitness conferring selective advantage to mutant
hematopoietic stem cells.[56]
The observation
that CHIP-related mutations involve a set of genes frequently altered
in leukemia, supports the view that these mutations may confer an
increased fitness to hematopoietic stem cells. Evolutionary models of
CHIP evolution in the time suggest that each specific mutation carries
a fixed fitness advantage, and this may explain the different relative
proportions and clonal sizes of CHIP driven by different mutations.[56]
The longitudinal analysis of CHIP clones over time in old individuals
showed that more than 90% of clones expanded at a stable exponential
rate over the analysis period, with different mutations driving clearly
different growth rates, ranging from 5% (DNMT3A or TP53) to more 50% for SRSF2.[57] Different patterns of lifelong clonal behavior were observed in different individuals.[56] DNMT3A and TP53
mutant clones preferentially expanded early in life and expanded slowly
in old age, while splicing gene mutations drive clonal expansion only
later in life and TET2-mutant clones emerged across all ages.[57]
A
large screening of a Japanese BioBank cohort comprising 11,234 healthy
individuals (672 with subsequent hematological malignancy) provided
important information about the frequencies of various gene mutations
and their tendency to generate an hematological malignancy. This study
was based on targeted sequencing of major CHIP-related genes in
blood-derived DNA to assess the frequency of driver mutations/indels
and copy number alterations (CNAs).[58] The frequency
on individuals with CHIP in this population of >60 years of age was
41.5%; in individuals with CHIP, 67% displayed either mutations alone
or CNAs alone, 21% two alterations (either mutations or CNAs alone, or
concomitantly both mutations and CNAs), 7.5% and 2.3% three or four
alterations, respectively (predominantly mutations and CNAs).[58] DNMT3A, TET2, ASXL1, PPM1D, TP53, SF3B1 and SRSF2 were the most frequently mutated genes in CHIP; CHIPs bearing TP53, JAK2, ASXL1, SF3B1, U2AF1 and DNMT3A
mutations have the greatest proportion of co-occurring alterations; the
proportion of subjects with CNAs within CHIPs harboring TP53, JAK2, ASXL1, SF3B1, U2AF1 and DNMT3A was higher compared to other gene mutations.[58] The most relevant association between CNAs and mutations were those represented by TP53/17pLOH, DNMT3A/2pLOH, TET2/4qLOH and JAK2/9pUPD:
these mutations/CNAs association leads to biallelic alterations and
were associated with higher mortality related to hematological
malignancies.[58] In this cohort of individuals a
hematological malignancy was observed in 8.2% of CHIP-positive
individuals compared to 4.45% in CHIP-negative individuals:
interestingly, the limitation of the analysis only to myeloid
malignancies showed a frequency of 3.48% in CHIP-positive individuals
compared to 0.82% in CHIP-negative subjects.[58] A
part of the subjects with CHIP display abnormalities of blood cell
counts or isolated cytopenia or multiple lineage cytopenias; compared
to individuals with SNVs or CNAs alone, CHIP individuals with both
mutations and CNAs display a higher clone size and more abnormal blood
counts.[58] In this context, individuals with JAK2 mutations display high platelet counts and those with U2AF1 mutations show cytopenias of any type; furthermore, there is a clear association between tp53, U2AF1 and SF3B1 mutations and lower hemoglobin levels and lower platelet counts.[58]
Other studies have shown the co-occurrence of gene mutations and
chromosomal abnormalities in a part of CHIPs; some gene mutations, such
as TP53, PPM1D, DNMT3A, SRSF2, JAK2 and ATM, have a pronounced tendency to be associated with chromosomal abnormalities.[59] TP53 mutations are associated with chromosome 3, 5, 7 and 17 abnormalities, PPM1D with chromosome 7 and 17 abnormalities.[59] The association of gene mutations with chromosome abnormalities define CHIPS at high risk of leukemic progression.[58]
The study of a large cohort of individuals of two BioBanks led to
define two types of CHIPs: one type of CHIP with myeloid drivers
(M-CHIP, with DNMT3A, TET2, ASXL1, TP53, PPM1D, SRSF2 and S3B1 as most recurrently mutated genes) and another type with lymphoid drivers (L-CHIP, with DUSP22, FAF1, KMT2D, SYNE1, ATM and KMT2C
as most recurrently mutated genes); these two different types of CHIPs
are also distinguished by different recurrent chromosome abnormalities.[60]
In both types of CHIPs, the association of mutational events with
chromosome abnormalities defines a subset of individuals with increased
risk of developing myeloid and lymphoid malignancies, respectively.[60]
A recent study based on an exome screening of a very large population
of 40,208 carriers of CHIP, through the analysis of genome-wide and
exome-wide associations, identified 24 loci, whose germline variation
affects predisposition to develop CHIP.[61]
CHIP
is a risk factor for blood malignancies and particularly for developing
AML; however, it is unclear while some individuals who harbor CHIP
driver mutations progress, while other ones do not progress to AML is
still unclear. A recent study modeled the interaction between positive
and negative selection mechanisms observed in deeply sequenced blood
samples derived from patients who subsequently progressed to AML,
compared to those observed in normal individuals, using deep learning
and population genetics methodology.[62] This study
evidenced the existence of purifying selection operating in all
individuals and preventing disease-predisposing clones from rising to
dominance and from inducing a pre-leukemic process.[62] The balance between evolutionary pressures ultimately drives mutation dynamics and health outcomes in aging blood elements.[62]
An initial study by Wong et al. carried out in 4 tAML patients bearing in their leukemic cells TP53
mutations, showed that the same mutations were present in 0.0003-0.7%
of mobilized blood leukocytes or bone marrow 3-6 years before the
development of tAML.[63] In mouse bone marrow chimeras containing both WT and TP53(+/-) HSCs/HPCs, the TP53(+/-) HSCs preferentially expanded after exposure to chemotherapy.[63] According to the results of this study, it was suggested that TP53-mutant HSCs resist cytotoxic therapy and expand preferentially after treatment generating tAML.[63]
Two large studies by Gillis et al[64] and Takahashi et al.[65]
provided evidence that patients with CHIP in pre-treatment PB samples
have a significantly increased probability to develop tAML after
treatment. CHIP can be detected in 70% of patients with cancer who
subsequently developed tMN.[65] Not only gene
mutations, but also chromosome arm-level copy-number alterations are
detectable as CHIP and preexist before exposure of patients to
chemotherapy or radiotherapy.[66]
Some mutations
are recurrently observed in tAMLs and are related to the previous
therapy to which these patients were exposed. Thus, Coombs et al. have
assessed in 8,810 cancer patients with solid tumors the occurrence of
CHIP: CHIP was identified in 25% of these patients, 4.5% with
presumptive leukemic driver mutations (CH-PD).[67] PPM1D and TP53 mutations were associated with prior exposure to chemotherapy[67]
CHIP was particularly frequent in some tumors such as thyroid cancer
(possibly because of radioactive iodine exposure) and with the lowest
frequency in germ cell cancers (probably because of the younger age of
the patients with this malignancy).[67] Among the
most common solid cancers, the occurrence of CHIP is more frequent in
patients with lung cancer, seemingly because of the enrichment for
smokers among lung cancer patients.[67]
Another study confirmed that mutations in the DNA damage response regulator PPM1D (protein phosphatase Mn2+/Mg2+-dependent 1D) present in CHIP, are observed in about 1/5 of tAML patients and are correlated with cisplatin exposure.[68]
Cell lines with hyperactive PPM1D mutations expand to outcompete normal
cells when exposed to cytotoxic DNA damaging agents such as cisplatin
and this mechanism could be responsible for their elevated frequency in
tAML.[68]
A recent study explored a very large
set of cancer patients (24,439 individuals) and observed CHIP in 30% of
these patients: 68% of these patients had one mutation in CHIP and 32%
two or more mutations; the most frequently mutated genes were the
epigenetic regulators DNMT3A and TET2 and the genes involved in DNA Damage Response (DDR) pathway, including PPM1D, TP53 and CHEK2; 90% of the mutations observed in CHIP were classified as driver myeloid mutations.[69]
The spectrum of gene mutations observed in CHIP was similar in
different cancer types, except for DDR gene mutations, particularly of
the PPM1D gene, which were enriched in ovarian and endometrial cancers.[69] The presence of specific gene mutations was associated with some pathogenic events: (i) mutations of the spliceosome genes SRSF2 and SF3B1 were less frequent than other CH mutations and are clearly associated with age; (ii) CHIP mutations in the DDR genes TP53, PPM1D and CHEK2 were strongly associated with prior oncologic therapy; (iii) CHIP mutations in ASXL1 gene were strongly associated with smoking.[69]
Furthermore, the fitness associated with mutations in epigenetic
regulators or splicing regulators was not markedly modulated by
oncologic therapy.[69] The environmental factors most
strongly associated with development of CHIP myeloid driver mutations
are represented by radiation therapy, platinum (mostly carboplatin)
chemotherapy and exposure to topoisomerase II inhibitors.[69]
The characterization of the clonal dynamics of evolution of CHIP
mutations in 525 cancer patients in a median lapse time of 23 months
provided evidence that 62% remained stable, 28% increased and 10%
decreased in clonal size; the growth rate was most pronounced for CHIP
mutations in DDR genes.[69] The incidence of CHIP far
exceeds that of tAML and the main determinants of the risk of a CHIP to
transform into a therapy-related myeloid neoplasia are related to the
type of CHIP mutations (mostly TP53 and spliceosome genes SRSF2, U2AF1 and SF3B1 mutations), the number of CHIP mutations and clonal size.[69] As above discussed, TP53 is one of the mutated genes frequently involved in tAML: the analysis of 34 tMN seemingly evolving from CHIP displayed TP53 mutations in 44% of cases; 73% of these TP53-mutant tMNs displayed pre-tMN TP53
mutations; 73% of TP53-mutated tMNs showed complex karyotype
alterations, an event acquired at the level of neoplastic
transformation, but absent in pre-neoplastic CHIPs.[69]
To understand the mechanisms through which TP53
mutations may promote clonal hematopoiesis and the development of tAMLs
it is fundamental to analyze its possible function in the physiology of
normal HSCs. P53 was shown to be an important regulator of HSC
quiescence through the modulation of the expression of its target gens
Gfi-1 and Necdin.[70] Necdin knockout in mice induced
less quiescence and more proliferative activity of the HSC compartment;
necdin-null HSCs/HPCs displayed enhanced sensitivity to chemotherapy.[71] These observations supported an important role for necdin as a regulator of DNA damage response in HSCs.[70-71] TP53 regulates the quiescence of HSCs also through induction of p21, an effect inhibited by CDK19.[72] Mutant TP53 enhances the repopulating activity of HSCs; furthermore, expressing mutant TP53 expand in response to chemotherapy and radiotherapy, thus indicating a key role for mutant TP53 in regulating the response of HSCs to genotoxic stresses.[73]
A more recent study by Chen et al. elucidated the mechanisms through
which mutant TP53 promotes expansion of HSCs and HPCs. Mutant TP53 confers
a competitive advantage to HSCs and HPCs following bone marrow
transplantation and induces HSC/HPC survival and expansion after stress
induced by radiation.[74] At transcriptional level, mutant TP53
promotes in HSCs/HPCs an enrichment of HSC and AML signatures, which
are different from gene expression signatures regulated by WT-TP53.[74] In HSCs/HPCs expressing mutant TP53, EZH2 target genes are downregulated and this effect is due to the capacity of mutant TP53
to interact with EZH2 and to enhance its association with the
chromatin, thus increasing the levels of methylated histones (H3K27me3)
in genes involved in the regulation of OSPC self-renewal and
differentiation; as expected, genetic and pharmacologic inhibition of
EZH2 led to a decrease of the repopulating capacity of HSCs.[74] These observations supported a major role for epigenetic mechanisms in the mechanism of TP53-mediated effects on clonal hematopoiesis.
A
recent study showed that in some patients tMNs are preceded by a
condition of clonal cytopenia (tCC). tCC develops earlier after primary
diagnosis compared to tMN (34 vs 79 months, respectively) and more
frequently received radiation therapy (30% vs 8%, respectively) and
less frequently chemotherapy (62% vs 82%, respectively) compared to
tMN.[75] tCCs displayed a low rate of cytogenetic abnormalities with absent complex karyotype and chromosomic monosomies.[75] At the level of mutational profile, tCCs were enriched in TET2 and SRSF2 mutations compared to tMNs and less frequently displayed TP53 mutations compared to tMN.[75] At tMN progression, 44% of tCC patients showed clonal evolution.[75]
TP53-mutated MDS and AML
De novo MDS.
The molecular abnormalities present in MDS patients have been explored
in detail in the last years. These studies have shown that TP53 is
mutated in about 7-10% of MDS patients and is more frequently mutated
in patients with high-risk MDSs; these studies showed also that TP53-mutated MDSs are characterized by the frequent association with complex karyotype abnormalities, del(5q) and 17qLOH.[76] About 24% of TP53-mutated MDSs are low-grade MDSs; in lower risk MDS, TP53 mutations showed a lower VAF.[77]
MDSs
are mainly observed in older adults with a median age at diagnosis of
greater than 65 years; however, more rarely, MDSs are also observed in
younger adults of age between 20-50 years. The number of mutations
increases linearly with age and on average patients >50 years of age
have more mutations (particularly, TET2, SRFSF2 and DNMT3A mutations) than those >50 years old.[78] However, TP53 mutations represent a notable exception, being observed in more than 20% of MDS patients >50 years old.[78,79] These observations suggest that TP53 mutations represent early onset ancestral events in the genesis of MDSs.
TP53-mutated MDSs and AMLs represent a peculiar subset of hematological tumors. The frequency of TP53 mutations in de novo MDSs or AMLs under the age of 65 years is evaluated in the order of 5-10%. In MDSs, according to the TP53 mutational status three sets of patients were identified: 82% had one TP53 mutation, while 3% displayed two TP53 mutations and 0.1% three mutations; about 54% of patients with one TP53 mutation had loss of the wild-type allele, while only 13% of those with more than one TP53 mutation had loss of the wild-type allele; according to the mutational status and to allelic imbalance, one third of TP53-mutant patients displayed monoallelic mutations (single hit) and two third displayed multiple allelic targeting (multi hit) (Figure 1).[80] In multi hit patients, no residual TP53
activity was maintained. Multi hit patients displayed several
associations with complex karyotype, few co-occurring mutations
(co-mutations occur in less than 25% of cases), high-risk presentation
and poor outcomes; furthermore, multi hit state predicted risk of
leukemic transformation and of death (Figure 2).[80] Monoallelic TP53
patients were less cytopenic and displayed a lower frequency of bone
marrow blasts compared to multi-hit patients; furthermore, mono-hit
TP53 patients were enriched in lower risk MDS patients according to
IPSS-R and WHO criteria of classification; MDS 5q- predominantly
showed TP53 mono-hit, while patients with MDS-EB2 predominantly displayed a TP53 multi-hit. Monoallelic patients displayed outcomes and response to therapy like those observed in WT-TP53 patients (Figure 2).[80] Monoallelic TP53 mutations more frequently display co-mutations in other genes, particularly TET2 (29%), SF3B1 (27%), ASXL1 (16%) and DNMT3A (16%), as subclonal events playing a variable impact on outcomes.[80] Finally, a remarka-ble difference between the two subtypes of TP53-mutant
MDSs is that in multi hit state TP53 muta-tions are predominantly found
in the dominant clone, while in monoallelic TP53-mutant MDSs are mainly subclonal.[80] The differential effect of monoallelic and biallelic TP53 mutations in MDS clinical presentation and outcome are seemingly related to a dose-dependent effect of TP53 inactivation on genomic instability, as supported by the observation that biallelic TP53
alterations are associated with an increased number of chromosomal
aberrations and an increased frequency of complex karyotypes compared
to monoallelic TP53 mutations. Finally, the outcome of monoallelic TP53-mutated
MDSs is strongly influenced by the concomitant presence of comutations;
in fact, while monoallelic patients with no other driver mutations have
a 5-yr mOS of 81%, it was 36% for patients with one or two mutations,
26% for patients with three or four co-mutations and only 8% for
patients with five or more co-mutations; in contrast, the outcomes of
patients with multi-hit TP53 alterations is poor and not influenced by the presence and by the number of additional mutations.[80]
 |
Figure 1. Main molecular properties of TP53-mutated MDS. A: proportion of MDS patients bearing 1, 2 or 3 TP53 mutations. B: MDS patients according to the number of TP53 abnormalities are classified as monoallelic or biallelic, following the involvement of one or both alleles: the types of TP53 mutations, defined as missense, truncated or other mutations, as well as the VAF of TP53 mutations and the frequency of complex karyotype are shown. C: proportion of TP53-mutant MDS patients classified into four subgroups following the presence of a single TP53 mutation (1mut) or of multiple TP53 mutations (>1 mut) or of TP53 mutations+chromosome 17 deletions at the level of TP53 locus (Mut+Del) or of TP53 mutations + cnLOH of TP53 detected only by NGS (Mut + cnLOH). D: Frequency of chromosome monosomies observed in MDS samples classified as above (mean±SEM). E: VAF of TP53 mutations (median value) observed in four subgroups of TP53-mutated MDS, classified as above. F: Frequency of different types of Chromosome 17 abormalities at TP53 locus into three subgroups of MDS with TP53 alterations: 0 TP53 mutations, a rare subgroup, with absent TP53 mutations but with structural alterations affecting TP53 expression, 1 and 2 TP53 mutations. The chromosome 17 status at TP53
locus is defined as normal, deleted, cnLOH or isoq17 (isochromosome 17q
rearrangement). The data reported in this figure are issued from
Bernard et al.[80] |
 |
Figure 2. Association between molecular features of TP53-mutated MDSs and clinical parameters. A: Percentage of bone marrow blasts in MDS without TP53 mutations (TP53-WT) and with 1 or >1 TP53 mutations (mean value ± SEM); B: median OS in TP53-WT, TP53 1 mut and TP53 >1 mut (mean value ± SEM); C: frequency of MDS patients with very poor prognosis among TP53-WT, TP53-1 mut and TP53 >1 mut patients; D: 5-yr mean OS in TP53 1 mut patients subdivided into four subgroups according to the number of co-mutations.
|
The presence of TP53 mutations
divides MDSs with complex karyotypes (CK-MDSs) into distinct prognostic
groups. In a cohort of 359 CK-MDS patients, TP53 mutations were identified into 55% of these patients. TP53-mutated CK-MDSs have fewer co-mutated genes, such as ASXL1, U2AF1 and RUNX1
but are enriched for some chromosome abnormalities, such as del(5q)
chromosomal abnormality, monosomal karyotype and high karyotype
complexity, identified by the concomitant presence of 4 or more
chromosomal abnormalities.[81] The presence of TP53 mutations into CK-MDSs significantly reduced OS[81] (Figure 3).
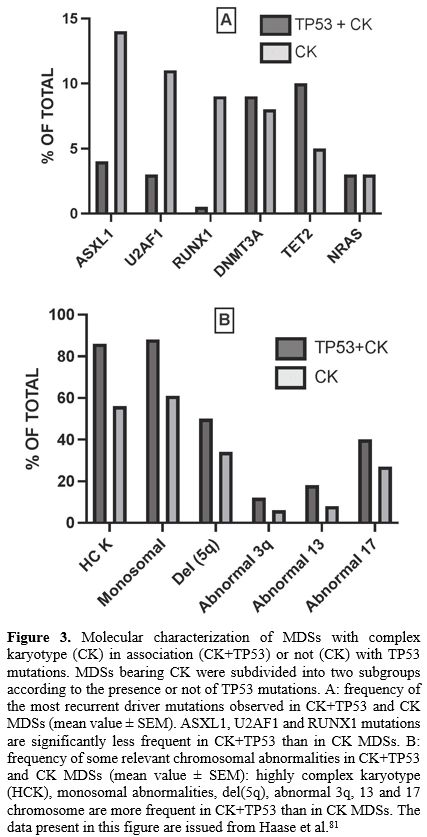 |
- Figure 3. Molecular characterization of MDSs with complex karyotype (CK) in association (CK+TP53) or not (CK) with TP53 mutations. MDSs bearing CK were subdivided into two subgroups according to the presence or not of TP53 mutations. A: frequency of the most recurrent driver mutations observed in CK+TP53 and CK MDSs (mean value ± SEM). ASXL1, U2AF1 and RUNX1 mutations are significantly less frequent in CK+TP53 than in CK MDSs. B: frequency of some relevant chromosomal abnormalities in CK+TP53
and CK MDSs (mean value ± SEM): highly complex karyotype (HCK),
monosomal abnormalities, del(5q), abnormal 3q, 13 and 17 chromosome are
more frequent in CK+TP53 than in CK MDSs. The data present in this figure are issued from Haase et al.[81]
|
TP53 mutations were detected in 18% of low-risk MDS with del(5q); among these patients, those with TP53 mutations had a significantly higher risk of AML evolution compared to those without TP53 mutations (50% vs 15%, respectively).[82] Crisà et al have evaluated TP53 mutations in MDS patients with isolated partial or total loss of chromosome 7 and observed a higher frequency of TP53 mutations among patients with 7q loss compared to those with 7 loss (9.8% vs 1.2%, respectively). The presence of TP53 mutations in these patients had a negative prognostic impact on overall survival.[83] TP53 mutations, together with ASXL1, RUNX1 and CBL mutations represent the mutations whose presence is associated with an increased risk of evolution to high-risk MDS or AML.[84]
Various studies have supported a prognostic role of TP53 VAF (variant allele frequency) in MDSs. In low-risk MDSs a TP53 VAF >6% was associated with shortened OS and inferior progression-free survival; in high-risk MDSs, the level of TP53 VAF clearly correlates with the occurrence of complex karyotype and a TP53 VAF >40% was an independent prognostic factor predicting reduced OS.[85,86] The study of a large cohort of 261 MDS patients with TP53 mutations confirmed the important prognostic role of TP53 VAF; 67% of these patients had 1 TP53 mutation, 29% had 2, 4% had 3 and 0.4% had 4; 37% of these patients had mutations in genes other than TP53; 83% of these patients had a complex karyotype and displayed a median TP53
VAF of 39%; the VAF of TP53 mutations in patients without a complex
karyotype was significantly lower than in those with complex karyotype
(5.1% vs 33.9%, respectively).[87] 32% of patients with TP53 mutations had concomitant TP53 deletions; patients with more than 1 TP53 mutations are less likely to have TP53 deletions than those with 1 mutation (9.3% vs 42.9%, respectively).[87] TP53 VAF level was associated with worse prognosis and patients with lower TP53
VAF respond better to therapy with hypomethylating agents (HMAs):
patients responding to treatment with HMAs showed a stable TP53 VAF just after therapy and a decreased TP53 VAF at the time of clinical response; patients not responding to HMAs showed an increased TP53 VAF after therapy.[87] The combination of TP53
VAF with the presence of complex karyotype defined a subgroup of MDS
patients with particularly poor prognosis. Increase in TP53 VAF was observed in 61% of patients at the time of leukemic transformation.[87]
A recent study reported the analysis of 2355 MDS patients, including 490 (21%) patients with TP53 mutations: of these, 78% were biTP53 and 22% maTP53. Median OS was worse for biTP53 subset compared ot the maTP53 subset (1 year vs 1.3 years, respectively); patients with maTP53 and those with biTP53 have a doubled and quadrupled risk of death, respectively compared to TP53-WT patients; compared to TP53-WT the risk of death was higher for TP53 with CK compared to TP53 without CK; among patients without CK, allelic TP53 mutational status had significant impact on outcomes (mOS of 2.8 years for maTP53 and 1.2 years for biTP53); among patients with CK, there was no survival differences between the maTP53 and the biTP53 subsets; among patients with low-risk MDS outcomes were worse for patients bearing TP53 mutations and there were no differences between maTP53 and biTP53.[88] These observations further supported the conclusion that TP53 mutant MDSs are a heterogeneous group, whose biological and clinical behaviour is influenced by TP53 allelic mutational status and cytogenetic architecture.[88]
The
use of the Evolutionary Action score (EAp53), a computationally derived
score to quantify the deleterious impact of different missense TP53
mutations on the basis of phylogenetic divergence of the mutated
sequence position and perturbation due to amino acid substitution,
allowed to define a scoring system ranging from 0 to 100, where a
higher score indicates a worse impact, and a 0 score indicates
wild-type function.[89] This analysis allowed the characterization of a large cohort of TP53-mutated MDSs with low-EAp53 score and a favorable prognosis.[89] Low-EAp53-MDSs have a lower frequency of multiple TP53 mutations and multi-allelic TP53 alterations, fewer cytogenetic alterations, and a lower frequency of complex karyotype and monosomal karyotype.[89] Furthermore, low-EAp53-MDS more frequently have co-mutations, involving partic-ularly NRAS and RUNX1 mutations.[89]
De novo AMLs. The pivotal study of TCGA on the molecular characterization of 200 de novo AML adult pa-tients, with an age of 55±16 years, reported a frequency of 8% of TP53
mutations, strongly associ-ated with unfavorable risk and with complex
cytogenetic abnormalities.[90] Bowen et al. explored 166 AML patients
with cytogenetic abnormalities and observed that 31% of these patients
had TP53 mutations; 97% of TP53-mutant AMLs had unfavorable cytogenetics and 53% of AML patients with complex cytogenetic abnormalities had TP53 mutations.[91] Rucker et al. explored 234 AMLs with complex karyotype for TP53 alterations: 60% of these patients had TP53 mutations and 40% had TP53 losses; in total, 70% of these patients displayed TP53 alterations. Furthermore, TP53-altered
AMLs more frequently exhibited a monosomal karyotype [-7/7q- (59%),
-5/5q- (77%), -11/11q- (13%), -12/12q- (32%), -18/18q- (34%) and -3/3p-
(29%)]. This study confirmed also that TP53
is the most frequently known altered gene in complex karyotype AMLs and
that patients with TP53 alterations were older and had significantly
lower remission rates, inferior event-free, relapse-free, and overall
survival.[92] Deletions in chromosome 7 (-7) or its long arm (7q-)
represent the most frequent adverse cytogenetic events in AML; TP53
and -5/5q are the most frequent co-occurring mutations and cytogenetic
abnormalities in this AML subset.[93]
TP53 aberrations
in AML include gene mutations, mostly involving the DNA binding domain
of the gene, and deletions of different sizes implying the TP53 locus at the level of chromosome 17p13. Functional studies on missense TP53
mutant variants commonly observed in AML indicate loss-of-function
effects and induction of effects comparable to those observed with
complete TP53 inactivation; these findings have suggested a dominant negative effect as the primary force of se-lection of TP53 mutations in myeloid malignancies.[94] In addition to somatic TP53 mutations, TP53 germline mutations are observed in a minority of AML patients and are more frequent in t-AML.[95]
The prognostic impact of different TP53 mutations is heterogeneous; in fact, Stengel et al. have explored a large cohort of TP53-mutated AMLs: TP53
mutations were detected in 13% of cas-es (mutation-only 7%; mutation +
deletion 5%; deletion - only 1%); all patients with TP53 mutations alone or in association with TP53 deletions, but not cases with TP53 deletions-only, were associated with a poor prognosis and reduced overall survival.[96]
A recent study reported the most extensive and detailed evaluation and molecular characteri-zation of more than 500 TP53-mutant AML patients.[97] About 75% of these patients harbored a TP53
missense variant, most frequently corresponding to mutations such as
R248, R273 and Y220; other genetic variants, including TP53 deletion, nonsense and frameshift mutations, were less frequent (Figure 4).[97] Furthermore, in 70% of cases a TP53 abnormality was associated with a TP53 copy-number loss.[97] The concomitant presence of a TP53 abnormality with a TP53 copy-number loss or of multiple TP53 mutations was associated with a worse prognosis[97]
Importantly, this study showed that mutant p53 protein expression
patterns by immunohistochemistry evaluated using digital-image-assisted
analysis provide an important tool integrating both TP53
mutation and allelic states in AML patients: some patients (44.5%)
displayed a mutant expression pattern characterized by high p53
expression (p53high) and a minority of patients (16.5%) showed a mutant expression pattern with absent p53 expression (p53truncated); other patients (39%), event in the presence of a mutant TP53 allele, displayed a normal p53 protein expression pattern (p53WT) (Figure 4).[97] These three groups greatly differed for their association with complex karyotype: 79% in p53high, 33% in p53truncated and 5% in p53WT; similarly, the response to therapy was also different with p53high achieving 18% of CRs, p53truncated 7% and 44% in p53WT.[97]
Genomic analysis of comutations in TP53-mutant AMLs shows a mutated
profile involving mainly mutations in genes involved in epigenetic
regulation such as DNMT3A and TET2, RAS-MPK signaling such as NF1, KRAS/NRAS and PTPN11 and RNA splicing such as SRSF2; this comutation profile was similar for frontline TP53-mutated patients and for those with therapy-secondary TP53-mutated AMLs and for those undergoing salvage treatment.[97] In patients with 1 TP53 mutation the most common co-mutations involved SRSF2, RUNX1 and ASXL1, while those with ≥2 TP53 mutations most com-monly displayed co-mutations involving KRAS/NRAS, PTPN11 and RUNX1.[97]
 |
- Figure 4. Main molecular properties of TP53-mutated AMLs. A: types of TP53 mutations present in TP53-mutated AMLs bearing 1 TP53 mutation; TP53 mutations were classified as missense, nonsense, frameshift, deletion and splice site. B: types of TP53 mutations present in TP53-mutant AMLs bearing >1 TP53 mutation. C: TP53-mutant AMLs were subclassified into two subgroups according to the presence or not of TP53 copy number alterations (TP53 copy number intact and TP53 CN loss): TP53 VAF was higher in TP53 CN loss than in TP53 CN intact AMLs; CK was markedly more frequent in TP53 CN loss AMLs than in TP53 CN intact; TP53 1 mutations are more frequent in TP53 CN loss than in TP53 CN intact; TP53 with >1 mutation are equally frequent in TP53 CN intact and TP53 CN loss AMLs. D: Immunohistochemical (IHC) classification of TP53-mutant AMLs, with the definition of three subgroups: p53high, p53truncated and p53WT. E: frequency of TP53 CN intact and of TP53 CN loss in p53high and p53truncated AMLs; frequency of AMLs with adverse prognosis and with complex karyotype in p53high and P53truncated AMLs. F: frequency of different types of TP53 mutants in p53high and p53truncated AMLs. The data present in this figure are issued from Takashori et al.[97]
|
Prochazka and coworkers have explored the clinical impact of subclonal TP53 mutations in AML patients.[98] These authors have explored 1537 AML patients (91.6% with de novo AML, 4% with sAML and 4.4% with tAML; 98 of these patients (6.4%) were found to harbor TP53 mutations: 62.2% of these TP53-mutant AMLs displayed a VAF (variant allele frequency) of >40%, 19.4% a VAF between 20% and 40% and 18.4% a VAF <20%.[98] The large majority of TP53 mutations in all three subgroups were missense mutations located in the DNA binding domain of the gene.[98] In either TP53-mutated
group, patients exhibited a lower rate of complete responses and
displayed a lower rate of event-free survival and of overall survival.[98] Another study confirmed the worse prognosis of TP53-mutant AML,
irrespective of the allele burden, including cases with VAF
<20%.[99] At the variance with the two previous studies, a more
recent study suggested a prognostic role of mutant TP53 VAF.[100] Thus, in a retrospective analysis on 202 de novo AML patients with a median age of 70 years it was shown that a TP53
threshold of 40% was predictive of a significant difference in OS, with
a median OS of 6.9 months in patients with VAF <40% and an OS of 5.5
months with VAF >40%.[100] Particularly, the TP53
VAF was predictive of response to cytarabine-based regimens, with a
median OS of 7.3 months in patients with VAF <40%, compared to a
median OS of 4.7 months in patients with VAF >40%.[100] The TP53VAF was also predictive of the response after HSCT.[100]
The prognostic role of TP53
allelic mutational status is reinforced also by the results of a
retrospective study on 983 adult AML patients enrolled in 3 different
clinical studies and treated with induction chemotherapy; 83 of these
patients displayed TP53 mutations, 14 moTP53 and 69 biTP53; biTP53 patients were associated with worse overall survival compared to moTP53 (2-year OS 4% vs 43%, respectively).[101] Importantly, moTP53
patients displayed an OS comparable to that observed in AML patients
classified as intermediate risk following the ELN 2017 risk
classification.[101]
It is important to note that many TP53-mutated
AMLs are classified as AML-MRC. Particularly, in the context of
AML-MRC, the AML-MRC-C subtype is particularly enriched in TP53 mutations (40-55%), while the AML-MRC-H and AML-MRC-M subtypes more rarely display TP53 mutations.[102,103] It is of interest to note that AML-MRC-C subgroup is heterogeneous in that it can be subdivided into TP53-mutant and TP53-WT cases: the TP53-mutant cases have a lower rate of mutations of RNA splicing genes and of ASXL1, BCOR and EZH2 genes compared to those TP53-WT.[102,103] TP53-mutant
AML-MRC-C are associated with cytogenetic abnormalities in 5q, 7q, 17p
and complex karyotype and are associated with poor outcome,
independently of their multi-hit or single-hit TP53 mutational status.[102,103]
There are some remarkable differences in the definition of mono-hit and multi-hit TP53 alterations following either the ICC classification [104] or Grob et al..[105] Multi-hit TP53 mutations were defined by ICC as ≥2 distinct TP53 mutations (VAF >10%) or a single TP53
mutation associated with either: (i) cytogenetic deletion involving
chromosome 17p (del(17p) or monosomy 17; (ii) a VAF >50%; any
complex karyotype.[104] Grob et al. defined multi-hit TP53 mutations as: (i) ≥2 TP53 gene variants irrespective of VAF; (ii) ≥1 TP53 gene variant co-occurring with a cytogenetic deletion involving chromosome 17; (iii) TP53 mutations with VAF >55%.[105] A recent study[106] evaluated the potential prognostic impact of TP53
mutations classified as multi-hit or mono-hit according to both the
criteria above reported in a cohort of AML/MDS patients randomized to
receive azacitidine + durvalumab (anti-PDL1 antibody) or azacitidine
alone; these 205 patients included 61 TP53-mutated MDS/AML cases [107-108]. Since there was no difference in the response to these two treatments,[107-108] the patients were pooled in an unique analysis.[106] The results of this analysis showed that outcomes of MDS/AML patients with TP53 mutations are worse compared to TP53-WT, without any significant difference between mono-hit or multi-hit status as defined by either the ICC or Grob et al..[106]
TP53 mutations
in AMLs are associated with some copy number alterations, allowing to
identify subsets of these patients associated with a very-high risk
condition. Ets-regulated gene (ERG) amplification is an event observed
in 4-6% of AMLs and is associated with unfavorable prognosis. ERG amplification was related to cytarabine resistance.[109] EGR amplification was found to be associated with some chromosome aberrations, including chromotripsis and with TP53 gene alterations.[110] The association of ERG amplification with biallelic loss of TP53 identified a high-risk subgroup of AMLs with a median overall survival of only 2.5-3.8 months.[111]
Chromotripsis
is a catastrophic event generating multiple genetic alterations
reflected by an oscillating pattern of DNA copy number levels in one or
few chromosomes. Chromotripsis is an event frequently observed in some
tumors. At the level of AMLs, chromotripsis was observed in AML with
complex karyotype (34.5% of cases) and was strongly associated with TP53 mutations, monosomal karyotype, -5/5q- abnormalities; particularly, CK-AML with chromotripsis displayed a frequency of TP53
mutations of 85%, compared to 53% in CK-AML without chromotripsis.[112]
The presence of chromotripsis was associated with a particularly poor
outcome.[112] Bochtler et al. observed the occurrence of chromotripsis
in about one third of AMLs associated with chromosomal abnormalities;
the chromotripsis-positive cases were characterized by a particularly
high degree of karyotype complexity, TP53
mutations, and dismal prognosis.[113] A screening of 395 newly
diagnosed AMLs showed the occurrence of chromotripsis in 6.6% of
cases, in association with typical features of chromosomal instability,
including TP53
alterations, 5q deletion, higher number of CNAs, complex karyotype,
alterations in DNA repair and cell cycle, and focal deletions on
chromo-somes 4, 7, 12, 16 and 17.[114]
A recent study reported the whole genome sequencing of 42 TP53-mutated AMLs and provided evidence that most cases (94%) display TP53
mutational events.[115] Furthermore, most of cases displayed aneuploidy
and chromotripsis.[115] Recurrent structural variants affected
chromosome regions that affect ETV6 on chr12p (45% of cases), RUNX1 on chr21, and NF1 on chr17q; interestingly, ETV6 transcript expression was low in TP53-mutated
myeloid malignancies with and without structural rearrangements
involving chr12p.[115] Finally, telomeric content was found to be
increased in TP53-mutated MDS/AML compared to other AML subtypes.[115]
Nguyen
et al. have analyzed the TCGA dataset on AML patients and through a
multi-omic clustering approach have identified three primary clusters;
one of these clusters was characterized as a very high-risk molecular
subgroup (HRMS), with only about 10% survival at 2 years.[116] At
mutational level, this subgroup was characterized by a high TP53 mutation rate (56%) and a low NPM1
mutation frequency (4%).[116] Furthermore, this high risk AML cluster
was characterized also by high expression of E2F4, CD34, CD109, MN1,
MMLT and CD200 genes.[109] Multi-omic pathway analysis using RNA
expression and CNA data identified in the HRMS group over-activated
pathways involving immune function, cell proliferation and DNA
damage.[116]
The frequency of TP53
mutations was higher in older AML patients compared to younger AML
patients. It is important to note that the mutational pattern of older
AML patients was consistently different from that observed in younger
AML patients, with: (i) some mutations, such as those of TET2, SRSF2, AXL1, RUNX1 and TP53 genes being more frequent in older than in young patients; (ii) other mutations, such as FLT3-ITD and WT1, being less frequent in older than in younger AML patients; (iii) other mutations, including NPM1 and DNMT3A mutations,
have similar frequencies in older and younger AML patients.[117-120]
Particularly, Tsai et al. reported a frequency of TP53 mutations of
4.2% in a group of patients with a mean age of 40 years, compared to
13% in a group of older AML patients with a mean age of 71 years.[117]
Prossek et al. reported frequencies of TP53 mutations of 5%, 14% and
14.5% in a group of patients of less than 60 years, 60-74 and >75
years old, respectively [120]. The increase of the frequency of TP53
mutations with the age of AML patients is not surprising given their
role in clonal hematopoiesis of indeterminate potential (CHIP); somatic
mutations of TP53 are among the five gene most frequently mutated in
CHIP and their presence contributes to the progression of CHIP.[74]
TP53 mutations in acute erythroid leukemia. Acute
erythroleukemia (AEL) is a rare subtype of AML, defined on the basis of
the presence of a high frequency of erythroblasts, including at least
30% of proerythroblasts, associated with recurrent TP53 mutations. In
the International Consensus Classification pure erythroid leukemia is
no more recognized as a separate entity and is instead included in a
broader category of AMLs with mutated TP53;[104] in the 5th
edition of the WHO classification of myeloid disorders, the WHO changed
the terminology of pure erythroid leukemia with acute erythroid
leukemia.[121] A key study by Iacobucci and coworkers reported the
first detailed molecular characterization of AEL and defined five
age-related subgroups: adult, including TP53 mutated, NPM1 mutated, KMT2A mutated/rearranged and DDX1 mutated; pediatric, including NUP98 rearranged.[122] The molecular characterization of TP53-mutated AEL, corresponding to 35.9% of all cases, showed that all but one of the TP53-mutated cases displayed alterations of both alleles; TP53 mutations were predominantly missense mutations occurring in the DNA binding domain and were associated with a poor prognosis.[122]
It is important to note that this study involved different types of
AMLs implying a significant involvement of the erythroid lineage. A
more recent study based on the molecular char-acterization of a similar
range of erythroid-associated AML subtypes confirmed the findings
observed in the previous study.[123] Importantly, this study showed
also that TP53-mutated AELs are characterized by the presence of gain/focal amplifications involving the EPOR, JAK2 and ERG/ETS,
with about 60% of these patients harboring one or more than one of
these lesions.[123] It is of interest that most of AELs
expressing TP53 mutations in association with EPOR, JAK2 and ERG/ETS CNAs correspond to cases of pure erythroid leukemia.[123] The association between TP53 mutations and EPOR/JAK2
gains and amplifications underlines a sensitivity to JAK2 inhibitors in
preclinical models.[123] In line with this last finding, two different
reports showed that virtually all cases of pure erythroid leukemia
display biallelic TP53 alterations.[124-125]
TP53 mutations in therapy-related MDS and AML. As
above reported, therapy-related myeloid neoplasms (tMN) represent a
dramatic conse-quence of cancer therapy and develop 3 to 7 years after
treatment with chemo- and radiation therapies (CRTs) and typically
present in a form of tAML or tMDS and are frequently associated with
poor prognostic features, such as complex karyotypes and TP53
mutations. tMDS and tAML are characterized by a peculiar mutational
profile: in tAMLs TP53 resulted to be the gene most frequently
mutated, in the range of 25% to 58% of cases.[46,63,126-129] Ok and
coworkers have characterized the TP53 mutational spectrum in 35
tAML/tMDS patients and observed that: TP53 mutations were mainly
clustered at the level of DNA-binding domains, with an allelic
frequency of 37%; missense mutations were the most frequent, followed
by frameshift and nonsense mutations.[129] This TP53 mutational pattern
was highly comparable to that observed in de novo AMLs/MDSs.[130]
Lindsley
et al. have evaluated 1514 MDS patients undergoing hematopoietic stem
cell transplantation and observed a remarkable difference in the
frequency of TP53 mutations among tMDS compared to pMDS (38% vs 14%,
respectively).[49] Interestingly, in tMDS patients it was identified
also a higher frequency of mutations of PPM1D, a regulator of TP53, in
tMDS compared to pMDS (15% vs 3%, respectively); thus, 46% of tMDS
patients display TP53 or PPM1D mutations.[49] PPM1D gene encodes a
serine-threonine phosphatase, involved in the cellular response to
environmental stress. PPM1D mutations alone in MDS did not show any
significant association with complex karyotype, while TP53 mutations
are strongly associated with complex karyotype.[49] The proportion of
patients with tMDS was about 12% among patients without TP53 and PPM1D
mutations, about 40% in patients with TP53 mutations without PPM1D
mutations, about 50% among patients without TP53 mutations and with PPM1D mutations and about 55% among patients with both TP53 and PPM1D
mutations.[49] TP53-mutated MDS patients, independently of their age,
have a clearly reduced overall survival after transplantation compared
to those without TP53 mutations.[49]
Hiwase and coworkers have
analyzed 245 tMDS and 132 tAML patients; 123 of these patients had TP53
mutations with VAF >10% or loss-of heterozygosity or copy neutral
LOH involving the TP53 locus: 21 of these patients were classified as
single-hit and 102 as multi-hit.[131] Overall survival was not
significantly different between single-hit and multi-hit TP53-mutant
patients; furthermore, there was no difference in the incidence of
progression in AML between single- versus multi-hit TP53-mutant
tMDS.[131] These observations suggest that the TP53 VAF of 10% is a
clinically useful threshold to identify patients with poor outcome.[131]
The
analysis of 118 tAML/tMDS patients with complex karyotype confirmed the
very strong association between this chromosomal abnormality and TP53
mutations (90%).[132] Conversely, patients with tAML/tMDS with normal
karyotype show distinct genomic and clinical characteristics compared
to their counterparts with abnormal karyotype, characterized by a
markedly lower frequency of TP53 mutations.[133,134]
Interestingly,
some recent studies have shown some remarkable differences in the
cellular and molecular mechanisms of development of tMN in pediatric
cancer patients. The mutational profile of pediatric tMN is different
from that described in adult TMN and is characterized by frequent
Ras/MAPK pathway mutations (KRAS, NF1 and NRAS mutations), RUNX1
mutations and KMT2A re-arrangements, while TP53 mutations are less
frequent (6%).[135] At variance with the results observed in adult tMN
patients, no evidence of pre-existing minor tMN clones, including also
those with TP53 mutations, was observed; in the majority of cases, tMN
development was related to the generation of mutant clones arising as a
consequence of cytotoxic therapy.[135] In line with this study, a
report on three pediatric neuroblastoma patients developing tMN showed
that clonal hematopoiesis, mainly consisting of platinum-induced
mutation and no driver myeloid genes, preceded the development of tMN
that arose after the acquisition of driver mutations.[136] A recent
study provided additional support to the role of chemotherapy in
promoting directly or indirectly mutations at the level of the HSPC
compartment, responsible for tMN development.[136] Thus, Bertrams and
coworkers have explored mutation accumulation in HSPCs before and after
cancer therapy in 24 children and observed that post-treatment HSPCs
have a considerable increase of mutation burden, comparable to what
treatment-naïve cells accumulate during 16 years of life.[136]
Particularly, chemotherapy may be mutagenic for hematopoietic cells
through three different mechanisms: directly to all exposed cells by
DNA cross-linking; directly to dividing cells by base analogue
incorporation; indirectly, by mimicking aging processes.[137] Drugs
such as platinum-based drugs induce clock-like processes, mimicking in
an accelerated way normal aging events.[137] Phylogenetic
reconstruction of tumor development in these children showed that tumor
tMN in children originate after the start of treatment and leukemic
clones become dominant after chemotherapy exposure.[137] It is
important to note that analysis of individual HSPCs purified from
human subjects from the birth to 81 years of age showed that these
cells spontaneously accumulate a mean of 17 mutations per year after
birth and lost 30 base pairs per year of telomere length.[138]
Interestingly,
TP53 germline pathogenic or likely pathogenic variants were identified
in 13 of 84 children with a tMN.[135] This observation suggests that
individual predisposition to cancer development may play a role in tMN.
In
conclusion, mutations of TP53 are the single most frequent molecular
abnormality of tMN and are frequently associated with complex
karyotype.[139] TP53 mutations represent one of the major challenges to
ameliorating outcome in t-MN and their presence in tMN is associated
with short duration of clinical responses.[139]
The role of TP53 mutations in the development of therapy-related myeloid neoplasias. Two
models have been proposed to explain the development and the role of TP53 mutations in tMN. Following one model, chemo-radiotherapy induces
the development of mutations at the level of HSPCs. However, following
the other model, chemo-radiotherapy promotes clonal selection of
pre-existing mutant HSPCs. Several observations support the second
model: the mutational burden of pAML and of tAML is comparable; somatic
mutations present in tMN are detectable years before chemo-radiation
exposure; somatic mutations in genes involved in the DNA damage
response, such as TP53 and PPM1D, are enriched in the blood of patients
exposed to chemo-radiotherapy.
Several studies carried out in
cancer patients undergoing autologous stem cell transplantation (ASCT)
or allogeneic stem cell transplantation (alloSCT) have provided
evidence in favour of tMN generated from CHIP bearing mutant genes.
Thus, Gibson and coworkers showed that in lymphoma patients undergoing
ASCT the presence of CHIP was associated with an increased risk of
developing tMN post-transplantation; in 8 of these patients developing
tMN, 4 displayed TP53 mutations and 2 PPM1D mutations and these
mutations were maintained in tMNs.[140] An analysis of 565 Danish
lymphoma patients undergoing ASCT showed that 25.5% of these patients
displayed CHIP; the global overall survival of patients with CHIP was
not significantly inferior to that of those without CHIP in a
multivariate analysis; however, patients with mutations in TP53 and PPM1D (corresponding to 35% of all patients with CHIP) had a
significantly lower overall survival, in part due to increased rates of
therapy-related leukemia.[141] Liu et al. have explored 362 patients
with diffuse large B-cell lymphoma (DLBCL) and observed that 29% of
these patients displayed CHIP.[142] 7 of these patients developed tMN
with a lapse of 6 to 30 months after therapy: all these patients have
evidence of CHIP with mutations observed also in tMN; 3/7 of these
patients showed CHIP bearing TP53 mutations expanding at the time of
tMN development.[142]
Several studies have explored the mechanism
of tMN in multiple myeloma (MM) patients undergoing ASCT. An initial
study carried out on 6 MM patients undergoing ASCT and developing tMN
showed the presence in the stem cell-enriched bone marrow fractions of
leukemia-associated mutations (TP53 mutations in 5/6 cases) years
before the development of tMN.[143] Mouhieddine evaluated a large
cohort of 629 MM patients undergoing ASCT: 21.6% of these patients
displayed CHIP and 19.8% of these CHIP-positive patients displayed TP53
or PPM1D mutations.[144] The longitudinal analysis on 14 patients who
developed tMN showed that 6 displayed CHIP with TP53 mutations and 9
showed TP53 mutations at the level of tMNs.[144]
It is important
to note that TP53 and PPM1D mutations have a negative impact in
lymphoma and MM patients undergoing ASCT not only because they confer
an increased risk of tMN develop-ment, but also because their presence
is associated with a reduced stem cell mobilization at the time of
pre-transplantation setting.[145] In line with these observations,
Berger and coworkers have studied 18 lymphoma/MM patients developing
tMN after ASCT; 70% of these patients displayed CHIP and in 85% of
cases tMN mutations were observed in CHIP.[146] Importantly, these
patients displayed an impaired stem cell mobilization at the time of
transplantation and a delayed regeneration after transplantation.[146]
7 patients were analyzed longitudinally after transplantation and
showed initial signs of clonal expansion in the lapse of time comprised
from 1 to 14 years after transplantation; in 4 of these 7 patients, the
expansion of TP53-mutated cells was observed.[146]
The study of
Bolton et al. provided evidence about the evolution of CHIP mutations
under the effect of anti-cancer therapy through the evaluation of two
large cohorts of untreated and treated cancer patients, showing that
the frequency of CHIPs bearing DNA damage response genes TP53, PPM1D
and CHEK2 is strongly associated with previous exposure to cancer
therapy.[68] In contrast, the frequency of CHIPs defined by mutations
in epigenetic modifiers, such as DNMT3A and TET2, and by spliceosome
regulators, such as SRSF2, SF3B1 and U2AF1, was not significantly
modified by anti-cancer treatments.[68] Mutations in TP53 and PPM1D
were most strongly associated previous exposure to platinum or
radionuclide or taxane therapy.[68] Serial sampling analysis clearly
showed that cancer therapy select for clones with mutations at the
level of TP53, PPM1D and CHEK2 and that these clones have lower
competitive fitness in the absence of cytotoxic or radiation
therapy.[68] The longitudinal analysis of 35 cancer-treated patients
developing tMN showed that in all these cases the CHIP mutation was
present at the time of tMN diagnosis; however, leukemic transformation
was associated with the acquisition of additional mutations (such as
FLT3, NRAS, KRAS mutations) and chromosomal abnormalities.[68] 40% of
tMN patients displayed TP53 mutations in 10/14 cases already present at
the time of CHIP testing; after therapy, the TP53 clone consistently
attained dominance by the time of tMN development and acquired
chromosomal abnormalities.[68] In conclusion, the hematopoietic cells
harboring TP53 mutations are positively selected when exposed to
anticancer therapy and may attain clinical dominance with acquisition
of additional mutational events and chromosomal aneuploidies.
The
study of gynecologic tumor patients undergoing chemo-radiation
treatments further sug-gested a key role of pre-existing TP53 mutant
CHIP clones in the development of tMN. Among gynecological cancer
patients, it was estimated that 24% of ovarian cancer, 23% of breast
cancer and 25% of endometrial cancer patients have CHIP.[67] Recently,
Weber-Lassalle reported the study of 448 ovarian cancer (OC) patients
(249 at primary OC diagnosis and 199 at platinum-sensitive
recurrence); 17.4% of these patients displayed at least one CHIP gene
mutation: DNMT3A (7.3%), PPM1D (6.6%), TET2 (2.6%), ASXL1 (1.8%) and
TP53 (1.5%) were the genes most frequently mutated.[147] TP53 and PPM1D
mutations were observed only in patients who received at least one
previous line of carboplatin treatment; TP53 mutations correlated with
the number of previous lines of platinum treatment but not with age or
BRCA mutational status.[147]
PARP inhibitors have been approved by
FDA as frontline maintenance for BRCA-associated advanced stage
ovarian cancer and have demonstrated an improvement in relapse-free
survival. Kwan et al. explored ovarian cancer patients enrolled in the
ARIEL2 and ARIEL3 trials involving treatment with the PARP inhibitor
Rucaparib.[148] 20 of these patients developed tMN after Rucaparib
treatment: 45% of these patients displayed CHIP in their blood,
compared to a frequency of 25% among patients not developing tMN.[147]
Interestingly, all tMN developing patients display TP53 missense
mutations; CHIP TP53 mutations were significantly less frequent in
patients not developing tMN (13.6% in those not developing tMN compared
to 45% in those developing tMN).[148] Patients with TP53 variant CHIP
have a significantly longer prior exposure to platinum-based therapy.
The longitudinal analysis of 5 tMN-developing patients showed a marked
increase of TP53 VAF after rucaparib treatment and before tMN
development.[148]
Other studies have shown an increased incidence
of tMN in gynecologic cancer patients treated with PARP inhibitors. In
a meta-analysis of 28 randomized controlled trials, Morice et al.
reported an incidence of myeloid malignancies with PARP inhibitors of
0.73% compared to 0.47% observed in controls.[149] The study of tMNs in
breast cancer or ovarian cancer patients treated with PARP inhibitors
showed a particularly high frequency of TP53 mutations estimated in the
order of 70-75%.[150-152] Martin et al. have compared the occurrence of
CHIP among ovarian cancer patients treated or not with PARP inhibitors
as maintenance therapy and observed a higher frequency in those treated
with PARP inhibitors compared to those treated without PARP inhibitors
(78% vs 18%, respectively); the frequency of TP53 mutations was higher
in CHIP of those treated with PARP inhibitors compared to those treated
without PARP inhibitors (64% vs 14%, respectively), while the
frequency of PPM1D mutations was similar in these two groups of
patients (50% vs 43%, respectively).[152] These
observations have been confirmed by Bolton and coworkers who explored
10,156 cancer patients for CHIP in their blood and found that patients
exposed to PARP inhibitors have an increased frequency of CHIP (33%)
compared to that observed in cancer patients undergoing other
anticancer treatments (18%) or not yet treated at the moment of blood
draw (16%).[153] Studies carried out in experimental models led to the
conclusion that the increased frequency of CHIP observed in PARP
inhibitor-treated patients could derive from the interaction of
previous platinum treatment with PARP inhibitor treatment and TP53
mutations or mutations of other DNA damage repair pathway genes.[154]
Khalife-Hachem
et al. have explored 77 patients with gynecologic and breast cancers
develop-ing tMN; 55/77 of these patients showed CHIP, while the
remaining 26/77 did not have CHIP; the most frequently mutated genes in
these patients at the level of CHIP were those related to aging
(DNMT3A, TET2 and ASXL1), while PPM1D and TP53 represent 4.6% and 3.3%
of CHIP-related mutations, respectively.[155] The analysis of the
mutational profile of tMNs (49 tAML and 28 tMDS) showed three different
mutational profiles with 36% of cases showing a TP53/PPM1D profile, 25%
a MDS-like profile and 39% a de novo/pan-AML profile; the TP53-PPM1D
subgroup displayed a limited number of co-mutation events and the very
frequent association with complex karyotype.[155] It is of interest to
note that all tMN cases classified as TP53-PPM1D are associated with
pre-existing CHIP at the time of cancer diagnosis, while tMN cases
classified as MDS-like or de novo/pan-AML are in part associated with
the absence or with the presence of pre-existing CHIP.[155]
2.6%
of patients with neuroendocrine tumors develop tMN after peptide
receptor radionuclide therapy.[156] Patients with neuroendocrine tumors
exposed to peptide receptor radionuclide therapy display an expansion
of pre-existing CHIP containing mutant DNA damage repair genes (TP53,
PPM1D and CHEK2) with development of cytopenia.[157]
Some patients
undergoing CAR-T cell therapy for non-Hodgkin lymphomas or for multiple
myeloma may develop tMN. Thus, Miller et al have explored 154 patients
with NHL (144) or with MM (10) undergoing treatment with CAR-T cell
therapy for the occurrence of CHIP. 48% of these patients have a CHIP
with a VAF >2%; PPM1D, DNMT3A, TP53 and TET2 were the gene more
frequently mutated at the level of CHIP.[158] 3 of these patients
developed a tMN during the follow-up period, 2 of whom harboured TP53
mutant CHIP and developed TP53-mutant AML.[159] Another study reported
the case of a patient with large B-cell lymphoma undergoing CAR-T cell
therapy and developing progression of tMDS starting from a TP53-mutated
CHIP, initially associated with cytopenia.[159]
Somatic
mutations in cancer cell genomes are caused by different mutational
processes, each of which generates a typical mutation signature. More
than 40 mutational signatures have been described in cancer cells
related either to endogenous or exogenous factors. Particularly, some
mutation signatures are related to exposure to exogenous carcinogens,
represented also by some anticancer drugs or radiations. Therefore,
some chemotherapies damage DNA and cause mutations in both cancer and
healthy cells; therefore, each chemotherapy causes a mutational
footprint.[160] The study of chemotherapy mutational footprints in
therapy-related AML represents a barcode to determine whether the
clonal expansion occurred before or after the beginning of exposure to
the drug.[161] In fact, cytotoxic agents introduce hundreds of unique
mutations in each surviving cancer cell, detectable by sequencing only
in cases of clonal expansion of a single cancer cell bearing the
mutational signature; therefore, a unique, single-cell genomic barcode
can link chemotherapy to a discrete time window in a patient’s
life.[161] Using this approach, it was possible to show that multiple
myeloma seeding at relapse is caused by the survival and expansion of
single multiple myeloma cells following treatment with high-dose
melphalan therapy and autologous stem cell transplantation.[161] Using
this approach, Pich and coworkers showed that tAMLs originated in
patients exposed to platinum-based chemotherapies exhibit a mutational
footprint associated with these drugs, related to the capacity of these
drugs to induce specific mutagenic events in non-malignant
hematopoietic cells. The platinum-based mutational signature was used
to determine the clonal expansion originating the secondary AMLs begins
after the start of cytotoxic treatment.[162] In cases associated with
clonal hematopoiesis the absence of this signature is consistent with
the start of the clonal expansion predating the exposure to
platinum-based drugs.[162]
Diamond et al. have explored the
occurrence of chemotherapy-related signatures by whole genome
sequencing in 39 tMNs; 16 of these patients developed tMN after
melphalan/ASCT.[163] Five single-base substitution mutational
signatures have been observed in these tMNs: SBS1 and SBS-HSC, related
to clock-like mutations that accumulate with age; SBS31 and SBS35,
related to mutations induced by platinum compounds; SBSM1 induced by
the alkylator drug melphalan. In contrast, primary de novo and
relapsed AMLs display only clock-like mutation processes, in that drugs
used in the induction chemotherapy are not linked to distinct
mutational signatures.[163] A clear dichotomy was observed, in which
tMNs with evidence of chemotherapy-induced mutagenesis from platinum
and melphalan are hypermutated and enriched for structural variants
deriving from events such as chromotripsis, while tMNs originated in
patients treated with non-mutagenic chemotherapies display a
mutational profile like that observed in de novo
AMLs. Pooling together all somatic events occurring in tMN genomes,
including SV, CNA and SNV, the cases classified as
chemotherapy-positive cases displayed a higher prevalence of TP53
alterations (62.5% of cases) compared to signature-negative cases
(13%); particularly, concerning cases receiving melphalan/ASCT, all six
cases with SBS-MM1 signature had an event involving TP53 compared to
20% in those without the signature.[163] Therefore, in patients with
prior MM who were treated with high-dose melphalan and ASCT, tMN can
develop from either a reinfused CHIP clone that escapes melphalan
exposure and is selected following reinfusion, or from TP53-mutant CHIP
that survives direct myeloablative conditioning regimen and acquires
melphalan-induced DNA damage.[163]
Sperling et al. have analyzed
416 patients with tMN (40% tAML and 60% tMDS) and observed that there
is an association between gene mutations and prior cancer treatment
exposures.[164] Particularly, significant associations were found
between TP53 mutations and proteasome inhibitors and lenalidomide
analogues; multivariate analysis showed the existence of a significant
association between TP53 mutations and prior exposures to thalidomide
analogues and vinca alkaloids and negative association with
topoisomerase inhibitors; at disease level, TP53 mutations were
particularly associated with multiple myeloma and ovarian cancer.[164]
According to these findings, the association between TP53 mutations and
lenalidomide treatment in multiple myeloma patients was explored.
Studies in experimental models have shown that TP53 loss promotes
resistance to lenalidomide and confers a selective advantage on TP53-mutant HSPCs, determining their outgrowth.[164] In conclusion,
these studies have shown the existence of an association between
certain drugs and TP53 mutations that confer resistance to these drugs
and TP53 mutations that confer re-sistance to these drugs and promote
clonal expansion under the selective effect of these drugs.[164]
TP53 mutations in relapsed/refractory AMLs. The
majority of AML patients with newly diagnosed disease achieve complete
remission following treatment with intensive induction chemotherapy.
However, about two-third patients relapse after frontline therapy and
this relapse usually occurs with first 18 months. Late relapses,
defined as those occurring after 5 years of remission, are more rarely
observed (1-3% of all cases). Basically, early and late relapses are
due to resistant clones or subclones of leukemic cells that survive to
induction chemotherapy.
Few studies have longitudinally
evaluated individual AML patients at primary disease and at relapse.
Stratmann et al. reported the longitudinal analysis of 48 adult and 25
pediatric AML patients at diagnosis and at relapse: the genomic
mutational landscape at diagnosis and at relapse was highly
comparable.[165] Particularly, the frequency of TP53 mutations was
higher in relapsed AMLs compared to primary AMLs; interestingly, ARID1A
and CSF1R mutations are recurrently gained at relapse.[165]
Alwash
et al. retrospectively analyzed 200 AML patients who relapsed and were TP53-WT at diagnosis; importantly, 29 of these patients developed a
newly detectable TP53 mutation in the context of relapsed/refractory
disease.[166] 66% of these patients acquired a detectable TP53 mutation
after the first-line of therapy, 21% after two lines and 14% after
three lines of therapy.[166] Some factors increase likelihood of
developing a newly detectable TP53 mutation; particularly, new TP53
mutations are more common among patients with a baseline chromosome 5
abnormality and with a baseline IDH2 mutation and among patients
treated with intensive therapy compared to those treated with lower
intensity.[165] In 45% of these patients, the emergence of TP53
mutations occurred in the context of complex cytogenetics.[166] In
patients who developed TP53 mutations, the most frequent co-mutations
were DDX41 (30%), DNMT3A (22%), IDH2 (22%) and NRAS (18%).[166] The
overall survival of these patients acquiring TP53 mutations was low
(4.6 months). Finally, the analysis of 555 AML patients responding to
frontline therapy showed that 5 of these patients acquired TP53
mutations during the remission phase.[166] The results of this study
support the monitoring of new emergent TP53 mutations during AML
therapy may have a clinical utility.[166]
Classification of TP53-mutant MDSs and AMLs and their comparison. The
International Consensus Classification (ICC) of myeloid neoplasms and
acute leukemias recently updated the classification of MDSs and placed
these disorders in the context of a broader group of clonal cytopenias,
including cytopenias of undetermined significance (CCUS).[167] The
presence of multi-hit TP53 mutations or of SF3B1 mutation in a
cytopenic patient are considered as MDS-defining; furthermore, MDS with
biallelic TP53 gene aberrations are considered a new genetic category
of MDSs.[167] Similarly, the ICC created new genetic categories of AML,
represented by AML with mutated TP53 and AMLs with
myelodysplasia-related cytogenetic abnormalities and
myelodysplasia-related gene mutations.[168]
The WHO defines a
single category of MDS with biallelic TP53 inactivation (MDS-biTP53)
irrespective of the blast counts but excludes single-hit TP53-mutant
MDSs with bone marrow blasts <20%.[121] Similarly, the International
Prognostic Scoring System-Molecular considered the poor outcome of
multi-hit TP53-mutated but excluded single-hit TP53-mutated.[169]
According
to the latest European Leukemia Network (ELN) 2022 guidelines, the
presence of a pathogenic TP53 mutation, at a VAF of at least 10%, with
or without loss of the wild-type TP53 allele, defines a new entity of
TP53-mutated AML.[51] In the prognostic hierarchy of AMLs, AMLs with
mutated TP53 constitute the entity with the most adverse prognosis.[51]
Similarly,
the ICC guidelines emphasize TP53-mutant variant allele frequency
>10% regardless of single- or multi-hit status for MDS and AML.[167]
The
remarkable differences observed in these different classifications of TP53-mutated myeloid neoplasms recently proposed reflect conflicting
results observed in different studies. Thus, Bernard and coworkers
through the analysis of a large cohort of de novo MDS patients reached
the conclusion that single-hit MDSs have outcomes similar to TP53-WT
MDSs, while multi-hit TP53-mutated MDSs, associated with complex
karyotype, have poor OS.[80] In contrast. Grab et al reported a
similarly poor survival for both AMLs and MDSs with excess of blasts,
irrespective of single-hit or multi-hit TP53-mutant status; however,
this study excluded MDS <10% of blasts.[105] Similarly, Weinber et
al showed that the survival of MDS and AML with complex karyotype is
equally poor independently of single- or multi-hit TP53-mutant
status.[132]
Recent comparative analysis of the molecular
abnormalities of TP53-mutated AMLs and of multi-hit TP53-mutated MDSs
suggests that these two entities probably represent a unique
condition.[105,132,170] Thus, Grob et al reported the characterization
of TP53-mutated cases observed among 2200 de novo
AMLs and MDS-EB (myelodysplasia with excess of blasts): the molecular
characteristics of TP53-mutant AML and MDS-EB resulted highly
comparable in terms of association with co-mutations and cytogenetic
abnormalities; particularly, monosomal karyotype and complex karyotype
were reported at frequencies highly comparable in TP53-mutant AML and
MDS; similarly, concurrent mutations (DNMT3A, ASXL1, TET2, RUNX1 and
SRSF2) were observed at frequencies very similar (Figure 5).[105]
In both TP53-mutant AML and MDS, detection of residual mutant TP53 was
not associated with survival.[105] Furthermore, the clinical outcomes
were highly comparable, with a median overall survival around 6
months.[105] In both groups, overall survival was negatively affected
by the association with complex karyotype.[105] These similarities
between TP53 aberrant MDS and AML were confirmed by Dunn et al. through
the analysis of 84 patients with TP53-mutated AML and MDS patients.[170]
 |
- Figure 5. Comparison of the mutational profile (A) and chromosomal abnormalities (B) in TP53-mutated
MDSs and AMLs. Top panel: the frequency of most recurrent driver
mutations in reported. Bottom panel: the frequency of most recurrent
chromosomal abnormalities is reported.
|
A
similar conclusion was reached by Weinberg et al. through the analysis
of 299 AML and MDS patients with complex karyotype; TP53 mutations were
observed in 83% of these patients (78% in AML patients and 86% in MDS
patients; the majority of these mutations were multihit TP53
mutations). A higher frequency of TP53 mutations were observed in
therapy-related cases.[132] Both in AML and MDS patients, the presence
of TP53 mutations predicted for worse outcome; the clinical features
and the response of both TP53-mutated or not, AMLs was like that
observed in the corresponding MDSs.[132] According to these findings
it was concluded that the presence of TP53 mutations in the context of
complex karyotype identifies a homogenously aggressive disease,
irrespective of the diagnosis of AML or MDS, of the blast cell count
at presentation or therapy-relatedness.[132]
According to all
these findings, it was proposed that TP53-altered MDS with excess
blasts and TP53-altered AML should be considered as a unique disease
for their treatment in clinical trials.[171]
In line with this
unification of TP53-mutated MDS and AML in an unique entity recent
studies have shown a similar impact of TP53 mutations on the prognosis
of MDS and of AML. Thus, Stengel and coworkers have analyzed a cohort
of newly diagnosed MDS (747 cases) and AML (772 cases), including 96 TP53-mutated MDS and 84 TP53-mutated AML; these patients were
classified as single-hit or multi-hit TP53-mutated MDS and AML.[172]
Overall survival was significantly shorter in patients with TP53
single-hit compared to patients without TP53 alterations both in AML
and MDS patients (sh vs no hit: AML, 8 months vs 21 months; MDS, 46 vs
70 months); in both MDS and AML, the presence of a multi-hit worsened
the prognosis markedly (mh vs no hit: AML, 1 vs 21 months; MDS, 11 vs
70 months).[172]
Shah et al. have explored a large cohort of 327
tMN patients, including 245 tMDS and 132 tAML and showed that patients
with TP53 mutations with VAF >10%, either classified as tMDS or
tAML, display a comparably negative prognosis, with similar OS for
mono-hit and multi-hit tMDS or tAML.[173] Furthermore, the number of
bone marrow blasts (either <5% 05 >5-9% or 10-19% or >20%)
does not affect the OS of the tMN.[173] A similar conclusion was
reached by Hiwase et al. showing that in TP53-mutated tMN patients the
OS in patients with TP53-mutant VAF >10%, but not ≤10%, was
significantly shorter than in TP53-WT patients.[131]
Importantly,
the recent classifications of myeloid neoplasms introduced important
changes in the classification of tMN. In the ELN[51] and the ICC[104]
classification the subcategory of tMN was changed with diagnostic
qualifiers instead. In the WHO classification, the tMN and secondary MN
were grouped and renamed as myeloid neoplasm after cytotoxic therapy,
considering that most of MDS and AML occurring post-cytotoxic therapy
have TP53 mutations and that multi-hit TP53-mutant have poor outcome
compared with single-hit.[121]
Treatment of TP53-mutated MDS and AML
As above discussed, TP53
mutations confer resistance mechanisms to DNA-damaging chemotherapeutic
agents, resulting in poor treatment outcomes. Studies carried out in
adult AML patients with TP53
mutations showed a lower complete remission rate, a significantly
inferior complete remission duration and overall survival, irrespective
of age or the type of treatment received (high-intensity or
low-intensity chemotherapy).[174] In the group of TP53-mutant AMLs, the TP53 mutational burden, defined according to the VAF is linked to inferior survival.[99-100]
Induction chemotherapy. TP53-mutated
AMLs have shown a low response rate to various chemotherapy induction
regimens, ranging from 20 to 40% and with a median OS of 4-9 months,
using regimens based either on a combination of cytarabine and an
anthracycline or cytarabine plus doxorubicin or mitoxantrone-based. The
use also of CPX-351, a liposomal form of cytarabine and daunorubicin
approved for the treatment of tAML and of AMLs with MRC, did not
improve the rate of responses in TP53-mutated AMLs with about 30-35% of
complete responses and with a very low rate of MRD-negativity (8%).[174-176] Furthermore, mOS was similar for TP53-mutant patients treated with CPX-351 compared to 7+3 standard induction chemotherapy.[175]
Since it is unclear what is the optimal induction regimen for AML patients with TP53
mutations either 7+3 standard induction chemotherapy in patients fit to
receive intensive chemotherapy, or, alternatively, venetoclax with
hypomethylating agents (VEN/HMA), CPX-351 or various high-dose
cytarabine containing regimens; all these treatments were associated
with variable results but none of these treatments was more efficacious
than the other ones.[176] The same therapeutic regimens are used in TP53-mutant
AML patients refractory to or relapsing after first-line treatment,
with a low rate of complete responses (24%) and with only 13% of
patients being able to receive allo-HSCT after achieving response.[177]
Although the response of TP53-mutant
AML patients to intensive chemotherapy is low, this treatment provides
a survival improvement compared to no treatment (8 months vs 1.7
months, respectively).[178]
Hypomethylating agents.
The hypomethylating agents include azacitidine (AZA) and decitabine
(DEC); these molecules are cytosine analogs that act as inhibitors of
methyltransferases, thus inhibiting hypermethylation events occurring
in leukemic cells, contributing to the silencing of the expression of
some genes, including tumor suppressor genes. These agents have been
used with some therapeutic efficacy for the treatment of MDS and of
elderly AML patients, not suitable for intensive chemotherapy
treatments.
A first prospective uncontrolled study reported
encouraging results using 10-day DEC in 113 patients with MDS or AML,
including 21 patients with TP53 mutations; the observed response rate
was higher in TP53-mutated patients compared to TP53-WT patients (100% vs 41% respectively, with no significant difference in the OS of these two different groups of patients).[179] However, a subsequent phase II prospective randomised study in part failed to confirm these results.[180] In this study 5-day and 10-day DEC dosing schedules were compared in elderly AML patients including also 24 TP53-mutated patients; a subgroup analysis of these TP53-mutated
patients showed response rates of 29% and 47%, in the 5-day and 10-day
dosing schedules, with a mOS of 4.7 and 4.9 months, not different from
those observed in other AML subgroups.[180] Finally,
a recent study explored the response to 10-day DEC in a group of
refractory/relapsed AML patients; although a part of patients achieved
a molecular response with a mOS around 400 days, long-term survival
remained poor.[181] In conclusion, although a significant proportion of TP53-mutant
MDS/AML patients respond to treatment with HMAs in monotherapy, these
responses are usually not durable and do not result in a significant
improvement of OS; only few patients who achieve either a marked
reduction or a clearing of TP53 mutations, display longer remissions.[182]
In
MDS patients with higher risk disease, the HMAs azacitidine and
decitabine are the standard of care due to their clinical activity and
the capacity to extend overall survival. The study of TP53-mutated
MDS showed a peculiar sensitivity to DEC. In fact, Chang et al.
evaluated the response of 109 MDS patients to DEC and 27.5% of these
patients displayed a complete response.[182] TP53 mutations in these patients predicted response to DEC therapy, with 66% of patients with TP53 mutations achieving a complete response.[183] 9/10 of these TP53-mutated responding patients displayed a complex karyotype. However, in spite the association with a higher response rate, TP53-mutated MDSs did not display an improved overall survival.[183] The longitudinal analysis of the mutational profiling of some of these patients showed that 5/7 TP53-mutated patients displayed the clearing of TP53 mutations but the maintenance of other mutations.[183]
The HOVON 135/SAKK30/15 trial compared the effect of the association of
the Bruton Kinase inhibitor Ibrutinib with DEC to DEC alone in older
AML and in high-risk MDS patients.[184] The results
of this study showed that Ibrutinib added to 10-day DEC does not
improve response or survival in AML and MDS patients compared to DEC
alone.[184] Molecular profiling of patients at diagnosis showed that patients with TP53 mutations had significantly higher response rates to DEC+Ibrutinib treatment.[184]
The TP53
mutational burden was evaluated in MDS patients undergoing treatment
with HMAs. Thus, Falconi et al. evaluated the VAF of a set of genes
recurrently mutated in MDS, including TP53, in response to standard treatment with HMAs as a bridge to alloHSCT.[184] This study showed that TP53
mutations were not predictive of AZA response and, while the allelic
frequency of most mutations did not change upon AZA treatment, a
significant decrease of TP53
mutational burden was observed with a decrease of mVAF from 29.5%
before treatment to 10.5% after treatment, which was independent of the
depth of response.[185] It is important to note that, although the TP53-mutant allelic burden significantly decreased upon AZA treatment, TP53 mutations never became undetectable, also in patients achieving a complete response.[185]
Hunter et reported the results of the serial molecular profiling of 108
MDS patients undergoing treatment with HMAs; this study included 35
patients with TP53 mutations, whose OS was shorter compared to the rest of patients.[186] 46% of patients exhibited clearance of TP53 mutations and displayed a better mOS (15.6 months) compared to those not achieving claering of TP53 mutations (7.7 months).[186] The pre-therapy TP53
mutant VAF of patients achieving mutational clearing was significantly
lower than that observed in patients not achieving mutational clearing
(12% vs 32%, respectively).[186] 16 TP53-mutated patients proceeded to alloHSCT: 7 patients achieving TP53
clearing before allo-HSCT displayed a trend toward improved OS compared
with patients with clonal persistence (25.2 months vs 11.7 months,
respectively).[186] These observations suggest that serial sequencing during treatment with HMAs is particularly valuable in TP53-mutated patients.
Interestingly, a recent study showed a promising activity of the DEC/cedazuridine (C-DEC) drug association in TP53-mutated
MDS. This drug association is based on the oral administration of DEC
with oral daministration of cedazuridine, a cytidine deaminase
inhibitor that blocks the rapid metabolism of DEC when orally
administered.[187] Recent studies have shown the
pharmacological equivalence of oral DEC with oral cedazuridine as
compared to intravenous DEC, with overall response rates of 60% and 43%
for high-risk MDS in phase II and III trials, respectively. The phase
III ASCERTAIN trial involved 133 with intermediate- or high-risk
MDS/myelomonocytic leukemia. The study evaluated OS in this patient
population, including 44 patients bearing TP53 mutations.[188]
The patients were randomized to receive either: (i) cycle 1 of oral
C-DEC followed by cycle 2 of intravenous DEC; or, (ii) cycle 1 of
intravenous DEC followed by cycle 2 of oral C-DEC.[188] Patients with TP53
mutations had worse mOS compared to those with WT-TP53 (25.5 months vs
33.7 months, respectively); the stratification of TP53-mutant into
mono-allelic and biallelic showed for those with biallelic alterations
a mOS of 13 months which compares favourably with historical results.[188]
BCL2 inhibitors.
The BCL2 inhibitor Venetoclax now represents the standard of care for
AML patients, newly diagnosed or relapsed/refractory, for elderly AML
patients and those who are unfit for intensive chemotherapy treatment,
conditions frequently observed among TP53 mutant AML patients. Therefore, several recent studies have evaluated the response of AML patients with TP53 mutations to VEN-based regimens.
Initial studies have supported a significant activity of VEN in association with DEC (VEN+DEC) in TP53-mutated AMLs. Thus, DiNardo and coworkers reported in TP53-mutated
treatment-naïve AMLs a CR/CRi rate of response of 47%, with a median
duration of response of 5.6 months and a mOS of 7.2 months, findings
that seemed favorable as compared to historical controls.[189] Another study retrospectively analyzed 32 TP53-mutated
AML patients and reported a rate of CR+CRi responses of 67% and 38% in
the frontline and in relapsed/refractory condition.[189]
Responses were observed either using a 5-day or a 10-day schedule and
responder and non-responder patients displayed a similar TP53 mutational status.[190]
Subsequent
studies failed to show a significant benefit of VEN when administered
together with DEC compared to DEC alone. Kim et al. performed a
post-hoc analysis of a phase II study involving 118 elderly AML
patients, including 35 TP53-mutated AMLs.[191] Outcomes were worse in patients who had TP53 mutations compared to those without TP53
mutations (overall response rate: 66% vs 89%, respectively); overall
survival: 5.2 months vs 19.4 months, respectively; relapse-free
survival: 3.4 months vs 18.9 months, respectively.[191] Outcomes with DEC+VEN were comparable to historical results with day-10 DEC alone.[191] In a retrospective analysis in 238 AML patients older than 65 years with newly diagnosed TP53-mutant
AML, patients who received VEN-based regimens had higher response rates
than those with non-VEN-based regimens (43% vs 32%, respectively), but
exhibited comparable OS with respect to patients treated with
non-VEN-based regimens (4.6 vs 5.5 months, respectively).[192]
These observations suggested that the addition of VEN to standard
treatment regimens did not improve outcomes in younger or older
patients who had TP53-mutant AML.[192]
The analysis of data from a phase III study comparing VEN+AZA or
placebo+AZA in poor-risk cytogenetics AML patients subdivided into TP53-mutant and TP53-WT
subgroups: VEN+AZA treatment improved remission rates but not duration
of response or overall survival compared to AZA alone in TP53-mutant AML patients.[193] In contrast, in TP53-WT
patients VEN+AZA treatment significantly improved overall survival
compared to AZA alone, with outcomes similar to those observed in
intermediate-risk AML patients undergoing a similar treatment.[193]
A propensity score cohort of 304 older AML patients treated with
DEC+VEN or DEC alone showed that DEC+ZEN significantly improved the
response rates and survival outcomes compared to DEC monotherapy;
however, some molecular subgroups, such as patients with TP53 mutations, displayed only a suboptimal response to VEN+DEC treatment (50% of responding patients).[194]
The
multiagent therapeutic regimen of fludarabine, cytarabine, granulocyte
colony-stimulating factor (G-CSF), and idarubicin is an affective
frontline treatment in AML patients suitable for intensive chemotherapy
induction. The comparative study of FLAG-IDA and CPX-351 as an
induction chemotherapy treatment in high-risk AML and MDS showed that
FLAG-IDA was unable to improve the outcomes of TP53-mutant AMLs compared to CPX-351.[195]
DiNardo and coworkers reported a consistent therapeutic efficacy of VEN
in combination with FLAG-IDA induction and consolidation in newly
diagnosed and relapsed/refractory AML patients, associated with deep
remissions and high rate of transition to successful HSCT.[196]
An update analysis of the response to VEN+FLAG-IDA in
relapsed/refractory patients showed a high ORR (60%) with 53% CR; 71%
of CR patients attained A MRD negative remission status and 68% of
responding patients proceeded to HSCT; however, these responses were
limited to TP53-WT patients: overall survival in TP53-WT and TP53-mutant AML patients was not reached compared to 5.4 months, with a 12 month overall survival and 17%, respectively.[197]
Daver
et al. recently published the results of a meta-analysis englobing 12
clinical trials involving the treatment of de novo TP53-mutated AML patients with either intensive chemotherapy or hypomethylating agents or VEN+HMA.[198]
The rate of complete remissions and the mean OS were low across the
three types of treatments; there was an improved response rate but not
in OS in VEN+HMA compared to HMA alone.[198] The median OS was uniformly poor across all three types of treatment: IC 6.5 months, HMA 6.1 months and VEN+HMA 6.2 months.[198] In another study Dover et al. have evaluated the response of 370 AML patients with TP53
mutations or chromosome 17p deletions to three different treatments:
VEN+HMA or HMA alone or intensive chemotherapy; poor OS is observed
with all these three treatments and only 8% of the patients can be
bridged to allo-HSCT.[199]
Mechanisms of resistance of TP53-mutated MDS and AML to venetoclax. Several studies have explored the cellular and molecular mechanisms mediating the resistance of TP53-mutant
MDS and AML to VEN. According to the response, DiNardo and coworkers
have categorized AML patients treated with VEN into three groups:
durable remission (0% with TP53 mutations); remission, then relapse (24% with TP53 mutations) and primary refractory (35% with TP53 mutations).[200] 32% of VEN-treated patients displayed an expansion of TP53-mutated cells after VEN treatment, thus suggesting that the presence of TP53 mutations reduced the sensitivity to VEN or increased relapse-initiating potential.[200]
The study of KO TP53
cells provided a fundamental contribution to define the mechanisms
through which TP53 deficiency may induce a reduced sensitivity to VEN.
Several BH3-only proteins, including BAK, BAX, PUMA and NOXA are TP53 target genes and lower levels of expression of these genes confer resistance to VEN.[201] Furthermore, TP53 KO resulted in reduced BCL2 and MCL1 levels that contribute to decrease the sensitivity to VEN.[201] TP53 KO induced some relevant changes in mitochondrial morphology and function: TP53
KO cells displayed less mitophagy when exposed to stress by a
mitochondrial uncoupler and increased cellular respiration, with
consequent higher production of cellular reactive oxygen species (ROS).[201]
Furthermore, TP53KO cells showed also a consistent metabolic
deregulation, with increased nucleotide synthesis, associated with
decreased glucose, pyruvate, amino acids, and urea cycle intermediate
levels, changes that suggest a metabolic shifting on preferential
carbon usage to support leukemic cell proliferation.[201] Further exploration of TP53KO cells showed that TP53
deficiency induces a reduced sensitivity not only to BCL2 inhibitors
but also to MCL1 inhibitors, when used in monotherapy as single
antitumor agent; particularly, BCL2 and MCL1 inhibitors induced only a
transient inhibitory killing effect on TP53KO cells and some surviving TP53 deficient cells outgrew TP53-WT cells over a longer period of exposure, thus suggesting a competitive survival advantage.[202]
Only the concomitant inhibition of both BCL2 and MCL1 increased
leukemia cell lethality and durably suppressed leukemia burden,
independently of TP53 mutation status.[202]
In line with these findings, Carter et al. showed that co-inhibition of
BCL2 with VEN and MCL1 with AMG176 synergistically targets AML cells
exhibiting intrinsic or acquired resistance to BH3 mimetics in vitro
and in vivo.[203] Particularly, they showed that
primary TP53-mutant AML blasts are scarcely sensitive to VEN or AMG176
added in monotherapy but are sensitive to these two drugs added in
combination; furthermore, in mouse models inoculated with TP53-mutant AML cells only the VEN+AMG176 drug combination was able to significantly prolong animal survival.[203]
However, at variance with this study, Mouijalled et al observed that
while BCL2 and MCL1 is an efficacious drug combination in many subtypes
of poor-risk AMLs, failed to induce an efficient killing of TP53-mutant primary AML cells with 7/8 cases resistant to this treatment.[204]
Chen
and coworkers showed that the mitochondrial chaperonin CLBP is
upregulated in human AML cells and particularly in those intrinsically
resistant to VEN; ablation of CLBP expression sensitizes AML cells,
including also TP53-mutated AML cells resistant to VEN.[205]
Schimmer et al. through the study of different engineered models of TP53 deficient cells reached the conclusion that either leukemic cells with TP53KO or bearing TP53
missense mutations equally display a reduced sensitivity to HMAs and
VEN, thus suggesting that loss of p53 function, rather than the
precise TP53 allelic configuration determines the inferior efficacy of HMAs and VEN.[206]
It
is important to note that a study carried out in different models of
resistant AML cells provided clear evidence that the concomitant p53
activation and BCL2 inhibition are synergistically lethal for leukemic
cells.[206] At functional level, p53 activation
negatively regulates the Ras/Raf/MEK/ERK pathway and activates GSK3
which induces MCL1 phosphorylation and promotes its degradation, thus
overcoming AML resistance to BCL2 inhibition; on the other hand, BCL2
inhibition overcomes apoptosis resistance to p53 activation by
modifying the cellular response from G1 arrest to apoptosis.[207]
These findings imply: (i) the necessity of restoring a p53 activity for
an efficient therapeutic response; (ii) the absolute importance of the
presence of a mutation in TP53 when using VEN-based therapy.[207]
Hematopoietic stem cell transplantation (HSCT) in TP53-mutated MDS and AML.
Allogeneic HSCT is the only potentially curative treatment for a
considerable number of MDS and AML patients. Retrospective studies have
shown that transplant efficiency is influenced by the genetic
alterations present in the various patients and particularly in TP53-mutated
MDS and AML patients. In these patients the outcomes of allo-HSCT are
considerably affected by the heterogeneous clinical conditions of TP53-mutated AML patients and by consistent heterogeneity of these leukemias (related to the TP53 allelic state, co-occurring somatic mutations, and the position within the clonal hierarchy).
Outcomes of HSCT for TP53-mutated AMLs are poor; in fact, a meta-analysis performed in 297 TP53-mutated AML patients undergoing allo-HSCT showed a 2-year OS of 29.7% with a relapse rate of 61.4% at 2 years.[208] Similarly, survival after allo-SCT is low for most TP53-mutated MDS patients.[209]
The fundamental study of Lindsley and coworkers reported the evaluation of 1514 MDS patients undergoing allo-HSCT.[49]
A significantly shorter OS was observed for TP53-mutated patients, with
an hazard ratio (HR) of 1.71 and shorter time to relapse, with HR of
2.10.[49] The impact of conditioning regimen was also
evaluated in these patients, showing that the median survival was
similar between myeloablative conditioning regimen (MAC) and
reduced-intensity conditioning regimens (RIC) (7.5 months vs 9.2
months, respectively).[49] Yoshizato et al have retrospectively analyzed 797 Japanese MDS patients; 295 of these patients have TP53 mutations and 98 of them have been transplanted. TP53-mutated
patients have been subdivided into two subgroups based on the
association or not with complex karyotype; the outcomes of these two
different subgroups were clearly different: The subgroup with TP53
mutations and CK (88% of cases) displayed a worse outcome, with a mOS
of 4.8 months and with >80% of deaths within 2 years after
transplantation; the other subgroup with TP53 mutations only had a markedly better survival posttransplant with 60% of patients alive at 60 months.[210] Ciurea and coworkers have reported the post-transplant outcomes of 83 MDS/AML TP53-mutated patients and median overall survival of less than one year and a 2-year overall survival rate of less than 30%.[211]
Three relevant prognostic factors were defined in these patients: the
median hematopoietic stem-cell transplantation comorbidity index
(HTC-CI) is 4, with a range from 0 to 9; the Karnofsy performance
status (KPS); the presence of a complete remission status in first-line
(CR1) or in second-line (CR2). A HCT-CI >4, the KPS <80% and the
absence of CR1 or CR2 correspond each to 1 point of risk score;
TP53-mutant patients with a risk 0 have a mean OS significantly better
than those with score 1 or score 2.[211]
The analysis of the long-term outcomes of 178 AML patients undergoing allo-HSCT showed that only TP53
mutations, but not the mutations of other genes, are associated with a
significantly shorter OS and RFS and with a higher relapse index.[212] Badar and coworkers have evaluated 370 AML patients with TP53 mutations: 49 patients received allo-HSCT after first-line therapy and 20 after second-line therapy.[213]
In the first-line group, 75% of patients were in complete remission and
70% were MRD-negative at the moment of allo-HSCT; in the second-line
group, 50% of patients were in CR and 43% were MRD-negative.[213]
The median OS in the first-line group was 30.5 months, compared to 20.2
months in the second-line group; the presence of a condition of CR at
day 100 post-transplantation favourably predicted an improved survival
post-transplantation.[213] Furthermore, patients in
CR with a MRD-negative status at the moment of allo-HSCT had a
significantly better OS than those in CR with a MRD-positive condition;
however, the presence or not of TP53-positivity at transplantation did not affect post-transplantation OS.[213]
Byrne et al. reported the retrospective analysis of 384 TP53-mutant
MDS/AML patients undergoing allo-HSCT: the post-transplant OS of MDS
and AML patients was similar; patients with chronic GVHD displayed a
significantly better OS and lower relapse rate than patients without
chronic GVHD; patients with biallelic TP53 disease or those with CK have a worse outcome compared to those with monoallelic TP53 disease or without CK, respectively; pre-transplantation TP53
mutations persistence by NGS predicted post-transplantation relapse,
whereas pre-transplantation CR and full donor BM chimerism were
associated with a lower rate of relapse.[214]
The
European society for Blood and Bone Marrow Transplantation
retrospectively analyzed the outcome of 179 AML patients with TP53 mutations and of 601 AML patients without TP53 mutations: in patients with TP53 mutations without CK or chromosome 17p loss the 2-year OS was 65%, while in patients with TP53 mutations, with either chromosome 17p loss or CK the 2-year OS was 24.6%.[215] Importantly, the 2-year OS of TP53 mutant patients without 17p loss or CK is like that observed for TP53-WT patients (65.2% vs 70.4%). These observations further support the conclusion that TP53 mutations with concomitant additional cytogenetic feature (CK or 17p-) determine a poor outcome in TP53-mutant AML patients.[215]
Three factors limit the efficiency of allo-HSCT in TP53-mutated AML patients. (i) Most of TP53-mutated
AML patients are old and receive reduced intensity conditioning (RIC)
to reduce cytotoxicity and RIC is unable in most of cases to induce
clearing of TP53
mutations and to induce a condition of MRD negativity before
transplantation, while myeloablative conditioning induces a much higher
rate of TP53 mutations clearing.[216]
(ii) In older AML patients molecular associations with MRD positivity
and transplant outcomes are driven primarily by baseline genetics, and
not by mutations present in remission and baseline TP53 mutations represent the most unfavourable genetic association.[217] (iii) The negative impact of TP53 mutations transplant outcomes is related to a very high risk of early relapse after transplantation, thus indicating that TP53
mutations induce rapid disease progression that outstrips functional
engraftment and development of graft-versus-leukemia (GVL) effect.[218]
The proportion of TP53-mutated
MDS/AML patients which can be transplanted is lower than that observed
for patients with other genetic abnormalities. In a recent study,
Marvin-Peek et al reported the results of a retrospective analysis of
352 patients with MDS/AML and 91 with TP53 mutations; the intention to transplant was similar for TP53-mutated and TP53-WT patients (50 vs 52%), but the real proportion of patients transplanted in the TP53-mutant group was significantly lower than in the TP53-WT group (19% vs 31%). The TP53-mutated
MDS/AML patients have an increased number of infections which likely
contributes to the lower rates of HSCT in these patients.[219]
The
anti-leukemic activity of allo-HSC is related to two main factors: (i)
the conditioning regimen; (ii) the immune-related graft-versus-leukemia
(GVL) effect. Thus, several studies, above reported, showed that the
occurrence of chronic GVHD correlated with improved EFS and OS in TP53-mutated MDS/AML patients undergoing allo-HSCT.
Very
interestingly, a recent study reported the first results on the
combination of a hypomethylating agent and Eprenetapopt administered as
maintenance therapy post allo-HSCT in TP53-mutated AML patients.[220] Eprenetapopt (APR-246) is a small molecule exerting a peculiar effect, restoring WT-TP53 in TP53-mutant AML cells and inducing apoptosis of TP53-mutant
leukemic cells. Studies that will be discussed below support the use of
this drug in combination with a hypomethylating agent in TP53-mutant
AML cells. A phase II study involved the treatment of 33 MDS/AML
patients with AZA+Eprenetapopt in post allo-HSCT: with a median
follow-up of 17.0 months, the median OS was 20.6 months and 1-year OS
probability was 78%; the observed OS outcomes were encouraging and
support prospective randomized studies to define the optimal schedule
and duration of this drug association and its therapeutic efficacy.[220]
In
conclusion, although there is a low probability of long-term cure and
the transplantation is associated with a substantial risk of morbidity
and mortality, allo-HSCT can be considered as an appropriate treatment
for MDS/AML patients with TP53 mutations.[218]
CD47 targeting in TP53-mutant AMLs.
CD47 is a membrane receptor ubiquitously expressed on the surface of
cells and plays a key role in self-recognition. Through interaction
with SIRPα, TSP-1 and integrins, CD47 acts as a modulator of cellular
phagocytosis by macrophages and of the activation of immune cells.[221]
The binding of CD47 to signal-regulated protein α (SIRPα) signals
cancer cells to escape from macrophage-mediated phagocytosis, thus
promoting tumor progression.[222] CD47 is
overexpressed on the surface of many malignant cell types and in some
tumors its level of expression is a negative prognostic factor.[222]
These observations have supported the rationale of blocking CD47 with
inhibitory monoclonal antibodies to promote macrophage anti-tumor
mechanisms. CD47 is heterogeneously expressed on AMLs, including the
fraction of leukemic stem cells, with 25-30% of these patients
displaying high levels of expression; high CD47 expression has been
shown to be an independent prognostic factor for poor overall survival
in AML patients.[223] CD47 expression was clearly
more elevated in AML than in MDS; furthermore, the level of CD47
expression is heterogeneous in the various molecular subtypes:
particularly, about 50% of TP53-mutant AMLs highly express CD47, while in the remaining cases it was lower.[224]
Preclinical
studies using monoclonal antibodies blocking CD47 have shown in in
vitro and in vivo mice leukemic models a consistent anti-leukemic
activity and have supported clinical studies. One of these antibodies,
Magrolimab was evaluated in clinical trials involving AML patients. In
monotherapy, Magrolimab was unable to induce any CR in patients with
refractory/relapsed AML. In subsequent studies, Magrolimab was
evaluated in association with other anti-leukemic drugs. A phase Ib
study involved the treatment of 52 treatment-naïve AML patients unfit
for intensive chemotherapy, with Magrolimab and Azacitidine.[225] In 21 TP53-mutant patients, 71% of patients achieved an objective response, 48% a CR; the median overall survival for TP53-mutant patients was 12.9 months compared to 18.9 months for TP53-WT patients.[225] A phase I/II study involved the enrolment of 18 newly diagnosed AML patients (8 with TP53 mutations) treated with Magrolimab plus VEN and AZA: in the 7 TP53-mutant
AML patients evaluable for response, a 100% CR/CRi response was
observed, with 57% achieving MRD negativity, as assessed by multicolor
flow cytometry assay.[226] A more advanced evaluation of the triplet drug combination showed in 22 frontline TP53-mutant AMLs (including 10 tAMLs) a CR+CRi of 63%, compared to 90% in TP53-WT AMLs and 1-year OS of 53% compared to 83% for TP53-WT AMLs; in 5 sAML patients with TP53 mutations a CR+CRi of 60% was observed.[226] 30% of TP53-mutant
AML patients proceeded to allo-HSCT. A phase III placebo-controlled,
randomized study to evaluate this drug triplet in newly diagnosed AMLs
has been initiated (ENHANCE-3 trial).[227] Finally, Daver and coworkers reported the results of a phase Ib study enrolling 72 frontline TP53-mutant
AML patients treated with Magrolimab plus azacitidine: a CR+CRi
condition was achieved in 41.6% of patients; the longitudinal TP53
VAF assessment in 8 patients who achieved a CR showed in 5 of these
patients A VAF decrease to <5%; the median OS for the 72 treated
patients was 10.8 months.[228] A phase III trial in TP53-mutant AML (ENHANCE-2) of this drug combination vs standard of care is ongoing.[228]
Johnson and coworkers have analyzed the depth of the molecular response in a group of TP53-mutant MDS and AML patients treated with Magrolimab and azacitidine.[229] In patients with TP53-mutated MDS, 38% of patients achieved a CR; in these patients, the initial median TP53 VAF was 0.38 and decreased to 0.07 by cycle 5 of treatment.[229] In patients with TP53-mutated AML, 63% of patients achieved a CR; in these patients, TP53 VAF <0.07 was observed in 54% of patients at cycle 3 and 75% at cycle 5 of treatment.[229]
The
ALX Oncology Holdings Inc developed a next generation CD47 blocker,
Evorpacept (ALX148): the CD47 binding domain of Evorpacept is an
affinity enhanced extracellular domain of SIRPα and its engineered Fc
binding domain does not provide the pro-phagocytic signal, but confers
to the molecule an antibody-like pharmacokinetic profile. Several
ongoing clinical studies are exploring Evorpacept in solid tumors and
in hematological malignancies. Recently, the clinical data from the
phase Ia (dose-escalation) study ASPEN-05 evaluating Evorpacept in
combination with azacitidine and venetoclax fort the treatment of
relapsed/refractory or newly diagnosed AML patients were presented.[230]
This study showed that: Evorpacept administered with AZA and VEN was
generally well tolerated; in 10 relapsed/refractory patients (including
8 patients that progressed after prior VEN treatment and 7 with TP53
mutations) reduction in marrow blasts was observed in 100% of patients,
with 40% objectives responses; in 3 newly diagnosed patients, all
with TP53 mutations, all achieved a response, with 2 complete responses.[230] Another ongoing clinical study (ASPEN-02) is evaluating the safety and the efficacy of Evorpacept in high-risk MDS patients.
Ligufalimab
(AK117) is a humanized IgG4 antibody against CD47. AK117 enhanced
macrophage-mediated phagocytosis of hematologic cancer and solid tumor
cells alone or in combination with other anti-tumor drugs.[231] Ligulifamab is under evaluation in phase I/II ongoing clinical trials.
Lemzopulimab
is a peculiar human IgG4 antibody targeting a unique CD47 epitope,
enabling CD47 epitope, enabling the sparing of red blood cells but
maintaining strong activity against tumor cells. A phase Ib,
dose-escalation trial is evaluating the safety and the efficacy of
Lemzopilimab in monotherapy in relapsed/refractory AML patients and in
high-risk MDS patients; this study showed a good tolerability of
Lemzoparlimab with no evident hematological toxicity; one of the five
treated patients achieved a morphologic leukemia-free state.[232]
Xiao et al. reported the clinical results on 53 newly diagnosed
high-risk MDS patients treated with Lemzoparlimab and AZA: the ORR was
82%; an increased pro-phagocytic signal in bone marrow-derived CD33
blasts, as well as an increased percentage of activated macrophages,
was observed in 23 responders, but not in 5 non responders; 4/4
patients with TP53-mutated MDS achieved a CR or a marrow CR, respectively.[233]
These observations preliminary support a promising activity of
Lemzoparlimab in high-risk MDS patients exhibiting a higher CALR
expression and immune infiltrates in bone marrow.[233]
Pevenodistat.
Pevenodistat (PEV) is an inhibitor of NEDD8-activating enzyme (NAE)
which is essential for the degradation of some cellular proteins
essential for tumor growth and survival. Preclinical studies have
supported the evaluation of PEV as a therapeutical agent for the
treatment of hematological malignancies. Particularly. These studies
supported the evaluation of PEV in combination with hypomethylating
agents.
Swords and coworkers have explored the safety and
efficacy of PEV administered together with AZA in elderly AML patients
unfit for intensive chemotherapy.[234] 5 of these patients displayed TP53 mutations and 4/5 of them were responders (CR+CRi+PR) to PEV+AZA treatment.[234] However, subsequent studies failed to confirm this high sensitivity of TP53-mutated
AMLs to PEV+AZA treatment. Thus Saliba et al. reported the results on
the response to PEV+AZA of 9 older AML patients with TP53
mutations enrolled in the phase II umbrella Beat AML Master trial;
these patients were selected according to the presence of TP53 mutations with a VAF >30%.[235] None of the 9 treated patients attained a CR and 2 patients exhibited a PR.[235] These authors argued that the lower sensitivity of TP53-mutated
patients observed in this study compared to the previous study of
Swords et al could be related to the criteria of selection of these
patients (with low TP53-mutation VAF in the study of Swords et al. vs with TP53-mutation VAF >30% in the study of Saliba et al.).[235]
The phase III PANTHER randomized trial explored the safety and efficacy
of PEV+AZA vs AZA alone in patients with newly diagnosed high-risk MDS
patients.[236] In the whole population of treated
patients no significant improvement of OS in the PEV+AZA arm vs AZA
alone was observed (21.6 months vs 17.5 months); however, patients
receiving >3 cycles or >6 cycles of treatment exhibited a
significant improvement in OS compared to AZA arm.[236] This study enrolled a high proportion of TP53-mutated MDS patients (28.9% in the PEV+AZA arm and 25.9% in the AZA arm); the ORR in TP53-mutated MDS was 25% with PEV+AZA and 28% with AZA alone.[236]
Preclinical
studies have supported the rationale of combining PEN+VEN+AZA, showing
that this triplet drug association induces a robust activity against
primary AML blasts, including also high-risk AML.[237] At mechanistic level, PEV+AZA act as inducers of NOXA expression which enhances VEN-mediated apoptosis.[236]
A phase I/II study evaluated the triplet combination of PEN, AZA and
VEN in patients with newly diagnosed sAML and MDS with hypomethylating
failure.[238] 32 AML patients were enrolled in this study and 34% of them displayed TP53 mutations: the median OS for patients with TP53 mutations was 8.1 months and 18 months for TP53-WT patients.[238]
Eprenetapopt (APR-246, PRIMA-1).
PRIMA-1, a small molecule compound, and its methylated analog known as
APR-246 or Eprenetapopt, acts as a suppressor of the growth of an
osteosarcoma cell line expressing the TP53 mutant R272H.[239]
This molecule displays the unique property of restoring the DNA binding
capacity of p53 mutant protein and, consequently, the growth and
tumor-suppressing activities of this protein.[240-241] The restoring capacity was observed for various TP53
mutants. Preclinical models have supported the anti-tumor activity of
APR-246 and its synergistic functional interaction with DNA-damaging
anticancer drugs.[242] The pharmacological activity
of APR-246 requires its conversion into a methylene quinuclidonone that
is able to covalently bind at the level of Cys 124 and 277 of mutant
p53 protein, inducing a shift in favour of the WT p53 conformation.[243]
A recent study suggested an additional mechanism of anti-tumor activity
of APR-246 through induction of oxidative stress mediated by
glutathione depletion and induction of ferroptosis.[244]
Preclinical studies have shown synergistic effects of APR-246 and AZA in TP53-mutated MDS and AML cells and have supported the clinical evaluation of this drug association[241]
Two phase Ib/II studies have evaluated the association of Eprenetapopt
with AZA; the first trial involved the enrollment of TP53-mutated MDS (with intermediate or high-risk) and AML (oligoblastic AMLs, with 20-30% of blasts);[245]
the second trial involved a similar population of patients, with the
exception of the admission of AML patients with any blast percentage
and the administration of the two drugs Eprenetapopt and AZA for up to
one year in the eventuality of a HSCT.[246] The pooled analysis of 100 patients enrolled in these two studies showed an ORR of 69%, a CR rate of 43%, a NGS TP53 mutation negativity of 40%, a MRD negativity rate of 6% and a median OS after allo-HSCT of 16.1 months.[247] Responding patients had significant reductions in TP53 VAF; responding patients had a significantly longer OS compared to non-responding patients.[247]
Patients who responded to treatment and proceeded to allo-HSCT had a
mOS not reached compared to 9.1 months for patients who did not respond
and undergo allo-HSCT.[247]
Other ongoing
clinical trials are evaluating Eprenetapopot in other clinical settings
and using other drug associations. Thus, Garcia-Manero and coworkers
reported the first results on 30 TP53-mutant
AML patients undergoing treatment with a triplet drug association based
on Eprenetapopt in combination with VEN and AZA.[248]
A CR rate of 30% and CR+CRi of 53% were observed and the Simon 2-stage
efficiency criteria supported future exploration of this drug
combination.[248]
A phase III clinical trial
comparing Eprenetapopt plus AZA to AZA alone in MDS patients failed to
meet its primary endpoint, as announced in a press release of APNEA
Company: although the results showed a higher rate of complete
responses of 33.3% in the Eprenetapopt+AZA arm compared to 22.4% in the
AZA monotherapy arm, the difference between the two arms did not meet
the predefined threshold for statistical significance.
A phase II
clinical trial evaluated the efficacy and safety of Eprenetapopt in
combination with AZA as a post HSCT maintenance therapy in TP53-mutated MDS and AML patients.[249]
This treatment was well tolerated with a good safety profile. With a
median follow-up of 17 months, the median OS was 20.6 months and 1-year
OS probability of 78.8%.[249] It is important to
note that 1-year relapse-free survival was of 60% with this treatment
that compares favourably with a previous report showing a 1-year
relapse-free survival of 30% for TP53-mutated MDS patients.[49]
Although
a phase I/II clinical trial combining Eprenetapopt with AZA showed an
ORR of 71%, 50% CR rate and 47% of molecular remissions, the duration
of these remissions was limited due to relapse that occurred with the
emergence of the same pre-treatment TP53
mutations, without secondary mutations, thus suggesting that relapse
was not related to the acquisition or selection of subclonal mutations.[247]
A recent study provided evidence that resistance to Eprenetapopt could
be related to the overexpression of the nuclear exportin XPO1,
resulting in shuttling to the cytoplasm of refolded p53, thus leading
to therapeutic resistance.[250]
Interestingly,
a recent study provided evidence that Eprenetapopt may stimulate
anti-immune tumor immunity through a peculiar mechanism, involving
increased p53 expression in tumor-associated macrophages.[251] This finding supports the therapeutic association of Epretapopt with immune checkpoint blockers.[251]
Immunotherapy with bispecific antibodies: Flotetuzumab.
Studies exploring the therapeutic activity of Flotetuzumab, a CD123xCD3
bispecific dual-affinity retargeting antibody (DART) molecule led to
define a significant sensitivity of TP53-mutated AMLs to immunotherapy
and to discover a peculiar immunological profile of TP53-mutated AMLs.
These studies were prompted by recent investigations suggesting that TP53,
in addition to its well-known function of tumor suppressor, plays also
a relevant role in the activation of genes involved in immune responses
and inflammation. Particularly, the analysis of transcriptomic data of
The Cancer Genome Atlas (TCGA) from 10,000 nonhematologic tumors showed
that TP53
mutations exhibit a correlation with increased leukocyte infiltration
and are enriched in wound healing and interferon-γ dominant immune
subtypes.[252]
Vadakekolathu et al., through
targeted immune gene expression profiling, identified two groups of
immune subtypes of AML cells: immune infiltrated and immune depleted.[253]
AMLs with immune-infiltrated profiles displayed higher expression of
IFN-stimulated genes and T-cell recruiting factors, T-cell markers and
cytolytic effectors, counter-regulatory immune checkpoints and
molecules involved in antigen presentation and processing; this
immunologic profile was associated with suppressed anti-tumor immune
reactivity and with response to immunotherapy in solid tumors and in
AML.[253] TP53-mutant
AMLs mostly correspond to immune-infiltrate AMLs. Overall protein
expression patterns identified four protein signatures (SIG1, SIG2,
SIG3 and SIG4); interestingly, all features of SIG3 group correlated
with TP53 mutational status.[253]
SIG3 signatures were enriched in biological processes related to T-cell
lineage commitment and T-cell homeostasis; deregulated genes in SIG3
include PD-L1, FoxP3, G2MB, PTEN and BCL2 and were predominantly
observed in AMLs with immune-infiltrated mRNA profiles.[253]
In parallel, the same authors explored the immune infiltration profiles
in AMLs corresponding to various mutational profiles: TP53
mutated AML cases showed higher immune infiltration, a higher number of
mutations and a higher fraction of genome altered, compared to other
AML subtypes without TP53 mutations, including FLT3-ITD or NPM1-mutant AMLs; concerning immune-related gene, TP53-mutated
AMLs expressed significantly higher levels of IFN-γ mRNA, CD8A mRNA,
PD-L1 mRNA, FoxP3 mRNA, G2MB mRNA and LAG3 mRNA than TP53-WT AMLs.[254] This immune gene expression profile suggests that the tumor microenvironment of TP53-mutant
AMLs is intrinsically proinflammatory and IFN-γ dominant and that these
features were associated with poor survival. These observations allowed
the discovery of a 34-gene immune classifier prognostic for survival in
independent cohorts of AML patients.[254]
The analysis of relapsed/refractory TP53-mutated
AML patients treated in the context of a clinical immunotherapy trial
involving Flotetuzumab provided some interesting information.
Flotetuzumab is a bispecific antibody targeting both CD123, a membrane
antigen preferentially expressed on leukemic blasts comparted to normal
hematopoietic cells and CD3: the use of this bispecific antibody aims
to drive an immune response (mediated by CD3) at the level of the sites
of leukemic cell development (mediated by CD123).[255] Flotetuzumab was evaluated in 88 adult AML patients with refractory/relapsed disease, showing a CR rate of 26.7%.[256] The analysis of the response of TP53-mutant patients enrolled in this study provided evidence of their sensitivity to Flotetuzumab treatment.[254] Particularly, 13 TP53-mutant
patients were enrolled and 10/13 displayed an increased immune
infiltration in tumor microenvironment, while 3/13-clustered in the
immune-depleted subgroup; the ORR in these patients was 60%, with 47%
of patients achieving a CR; interestingly, the ORR to Flotetuzumab was
higher in TP53-mutant than in TP53-WT patients (60% vs 33.3%); the mOS in TP53-mutant patients achieving a CR was 10.3 months.[254] These observations strongly support additional studies based on the treatment of TP53-mutated AML patients with Flotetuzumab and with other immunotherapeutic approaches.
Interestingly,
a recent study showed that Flotetuzumab enhances major
histocompatibility class II (MHC-II) in AML cells of patients treated
with this antibody and this effect is mediated by local production of
IFN-γ.[257]
Additional studies further characterized the abnormalities of immune response observed in TP53-mutated AMLs. The degree of CD8+
T cell infiltration in AMLs inversely correlates with overall survival,
a finding explained by the high dysfunctional state of these cells; in
fact, phenotypic and transcriptomic studies have shown that CD8+ T cells present in AML patients display features of exhaustion and senescence.[258]
Exhausted T cells express inhibitory receptors (PD-1, CTLA4, TIM3,
CD160, CD244) and show a reduced capacity to secrete cytokines and to
exert cytotoxic functions. Senescent T cells downmodulate
co-stimulatory molecules (CD27 and CD28), express senescence
membrane-associated markers, remain metabolically active and secrete
cytokines. Following chemotherapy treatment, the phenotypic and
transcriptomic profile of CD8+
T-cells diverge from responders and nonresponders, with upregulation of
costimulatory pathways and downregulation of apoptotic and inhibitory
T-cell signalling pathways in responsders.[258] Senescent-like CD8+ T-cells are unable to kill autologous AML blasts and their proportion negatively correlates with OS.[258]
From RNA-sequencing data, an immune effector dysfunction (IED)
signature was identified, whose scores correlate with adverse-risk
molecular lesions, including TP53 mutations, stemness and poor outcomes.[259]
Other studies have shown the peculiar immunological features of TP53-mutated
AML, such as an enrichment of resting memory CD4 T cells and resting NK
cells, a high CD8+ T-cell infiltration, a high expression of some
immune-related pathways, such as IL2 signal transducer signaling and
inflammatory response.[260]
Sallman et al. have
explored the immunological phenotype of the malignant clone and
alterations of the immune microenvironment of TP53-mutant MDS/AML and
observed that: (i) PD-L1 expression is significantly increased in stem
cells (CD34+/CD38- cells) of TP53-mutant MDS/AML compared to TP53-WT MDS/AML; (ii) patients with TP53 mutations exhibit reduced numbers of BM-infiltrating OX40+ cytotoxic cells and helper T lymphocytes; (iii) highly immunosuppressive regulatory T cells, such as ICOShigh/PD-1- and myeloid-derived suppressor cells (PD-1low) are expanded in BM of TP53-mutant patients; (iv) a higher proportion of ICOShigh/PD-1- Treg cells is a highly significant independent predictor of overall survival.[261] According to these observations it was concluded that TP53-mutant
MDS/AMLs have an immunosuppressive and immuno-evasive environment that
favor their development and resistance to therapy and that
immunomodulatory therapeutic strategies may provide some benefit.
TIM-3 targeting.
T-cell immunoglobulin and mucin domain 3 (TIM-3) is a type I
trans-membrane glycoprotein expressed on IFN-γ-producing T-lymphocytes,
FoxP3 Tregs and innate immunity cells. It is expressed on leukemic
myeloid cells, but normal hematopoietic stem cells lack expression: AML
cells overexpress both TIM-3 and its ligand galectin-9, thus generating
an autocrine loop that promotes self-renewal of leukemic stem cells.[262]
TIM-3 overexpression on leukemic blasts inhibits their recognition by CD8+
T cell and their destruction by these cells. Sabatolimab is a humanized
monoclonal antibody specific for TIM-3; sabatolimab was selected for
its binding and inhibitory capacities of TIM-3 and its administration
enhances T-cell killing and inflammatory cytokine production by
dendritic cells, facilitates the phagocytic uptake and removal of
TIM-3-expressing target cells and blocks the interaction between TIM-3
and its ligand galectin-9.[263] Sabotolimab is under
evaluation as an agent able to target TIM-3 in both immune and myeloid
cells in combination with HMAs in patients with AML and high-risk MDS.
A phase Ib study of sabatolimab in combination with HMAs involved the
enrollment of 51 high-risk and very-high-risk MDS patients and 40 de novo AML patients, showing an ORR of 33% in MDS and of 40% in AML patients.[264]
Interestingly, sabatolimab appeared efficacious in TP53-mutated
patients: 71.4% of ORR in 14 patients with MDS, with a median duration
of response of 21.5 months and with 24.5% of these patients proceeding
to allo-HSCT; 40% ORR in patients with AML.[264]
Based on the promising results observed in this phase I study, the
STIMULUS clinical trial program was developed to evaluate the safety
and the effectiveness of sabatolimab in various combinations with other
drugs in MDS and AML patients. Thus, a phase II clinical trial of
sabatolimab in combination with AZA and VEN in newly diagnosed AML
patients not suitable for intensive chemotherapy is ongoing
(STIMULUS-AML1) and the safety data were recently reported.[265]
Immune checkpoint inhibitors-based regimens.
As above discussed, the immune dysregulation observed in the tumor
microenvironment implies also an increased PD-L1 expression and a state
of immunosuppression, conditions that provide a rationale for
evaluating immune checkpoint inhibitors in the therapy of TP53-mutated MDS/AML patients.
Nivolumab is an anti-PD1 monoclonal antibody and was evaluated in 70 relapsing/refractory AML patients (16 of whom had TP53 mutations): only 3 patients with TP53 mutations responded to this treatment.[266]
Another study evaluated the association of nivolumab with induction
chemotherapy, based on idarubicin and cytarabine regimen, in 44
patients with AML and high-risk MDS, including 8 cases TP53-mutated.[267]
At median follow-up of 17 months there was a mOS of 18 months, with 43%
of patients achieving a response and proceeding to allo-HSCT.[267] The analysis of mutational profile in responders and non-responders showed that non-responders have more TP53 mutations than responders (40% vs 12%, respectively).
Other
studies have explored the safety and the efficacy of nivolumab as
maintenance therapy in high-risk AML patients in remission: however,
these studies showed a scarce effect of nivolumab as single agent in
eradicating MRD and in extending remission.[268-269]
Similarly, the addition of an anti-CTLA4 antibody, Ipilimumab, to AZA
and nivolumab failed to significantly improve the response of
relapsed/refractory AML patients compared to VEN+AZA or AZA+nivolumab.[270]
An
ongoing clinical trial is evaluating the triplet combination of
decitabine, VEN and nivolumab. Preclinical studies have shown that PD1
inhibition potentiated the anti-leukemia response in
decitabine/VEN-treated AML samples.[271] An initial
observation on one patient responding to the triplet drug association
showed the clearing of leukemic blasts and of leukemic stem/progenitor
cells and the expansion of CD8-positive memory T cells.[271]
Pembrolizumab
is another anti-PD1 monoclonal antibody and it was evaluated in
combination with high-dose cytarabine in 37 relapsed/refractory AML
patients showing a CR+CRi rate of 38% in all patients and in 2/5
(40%) TP53-mutated AML patients.[272] Pembrolizumab was evaluated also in combination with azacitidine[273] or decitabine[274]
in relapsed/refractory AML patients with a promising efficacy; however,
these studies did not provide a specific report on the response of TP53-mutated AML patients.
Other
studies have evaluated the therapeutic efficacy of durvalumab, an
anti-PD-L1 mAb, in high-risk MDS and AML patients. No significant
improvement in CR+CRi rates or in OS was observed in 84 first-line
high-risk MDS patients[107] or in 129 older/unfit AML patients treated with durvalumab plus azacitidine compared to azacitidine alone.[108] Particularly, in the MDS trial the TP53 mutant patients experienced poorer outcomes compared to TP53-WT patients (41% ORR vs 61% ORR, respectively),[107] in the AML trial, the ORR of both TP53-mutant and TP53-WT patients was similar (35% vs 34%, respectively).[108]
A pooled analysis of the results of these two studies showed that the
outcomes of MDS/AML patients with TP53 mutations are worse compared
to TP53-WT, without any significant difference between monohit and multihit TP53 mutational status.[106]
 |
- Figure 6. New therapeutic strategies under clinical investigation form the treatment of TP53-mutated MDS/AML.
|
Conclusions
Studies carried out in the last years have considerably improved our understanding of TP53-mutated myeloid malignancies. TP53-mutated MDS and AML have been recognized as distinct stem cell disorders; furthermore, recently it was proposed to unify TP53-mutated MDS and AML in a unique entity. The identification of TP53-mutated
MDS/AML as a separate and unique entity is important because it will
represent a fundamental condition for the development of dedicated
clinical trials.
The molecular characterization of TP53-mutated
MDS/AML based on the study of large cohorts of cases was of key
importance to define the major features of these myeloid malignancies,
related either to the characterization of TP53 alterations
(either mutations or gene deletions) or to the associated chromosomal
abnormalities (complex karyotype, chromosome monosomy) in the context
of a condition of genomic instability and associated co-mutations.
These studies have clearly shown that allelic involvement (monoallelic
or biallelic), the concomitant presence of chromosome abnormalities,
the presence of single or multiple TP53 mutations and the clonal size of the TP53
mutant clone and the number of co-mutations at the level of other
driver genes are key determinants of the clinical severity of these
hematologic malignancies. Therefore, these studies have shown that it
is the loss of both copies of TP53 gene that drives the dismal outcomes of TP53-mutated MDS/AML patients rather than the underlying mutation types. These studies underscore the importance of assessing TP53-mutant AML/MDS patients through an evaluation of TP53 mutational status, TP53 copy number, occurrent of concomitant chromosomal abnormalities and of co-mutations of other driver genes.
TP53 mutated
MDS/AMLs are associated with resistance to standard treatments and poor
outcomes. Standard treatments, including intensive chemotherapy, HMAs
and VEN, induce only a poor survival of newly diagnosed TP53-mutated
MDS/AML patients. Allo-HSCT is the only treatment capable of achieving
a significant improvement of overall survival of these patients.
However, the proportion of TP53-mutant MDS/AML patients suitable for allo-HSCT is low. The outcomes of TP53-mutated MDS/AML patients is related to some parameters TP53-related, such the allelic status of TP53 abnormalities and the presence of chromosome abnormalities and the achievement of a MRD negativity at transplantation and TP53-not related such as the intensity of the conditioning regimens and the comorbidity index.
Recent studies have identified some peculiar immunological features of TP53-mutant
MDS/AMLs, predicting their potential sensitivity to immunotherapy.
Thus, a promising therapeutic response to immunotherapies using agents
that improve macrophage anti-leukemia activity (Magrolimab or other
CD47-targeting agents) or T lymphocyte anti-leukemia activity
(Flotetuzumab or Sabatolimab) was reported in initial clinical studies.
Furthermore, Eprenetapopt, a drug promoting the refolding of mutant p53
protein, showed therapeutic activity in TP53-mutant
AMLs. Future phase III clinical trials are required to corroborate the
clinical efficacy of these new therapeutic strategies, with the
specific aim of improving the survival of patients not suitable for
allo-HSCT and of increasing the number of patients suitable for
allo-HSCT.
References
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M.
Epidemiology of acute myeloid leukemia: recent progress and enduring
challenges. Blood Rev. 2019, 36, 70-87.
https://doi.org/10.1016/j.blre.2019.04.005 PMid:31101526
- Juliusson, G.; Abrahamsson, J.; Lazarevic, V.; Antonovic, P.; Derolf,
A.; Gorelius, H.; Lehmanns, S.; Myhr-Eriksson, K.; Mollgord, L.; Uggla,
B.; et al. Prevalence and characteristics of survivors from acute
myeloid leukemia in Sweden. Leukemia 2017, 31, 728-731.
https://doi.org/10.1038/leu.2016.312 PMid:27795559
PMCid:PMC5339425
- Roman, E.; Smith, A.; Appleton, S.; Crouch, S.; Kelly, R.; Kinsey, S.;
Corgo C.; Patmore, R. Myeloid malignancies in the real-world:
occurrence, progression and survival in the UK's population-based
Haematological Malignancy Research Network 20094-15. Cancer Epidemiol.
2016, 42, 186-198. https://doi.org/10.1016/j.canep.2016.03.011
PMid:27090942 PMCid:PMC4911595
- Vardiman, J.W.; Harris, N.L.; Brunning, R.D. The World Health
Organization (WHO) classification of the myeloid neoplasms. Blood 2002,
100, 2292-2302. https://doi.org/10.1182/blood-2002-04-1199
PMid:12239137
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitrz,
M.T.; Porwit, A.; Harris, N.L.; LeBeau, M.M.; Hallstrom-Lindberg, E.;
Tefferi, A.; et al. The 2008 revision of the World Health Organization
(WHO) classification of myeloid neoplasms and acute leukemia: rationale
and important changes. Blood 2009, 114, 937-951.
https://doi.org/10.1182/blood-2009-03-209262 PMid:19357394
- Cancer Genome Atlas Research, Ley, T.J.; Miller, C.; Ding, L.; Raphael,
B.J.; Mingall, A.J.; Robertson, A.G.; Hoadley, AS.K.; Triche, T.J.;
Laird, P.W.; et al Genomic and epigenomic landscapes of adult de novo
acute myeloid leukemia. N. Engl. J. Med 2013, 368, 20959-2074.
https://doi.org/10.1056/NEJMoa1301689 PMid:23634996 PMCid:PMC3767041
- Hou, H.A.; Lin, C.C.; Chou, W.C.; Liu, C.Y.; Chen, C.Y.; Tang, J.L.;
Lai, Y.J.; Tseng, M.H.; Huang, C.F.; Chiang, Y.C.; et al. Integration
fo cytogenetic and molecular alterations in risk stratification of 318
patients with de novo non-M3 acute myeloid leukemia. Leukemia, 2014,
28, 50-58. https://doi.org/10.1038/leu.2013.236 PMid:23929217
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, K.; Paschka,
P.; Roberts, N.D.; Potter, N.E.; Hauser, M.; Thol, F.; Bolli, N.; et
al. Genomic classification and prognosis in acute myeloid leukemia. N.
Engl. J. Med. 2016, 374, 2209-2221.
https://doi.org/10.1056/NEJMoa1516192 PMid:27276561
PMCid:PMC4979995
- Bullinger, L.; Dohner, K.; Dohner, H. Genomics of acute myeloid
leukemia diagnosis and pathways. J. Clin. Oncol. 2017, 35, 934-946.
https://doi.org/10.1200/JCO.2016.71.2208 PMid:28297624
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le
Beau, M.M.; Blumfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016
revision of the World Health Organization (WHO) classification of
myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391-2405.
https://doi.org/10.1182/blood-2016-03-643544 PMid:27069254
- Kishtagari, A.; Levine, R.L.; Viny, A.D. Driver mutations in acute
myeloid leukemia. Curr. Opin. Hematol. 2020, 27, 49-57.
https://doi.org/10.1097/MOH.0000000000000567 PMid:31972687
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, J.;
Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, R.; Pich, O.; Bonet, J.;
Kranas, A.; et al. A compendium of mutational cancer driver genes. Nat.
Rev. Cancer 2020, 20, 555-570.
https://doi.org/10.1038/s41568-020-0290-x PMid:32778778
- Metzler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland
M.C.; Gorlich, D.; Schneider, S.; Kostandin, N.P.; Dufour, A.; Braundl,
K.; et al. Spectrum and prognostic relevance of driver gene mutations
in acute myeloid leukemia. Blood 2016, 128, 686-698.
https://doi.org/10.1182/blood-2016-01-693879 PMid:27288520
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.;
Borthakur, G. Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood 2017, 129,
424-447. https://doi.org/10.1182/blood-2016-08-733196 PMid:27895058
PMCid:PMC5291965
- Herold, T.; Rothenberg-Thurley, M.; Grumwald, V.V.; Janke, H.;
Goerlich, D.; Sauerland, M.S.; Kostandin, N.P.; Dufour, A.; Schneider,
S.; Neusser, M.; et al. Validation and refinement of the revised 2017
European Leukemia Net genetic risk stratification of acute myeloid
leukemia. Leukemia 2020, in press.
https://doi.org/10.1038/s41375-020-0806-0 PMid:32231256
PMCid:PMC7685975
- Fleming, S.; Tsai, C.H.; Dohner, H.; Dohner, K.; Papaemmanuil, E.;
Tien, H.F.; Reynolds, J.; Wei, A.H.; Hou, H.A. Use of machine-learning
in 2074 cases of acute myeloid leukemia for genetic risk profiling.
Blood 2019, 134, suppl.1, abst. 1392.
https://doi.org/10.1182/blood-2019-128243
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.;
Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al.
Functional genomic landscape of acute myeloid leukaemia. Nature 2018,
562, 526-531.
- Bottomly, D.; Long, N.; Schultz, S.E.; Kurtz, S.E.; Tognon, C.E.;
Johnson, K.; Abel, M.; Agarwal, A.; Avaylon, S.; Benton, E.; et al.
Integrative analysis of drug response and clinical outcome in acute
myeloid leukemia. Cancer Cell 2022, 40, 850-864.
https://doi.org/10.1016/j.ccell.2022.07.002 PMid:35868306
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes,
A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified
classification and risk-stratification in acute myeloid leukemia. Nat
Commun 2022, 13, 4622. https://doi.org/10.1038/s41467-022-32103-8
PMid:35941135 PMCid:PMC9360033
- Gao, Y.; Jia, M.; Mao, Y.; Cai, H.; Jiang, X.; Cao, X.; Zhou, D.; Li,
J. Distinct mutation landscapes between acute myeloid leukemia with
myelodysplasia-related changes and de novo acute myeloid leukemia. Am J
Clin Pathol 2022, 157, 691-700. https://doi.org/10.1093/ajcp/aqab172
PMid:34664628
- Kang, D.; Jung, J.; Park, S.; Cho, B.S.; Kim, H.J.; Kim, Y.; Lee, J.M.;
Kim, H.S.; Ahn, A.; Kim, M.; et al. Genetic characteristics according
to subgroup of acute myeloid leukemia with myelodysplasia-related
changes. J Clin Med 2022, 11, 2378. https://doi.org/10.3390/jcm11092378
PMid:35566503 PMCid:PMC9105081
- Qin, G.; Han, X. The prognostic value of TP53 mutations in adult acute
myeloid leukemia: a meta-analysis. Transf Med Hemother 2022, in press.
https://doi.org/10.1159/000526174
- Lipilkin, P.V.; Kulaeva, E.D.; Mashkina, E.V. Prognostic value of ASXL1
mutations in acute myeloid leukemia: a meta-analysis. Leuk Res 2022,
120, 106910. https://doi.org/10.1016/j.leukres.2022.106910
PMid:35785697
- Pan, X.; Mengge, G.; Wang, K.; Wang, Y.; Kong, J.; Sun, Y.; Zhao, X.;
Huang, X.J. Prognostic impact of WT1 mutation on AML of different risk
groups based on 2022 European Leukemianet (ELN) risk classification.
Blood 2022, 140, 3216-3217. https://doi.org/10.1182/blood-2022-166323
- Gardin, C.; Pautas, C.; Fournier, E.; Itkynson, R.; Lamasle, E.;
Bourhis, J.H.; Ades, L.; Marolleau, J.P.; Malfuson, J.V.; Gastaud,
L.; et al. Added prognostic value of secondary AML-like gene mutations
in ELN intermediate-risk older AML: ALFA-1200 study results. Blood Adv
2020, 4, 1942-1951. https://doi.org/10.1182/bloodadvances.2019001349
PMid:32380535 PMCid:PMC7218423
- Stahl, M.; Derkach, A.; Farnoud, N.; Bewersdorf, J.P.; Robinson, T.;
Famulare, C.; Cho, C.; Devlin, S.; Menghrajani, K.; Patel, M.A.; et al.
Molecular predictors of immunophenotypic mesurable residual disease
clearance in acute myeloid leukemia. Am J Hematol 2023, 98, 79-89.
https://doi.org/10.1002/ajh.26757 PMid:36251406
- Wang, M.; Lindberg, J.; Klevebring, D.; Nilsson, C.; Lehmann, S.;
Gronberg, H.; Rantailainen, M. Developemnt and validation of a novel
RNA sequencing-based prognostic score for acute myeloid leukemia. J
Natl Cancer Dev 2018, 110, 1094-1101.
https://doi.org/10.1093/jnci/djy021 PMid:29506270
PMCid:PMC6186516
- Arindrarto, W.; Borràs, D.M.; de Groen R. ; van den Berg, R.; Locher,
I.J.; van Diessen, S.; van der Holst, R. ; van der Meijden, E. ;
Honders, W.; de Leueuw, R. ; et al. Comprehensive diagnostics of acute
myeloid leukemia by whole transcriptome RNA sequencing. Leukemia 2021,
35, 47-61. https://doi.org/10.1038/s41375-020-0762-8 PMid:32127641
PMCid:PMC7787979
- Kong, W.; He, L.; Zhu, J.; Bruck, O.; Porkka, K.; Heckman, C.A.; Zhu,
S.; Aittokallio, T. An immunity and pyroptosis gene-pair signature
predicts overall survival in acute myeloid leukemia. Leukemia 2022, 36,
2384-2395. https://doi.org/10.1038/s41375-022-01662-6 PMid:35945345
PMCid:PMC9522598
- Nehme, A.; Dakik, H.; Picou, F.; Cheok, M.; Preudhomme, C. ; Dombret,
H. ; Lambert, J. ; Gyan, E. ; Pigneux, A. ; Récher, C. ; et al.
Horizontal meta-analysis identifies common deregulated genes, across
AML subgroups providing a robust prognostic signature. Blood Adv 2020,
4, 5322-5330. https://doi.org/10.1182/bloodadvances.2020002042
PMid:33108456 PMCid:PMC7594391
- Wagner, S.; Vadakekolathu, J.; Tasian, S.K.; Altmann, H.; Bornhauser,
M.; Pockley, A.G.; Ball, G.R.; Rutella, S. A parsimonius 3-gene
signature predicts clinical outcomes in an acute myeloid leukemia
cohort. Blood Adv 2019, 3, 1330-1342.
https://doi.org/10.1182/bloodadvances.2018030726 PMid:31015209
PMCid:PMC6482359
- Docking, T.R.; Parker, J.; Jadersten, M.; Duns, G.; Chang, L.; Jiang,
J.; Pilsworth, J.A.; Swanson, L.A.; Chan, S.K.; Chiu, R.; et al. A
clinical transcriptome approach to patient stratification and therapy
selection in acute myeloid leukemia. Nat Commun 2021, 12, 2474.
https://doi.org/10.1038/s41467-021-22625-y PMid:33931648
PMCid:PMC8087683
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.;
Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et
al. A 17-gene stemness score for rapid determination of risk of acute
leukemia. Nature 2016, 540, 433-437.
https://doi.org/10.1038/nature20598 PMid:27926740
- Bill, M.; Nicolet, D.; Kohlschmidt, J.; Walker, C.J.; Mrozek, K.;
Eisfeld, A.K.; Papaioannou, D.; Rong-Mullins, X.; Brannan, Z.; Kolitz,
J.E.; et al. Mutations associated with a 17-gene leukemia staem cell
score and the score's prognostic relevance in the context of the
European LeukemiaNet classification of acute myeloid leukemia.
Haematologica 2020, 105, 721-729.
https://doi.org/10.3324/haematol.2019.225003 PMid:31413100
PMCid:PMC7049376
- Ng, S.W.; Murphy, T.; King, I.; Zhang, T.; Mah, M.; Lu, Z.; Stickle,
N.; Ibrahimova, N.; Arruda, A.; Mitchell, A.; et al. A clinical
laboratory development LSC17 stemness score assay for rapid risk
assessment of patients with acute myeloid leukemia. Blood Adv 2022, 6,
1064-1070. https://doi.org/10.1182/bloodadvances.2021005741
PMid:34872104 PMCid:PMC8945314
- Huang, B.J.; Smith, J.L.; Farrar, J.E.; Wang, Y.C.; Umeda, M.; Ries,
R.E.; Leonti, A.R.; Crowgei, mE.; Furlan, S.N.; Tarlock, K.; et al.
Integrated stem cell signature and cytomolecular risk determination in
pediatric acute myeloid leukemia. Nature Commun 2022, 13, 5487.
https://doi.org/10.1038/s41467-022-33244-6 PMid:36123353
PMCid:PMC9485122
- Cheng, W.Y.; Li, J.F.; Zhu, Y.M.; Lin, X.J.; Wen, L.J.; Zhang, F.;
Zhang, Y.L.; Zhao, M.; Fang, H.; Wang, S.Y.; et al. Transcriptome-based
molecular subtypes and transcription hierarchies improve the
classification framework of acute myeloid leukemia. Proc Natl Acad Sci
USA 2022, 119, e2211429119. https://doi.org/10.1073/pnas.2211429119
PMid:36442087
- Van Galen, P.; Hovestadt, V.; Wadsworth II, M.H.; Hughes, T.K.;
Griffin, G.K.; Battaglia, S.; Verga, J.A.; Syephansky, J.; Pastika,
T.J.; Lombardi Story, J.; et al. Single-cell RNA-Seq reveals AML
hierarchies relevant to disease progression and immunity. Cell 2019,
176, 1265-1281. https://doi.org/10.1016/j.cell.2019.01.031
PMid:30827681 PMCid:PMC6515904
- Zeng, A.; Bansal, S.; Jin, L.; Mitchell, A.; Chen, W.C.; Abbas, H.A.;
Chan-Seng-Yue, M.; Voisin, V.; van Galen, P.; Tierens, A.; et al. A
cellular hierarchy framework for understanding heterogeneity and
predicting drug response in acute myeloid leukemia. Nat Med 2022, 28,
1212-1223. https://doi.org/10.1038/s41591-022-01819-x
PMid:35618837
- Swerdlow, S.H.; Campo, E.; Harris, N.L. WHO classification of tumours
of haematopoietic and lymphoid tissues. Geneva, Switzerland, WHO,
2008.
- Friis, L.S.; Kjeldsen, E.; Werenbeg Marcher, C.; Preiss, B., et al.
Epidemiology and clinical significance of secondary and therapy-related
acute myeloid leukemia: a national population-based cohort study. J.
Clin. Oncol. 2015, 33, 3641-3649.
https://doi.org/10.1200/JCO.2014.60.0890 PMid:26304885
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.;
Allen, S.L:; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P., et al.
Acute myeloid leukemia ontogeny is defined by distinct somatic
mutations. Blood 2015, 125, 1367-1376.
https://doi.org/10.1182/blood-2014-11-610543 PMid:25550361
PMCid:PMC4342352
- Nazha, AS.; Al-Issa, K.; Radivoyevitch, T.; Carraway, H.E.; Sanikommu,
S.; Kalaoyo, M.; et al. The complexity of interpreting genomic data in
patients with acute myeloid leukemia. Blood Cancer J. 2016, 6, e510.
https://doi.org/10.1038/bcj.2016.115 PMid:27983727
PMCid:PMC5223150
- Kayser, S.; Dohner, K.; Krauter, J.; Kohne, C.H.; Horst, H.A.; Held,
G.; von Lillienfeld-Tost, M.; Wilhelm, S.; Kundgen, A.; Gotze, K.; et
al. The impact of therapy-related acute myeloid leukemia (AML) on
outcome in 2853 adult patients with newly diagnosed AML. Blood 2011,
117, 2137-2145. https://doi.org/10.1182/blood-2010-08-301713
PMid:21127174
- Kuzmanovic, T.; Patel, B.J.; Sankommu, S.R.; Nagata, Y.; Awada, H.;
Kerr, C.M.; Przychodzen, B.P.; Iha, B.K.; Hiwasa, D.; Singhal, D.; et
al. Genomics of therapy-related myeloid neoplasms. Haematologica 2020,
105, e98-e101. https://doi.org/10.3324/haematol.2019.219352
PMid:31413096 PMCid:PMC7049337
- McNermey, M.E.; Godley, L.A.; LeBeau, M.M. Therapy-related neoplasms:
when genetics and environment collide. Nature Rev. Cancer 2017, 17,
503-527. https://doi.org/10.1038/nrc.2017.60 PMid:28835720
PMCid:PMC5946699
- Kuendgen, A.; Nomdedeu, M.; Tuechler, H.; Garcia-Manero, G.; Korokji,
R.S.; Sekeres, M.A.; Della Porta, M.G.; Cazzola, M.; DeZern, A.E.;
Roboz, G.J.; et al. Therapy-related myelodysplastic syndromes deserve
specific diagnostic sub-classification and risk-stratification - an
approach to classification of patients with t-MDS. Leukemia 2021, 35,
835-849. https://doi.org/10.1038/s41375-020-0917-7 PMid:32595214
PMCid:PMC7932916
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Fu, B.; Tang,
G.; Goswami, M.; Singh, R.; Kanagal-Shamanna, R.; Pierce, S.A.; et al.
Mutational profiling of therapy-related myelodysplastic syndromes and
acute myeloid leukemia by next generation sequencing, a comparison with
de novo diseases. Leuk Res 2015, 39, 348-354.
https://doi.org/10.1016/j.leukres.2014.12.006 PMid:25573287
PMCid:PMC5548131
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson,
W.; Grauman, P.V.; Hu, Z.H.; Spellman, S.R.; Lee, S.J.; et al.
Prognostic mutations in myelodysplastic syndrome after stem cell
transplantation. N Eng J Med 2017, 376, 536-547.
https://doi.org/10.1056/NEJMoa1611604 PMid:28177873
PMCid:PMC5438571
- Leone, G.; Fabiani, E.; Voso, M.T. De novo and therapy-related
myelodysplastic syndromes: analogies and differences. Med J Hematol
Infec Dis 2022, 14, e2022030. https://doi.org/10.4084/MJHID.2022.030
PMid:35615324 PMCid:PMC9083943
- Dohner, H.; Wei, A,H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.;
Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.;
Diagnosis and management of AML in adults: 2022 ELN recommendations
from an internaitonal panel. Blood 2022, 140, 1345-1377.
https://doi.org/10.1182/blood.2022016867 PMid:35797463
- Jaiswal, S.; Fontanillas, P.; Flannick, J. Age-related clonal
hematopoiesis associated with diverse outcomes. N. Engl. J Med. 2014,
371, 2488-2498. https://doi.org/10.1056/NEJMoa1408617 PMid:25426837
PMCid:PMC4306669
- Abelson, S.; Collord, G.; Ng, S.; Weissbrod, O.; Cohen, M.N.; Niemer,
E.; Barda, N.; Zuzarte, P.C.; Heisler, R.; Sundravadanam, Y.; et al.
Prediction of acute myeloid leukemia risk in healthy individuals.
Nature 2018, 559: 400-404. https://doi.org/10.1038/s41586-018-0317-6
PMid:29988082 PMCid:PMC6485381
- Desai, P.; Mencia-Trinchant, N.; Savenkov, O.; Simon, M.S.; Cheang, G.;
Lee, S.; Samuel, M.; Ritchie, E.K.; Guzman, M.L.; Baliman, K.V.; et al.
Somatic mutations precede acute myeloid leukemia years before
diagnosis. Nat. Med. 2018, 24: 1015-1023.
https://doi.org/10.1038/s41591-018-0081-z PMid:29988143
PMCid:PMC6849383
- Young, A.L.; Tong, R.S.; Birmann, B.M.; Druley, T.E. Clonal
hematopoiesis and risk of acute myeloid leukemia. Haematologica 2019,
104, 2010-2017. https://doi.org/10.3324/haematol.2018.215269
PMid:31004019 PMCid:PMC6959179
- Watson, C.J.; Papula, A.; Poon, Y.; Wong, W.H.; Young, A.L., Druley,
T.E.; Fisher, D.S.; Blundell, J.R. The evolutionary dynamics and
fitness landscape of clonal hematopoiesis. Science, 2020, 367,
1449-1454. https://doi.org/10.1126/science.aay9333
PMid:32217721
- Fabre, M.A.; de Almeida, J.G. ; Fiorillo, E.; Mitchelkl, E.; Damaskou,
A.; Rak, J.; Orrù, V.; Marongiu, M.; Spencer Chapman, M.; Vijayabaskar,
M.S.; et al.The longitudinal dynamics and natural history of clonal
hematopoiesis. Nature 2022, 606, 335-342.
https://doi.org/10.1038/s41586-022-04785-z PMid:35650444
PMCid:PMC9177423
- Saiki, R.; Momozawa, Y.; Nannya, Y.; Nakagawa, M.M.; Ochi, Y.;
Yoshizato, T.; Terao, C.; Kuroda, Y.; Shiraishi, Y.; Chiba, K.; et al.
Combined landscape of single-nucleotide and copy number alterations in
clonal hematopoiesis. Nature Med 2021, 27, 1239-1249.
https://doi.org/10.1038/s41591-021-01411-9 PMid:34239136
- Gao, T.; Ptashkin, R.; Bolton, K.L.; Sirenko, M.; Fong, C.; Spitzer, B.;
Menghrajami, K.; Arango Ossa, J.E.; Zhou, Y.; et al. Interplay between
chromosomal alterations and gene mutations shapes the evolutionary
trajectory of clonal hematopoiesis. Nature Commun 2021, 12, 338.
https://doi.org/10.1038/s41467-020-20565-7 PMid:33436578
PMCid:PMC7804935
- Niroula, A.; Sekar, A.; Murakami, M.A.; Trinder, M.; Agrawal, M.; Wong,
W.J.; Bick, A.G.; Uddin, M.M.; Gibson, D.J.; Griffin, G.K.; et al.
Distinction of lymphoid and myeloid clonal hematopoiesis. Nature Med
2021, 27, 1921-1927. https://doi.org/10.1038/s41591-021-01521-4
PMid:34663986 PMCid:PMC8621497
- Kessler, M.D.; Damsk, A.; O'Keaffe, S.; Banerejee, N.; Li, D.;
Watanabe, K.; Marketta, A.; Van Meter, M.; Semrau, S.; Horowitz, J.; et
al. Common and rare variant associations with clonal hematopoiesis
phenotypes. Nature, 2022, 612, 301-310.
https://doi.org/10.1038/s41586-022-05448-9 PMid:36450978
PMCid:PMC9713173
- Skead, K.; Houle, A.A.; Abelson, S.; Agbessi, M.; Bruat, V.; Lin, B.;
Soave, D.; Shlush, L.; Wright, S.; Dick, J.; et al. Interacting
evolutionary pressures drive mutation dynamics and health outcomes in
aging blood. Nat Commun 2021, 2, 4921.
https://doi.org/10.1101/2020.04.25.058677 PMCid:PMC7666524
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch,
J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role
of TP53 mutations in the origin and evolution of therapy-related acute
myeloid leukemia. Nature 2015, 518: 552-555.
https://doi.org/10.1038/nature13968 PMid:25487151
PMCid:PMC4403236
- Gillis, N.K.; Ball, M.; Zhang, Q.; Ma, Z.; Zhao, Y.; Yoder, S.J.;
Balasis, M.E.; Masa, T.E.; Sallman, D.A.; Lancet, J.E.; et al. Clonal
haemopoiesis and therapy-related myeloid malignancies in elderly
patients: a proof-of-concept, case-control study. Lancet Oncol 2017,
18, 112-121. https://doi.org/10.1016/S1470-2045(16)30627-1
PMid:27927582
- Takahashi, K.; Wang, F.; Kantarjian, H.; Doss, D.; Khanna, K.;
Thompson, E.; Zhao, L.; Patel, K.; Neelapu, S.; Gumbs, C.; et al.
Pre-leukemic clonal hematopoiesis and the risk of therapy-related
myeloid neoplasms: a case-control study. Lancet Oncol. 2017, 18:
100-111. https://doi.org/10.1016/S1470-2045(16)30626-X PMid:27923552
- Takahashi, K.; Wang, F.; Kantarjian, H.; Song, X.; Patel, K.; Neelapu,
S.; Gumbs, C.; Little, L.; Tippen, S.; Thornton, R.; et al. Copy number
alterations detected as clonal hematopoiesis of indeterminate
potential. Blood Adv. 2017, 1, 1031-1036.
https://doi.org/10.1182/bloodadvances.2017007922 PMid:29296745
PMCid:PMC5728325
- Coombs, C.C.; Zehir, A.; Devlin, S.M.; Kishtagari, A.; Syed, A.;
Jonsson, P.; Hyman, D.M.; Solit, D.B.; Robson, M.E.; Baselga, J.; et
al. Therapy-related clonal hematopoiesis in patients with
non-hematologic cancers is common and associated with adverse clinical
outcomes. Cell Stem Cell 2017, 21, 374-382.
https://doi.org/10.1016/j.stem.2017.07.010 PMid:28803919
PMCid:PMC5591073
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer. E.; Jeong, M.; Wang,
F.; Zhang, J.; Heffernan, T.; Gera, S.; Kovacs, J.J.; et al. PPM1D
mutations drive clonal hematopoiesis in response to cytotoxic
chemotherapy. Cell Stem Cell 2018, 23, 700-713.
https://doi.org/10.1016/j.stem.2018.10.004 PMid:30388424
PMCid:PMC6224657
- Bolton, K.L.; Ptaskin, R.N.; Gao, T.; Braunstein, L., Devlin, S.M.;
Kelly, D.; Patel, M.; Barthon, A.; Syed, A.; Yabe, M.; et al. Cancer
therapy shapes the fitness landscape of clonal hematopoiesis. Nat.
Genet. 2020, 52, 1219-1226. https://doi.org/10.1038/s41588-020-00710-0
PMid:33106634 PMCid:PMC7891089
- Liu, Y.; Elf, S.E.; Miyata, Y. ; Sashida, G.; Liu, Y.; Huang, G.; Di
Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; Antipin, J.;
et al. P53 regulates hematopoietic stem cell quiescence. Cell Stem Cell
2009, 4, 37-48. https://doi.org/10.1016/j.stem.2008.11.006
PMid:19128791 PMCid:PMC2839936
- Asai, T.; Liu, Y.; Di Giandomenico, S.; Bae, N.; Ndiaye-Lobry, D.;
Deblasio, A.; Menendez, S.; Antipin, Y.; Reva, B.; Wewrick, R.; et al.
Necdin, a p53 gene, regulates the quiescence and response to genotoxic
stress of hematopoietic stem/progenitor cells. Blood 2012, 120,
1601-1612. https://doi.org/10.1182/blood-2011-11-393983 PMid:22776820
PMCid:PMC3429304
- Zhang, Z.; Lu, Y.; Qi, Y. ; Xu, Y. ; Wang, S.; Chen, F. ; Shen, M.;
Chen, M.; Chen, N.; Yang, L.; et al. CDK19 regulates the proliferation
of hematopoietic stem cells and acute myeloid leukemia cells by
suppressing p53-mediated transcription of p21. Leukemia 2022, 36,
956-969. https://doi.org/10.1038/s41375-022-01512-5
PMid:35110726
- Chen, S.; Gao, R.; Yao, C.; Kobayashi, M.; Liu, S.Z.; Yoder, M.C.;
Broxmeyer, H.; Kapur, R.; Boswell, H.S.; Mayo, L.D.; et al. Genotoxic
stresses promote clonal expansion of hematopoietic stem cells
expressing mutant p53. Leukemia 2018, 32, 850-854.
https://doi.org/10.1038/leu.2017.325 PMid:29263439
PMCid:PMC5842141
- Chen, S.; Wang, Q.; Yu, H.; Capitano, M.L.; Vemula, S.; Nabinger, S.C.;
Gao, R.; Yao, C.; Kobayashi, M.; Geng, Z.; et al. Mutant p53 drives
clonal hematopoiesis through modulating epigenetic pathway. Nat Commun
2019, 10, 5649. https://doi.org/10.1038/s41467-019-13542-2
PMid:31827082 PMCid:PMC6906427
- Shah, M.V.; Mangaonkar, A.A.; Begna, K.H.; Alkhateeb, H.B.; Greipp, P.;
Nanaa, A.; Elliott, M.A.; Hogan, W.J.; Litzow, M.R.; McCullough, K.; et
al. Therapy-related clonal cytopenia as a precusror to therapy-related
myeloid neoplasms. Blood Cancer J 2022, 12, 106.
https://doi.org/10.1038/s41408-022-00703-8 PMid:35803921
PMCid:PMC9270475
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049-1059.
https://doi.org/10.1182/blood-2018-10-844621 PMid:30670442
PMCid:PMC6587668
- Wang, W.; Routbort, M.J.; Tang, Z. ; Ok, C.Y.; Patel, K.P.; Daver, N.;
Garcia-Manero, G.; Medeiros, L.J.; Wang, S.A. Characterization of TP53
mutotions in low-grade myelodysplastic syndromes with a non-complex
karyotype. Eur J Haematol 2017, 99, 536-543.
https://doi.org/10.1111/ejh.12971 PMid:28926144
- Hirsch, C.M.; Przychodzen, B.P.; Radivoyevitch, T.; Patel, B.; Thota,
S.; Clemente, M.J.; Nagata, Y.; LaFramboise, T.; Carraway, H.E.; Nazha,
A.; et al. Molecular features of early onset adult myelodysplastic
syndrome. Haematologica 2017, 102, 1028-1034.
https://doi.org/10.3324/haematol.2016.159772 PMid:28255022
PMCid:PMC5451334
- Epstein-Peterson, Z.D.; Spitzer, B.; Derkach, A.; Arango, J.E.;
McCarter, J.; Medina-Martinez, J.S.; McGovern, E.; Farnoud, N.R.;
Levine, R.L.; Talmman, M.S. De novo myelodysplastic syndromes in
patients 20-50 years old are enriched for adverse risk features. Leuk
Res 2022, 117, 106857. https://doi.org/10.1016/j.leukres.2022.106857
PMid:35598475
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devin, S.M.; Tuecher, H.;
Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.;
Malcovati, L.; et al. Implications of TP53 allelic state for genome
stability, clinical presentation and outcomes in myelodysplastic
syndromes. Nat Med 2020, 26,n 1549-1556.
https://doi.org/10.1038/s41591-020-1008-z PMid:32747829
PMCid:PMC8381722
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.;
Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.;
Haferlach, T.; et al. TP53 mutation status divides myelodysplastic
syndromes with complex karyotypes into distinct prognostic subgroups.
Leukemia 2019, 33, 1747-1758. https://doi.org/10.1038/s41375-018-0351-2
PMid:30635634 PMCid:PMC6609480
- Jadersten, M.; Saft, L., Smith, A.; Kulasekararay A.; Panplun, S.;
Gohring, G.; Hedland, A.; Hast, R.; Schlegelberger, B.; Porwit, A.;
Hellstrom-Lindberg, E. TP53 mutations in low-risk myelodysplastic
syndromes with del(5q) predict disease progression. J Clin Oncol 2011,
29, 1971-1979. https://doi.org/10.1200/JCO.2010.31.8576 PMid:21519010
- Crisà, E.; Kalesekaray, A.G.; Adema, V.; Such, E.; Schenz, J.; Haase,
D.; Shirneshen, K.; Best, S.; Mian, S.A.; Kizilers, A.; et al. Impact
of somatic mutations in myelodysplastic patients with isolated partial
or total loss of chromosome 7. Leukemia 2020, 34, 2441-2450.
https://doi.org/10.1038/s41375-020-0728-x PMid:32066866
- Jain, A.G.; Ball, S.; Aguirre, L.E.; Al Ali, N.; Kaldas, D.;
Tinsley-Vance, S.; Kuykendall, A.T.; Chan, O.; Sweet, K.; Lancet, J.E.;
et al. The natural history of lower risk MDS: factors predicting
progression to high-risk myelodysplastic syndrome an acute myeloid
leukemia in patients with very low and low risk MDS according to the
R-IPSS criteria. Blood 2021, 138, suppl.1, 2600.
https://doi.org/10.1182/blood-2021-149708
- Belickova, M.; Vesela, J.; Josnasova, A.; Pejsova, B.; Votavova, H.;
Merkerova, M.D. TP53 mutation variant allele frequency is a potential
predictor for clinical outcome of patients with lower-risk
myelodysplastic syndromes. Oncotarget 2016, 7, 36266-36279.
https://doi.org/10.18632/oncotarget.9200 PMid:27167113
PMCid:PMC5094999
- Sellman, D.A.; Kromrokji, R.; Vaupel, C.; Cluzeau, T. ; Geyer, S.M.;
McGrow, K.L. Impact of TP53 mutation variant allele frequency on
phenotype and outcomes in myelodysplastic syndromes. Leukemia 2016, 30,
666-673. https://doi.org/10.1038/leu.2015.304 PMid:26514544
PMCid:PMC7864381
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Benton, C.B.; Class, C.A.;
Chien, K.S.; Naqvi, K.; Alvarado, Y.; Kadia, T.M.; Ravandi, F.; Daver,
N.; et al. Genomic context and TP53 allele frequency define clinical
outcomes in TP53-mutated myelodysplastic syndromes. Blood Adv 2020, 4,
482-490. https://doi.org/10.1182/bloodadvances.2019001101 PMid:32027746
PMCid:PMC7013259
- Aguirre, L.E.; Al Ali, N.; Jain, A.G.; Chan, O.; Ball, S.; Kuykendall,
A.; Sweet, K.; Lancet, J.E.; Padron, E.; Komrokji, R.S.; Sallman, D.A.
Characterization of TP53-mutated myelodysplastic syndromes and impact
of allelic status and concurrent cytogenetic abnormalities on survival
outcomes. Blood 2022, 140 (suppl.1), 1755.
https://doi.org/10.1182/blood-2022-171093
- Kanagal-Shamanna, R.; Montalban-Bravo, G.; Katsonis, P.; Sasaki, K.;
Class, C.A.; Jabbour, E.; Sallman, D.; Hunter, A.M.; Benton, C.; Chien,
K.S.; et al. Evolutionary action score identifies a subset of TP53
mutated myelodysplastic syndrome with favorable porgnosis. Blood Cancer
J 2021, 11, 52. https://doi.org/10.1038/s41408-021-00446-y
PMid:33677472 PMCid:PMC7936977
- The Cancer Genome Atlas Res Net. Genomic and epigenomic landscapes of
adult de novo acute myeloid leukemia. N Engl J Med 2013, 368,
2059-2074. https://doi.org/10.1056/NEJMoa1301689 PMid:23634996
PMCid:PMC3767041
- Bowen, D.; Graves, M.J.; Burnett, M.K.; Patel, Y.; Allen, C.; Green,
C.; Gale, R.E.; Hills, R.; Linch, D.C. Tp53 gene mutation is frequent
in patients with acute myeloid leukemia and complex karyotype, and is
associated with very poor prognosis. Leukemia 2009, 203-206.
https://doi.org/10.1038/leu.2008.173 PMid:18596741
- Rucker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Toleanu, V.;
Kett, H.; Hadbank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et
al. TP53 alterations in acute myeloid leukemia with complex karyotype
correlates with specific copy number alterations, monosomal karyotype,
and dismal outcome. Blood 2012, 119, 2114-2121.
https://doi.org/10.1182/blood-2011-08-375758 PMid:22186996
- Abbas, H.A.; Ayoub, E.; Sun, H.; Kanagal-Shamanna, R.; Shor, N.J.;
Issa, G.; Yilmaz, M.; Pierce, S.; Rivera, D.; Cham, B.; et al. Clinical
and molecular profiling of AML patients with chromosome 7 or 7q
deletions in the context of TP53 alterations and venetoclax treatment.
Leuk Lymphoma 2022, 63, 3105-3116.
https://doi.org/10.1080/10428194.2022.2118533 PMid:36089905
- Boettcher, S.; Miller, P.G.; Sharma, R.; McConkey, M.; Leventhal, M.;
Krivstov, A.V.; Giacomelli, A.O.; Wong, W.; Kim, J.; Chao, S.; et al. A
dominant-negative effect drives selection of TP53 missense mutations in
myeloid malignancies. Science 2019, 365, 599-604.
https://doi.org/10.1126/science.aax3649 PMid:31395785
PMCid:PMC7327437
- Zebisch, A.; Lai, R.; Muller, M.; Lind, K.; Kashofer, K.;
Girschikofsky, M.; Fuchs, D.; Wolfler, A.; Geigl, J.B.; Sill, H. Acute
myeloid leukemia with TP53 germ line mutations Blood 2016, 128,
2270-2272. https://doi.org/10.1182/blood-2016-08-732610 PMid:27621308
PMCid:PMC5095760
- Stengel, A.; Kern, W.; Haferlach, T.; Meggendorfer, M.; Fasan, A.;
Haferlach C. The impact of TP53 mutations and TP53 deletions on
survival varies between AML, ALL, MDS and CLL: an analysis of 3307
cases. Leukemia 2017, 31, 705-711. https://doi.org/10.1038/leu.2016.263
PMid:27680515
- Tashakori, M.; Kadia, T.; Loghavi, S.; Daver, N.; Kanagal-Shamanna, R.;
Pierce, S.; Sui, D.; Wei, P.; Khodakarami, F.; Tang, Z.; et al. TP53
copy number and protein expression inform mutation status across risk
categories in acute myeloid leukemia. Blood 2022, 140, 58-72.
https://doi.org/10.1182/blood.2021013983 PMid:35390143
PMCid:PMC9346958
- Prochazka, K.T.; Pregartner, G.; Rucker, F.G., Heitzer, E., Pabst, G.;
Wolfer, A.; Zebisch, A.; Berghold, A.; Dohner, K.; Sill, H. Clinical
implications of subclonal TP53 mutations in acute myeloid leukemia.
Haematologica 2019, 104, 516-523.
https://doi.org/10.3324/haematol.2018.205013 PMid:30309854
PMCid:PMC6395341
- Goel, S.; Hall, J.; Pradhan, K.; Hirsch, C.; Przichodzen, B.; Shastri,
A.; Mantzaris, I.; Janakiram, M.; Battini, R.; Kornblum, N., et al.
High prevalence and allele burden-independent prognostic importance of
p53 mutations in an inner-city MDS/AML cohort. Leukemia 2016, 30,
1793-1795. https://doi.org/10.1038/leu.2016.74 PMid:27125205
- Short, N.J.; Montalban-Bravo, G.; Hwang, H.; Ning, J.; Iranquiz, M.J.;
Kanagal-ShamannaR. Prognostic and therapeutic impacts of mutant TP53
variant allelic frequency in newly diagnosed AML. Blood 2020, 4,
5681-5689. https://doi.org/10.1182/bloodadvances.2020003120
PMid:33211826 PMCid:PMC7686900
- Fenwarth, L.; Vasseur, L.; Duployez, N.; Gardin, C.; Terré, C.;
Lambert, J.; de Botton, S.; Celli-Lebras, K.; Turlure, P.; Cluzeau,
T ; et al. Prognostic factor of monoallelic versus biallelic TP53
alterations in intensively-treated adult AML patients: a retrospective
study from the ALFA group. Blood 2022, 140 (suppl.1), 303.
https://doi.org/10.1182/blood-2022-163044
- Zhao, D.; Eladl, E.; Zarif, M.; Capo-Chichi, J.M., Schuh, A.; Atenafu,
E.; Minden, M.; Chang, H. Molecular characterization of AML-MRC reveals
TP53 mutation as an adverse prognostic factor irresespective of
MRC-defining criteria, TP53 allelic state, or TP53 variant allele
frequency. Cancer Medicine 2022, 00, 1-12.
https://doi.org/10.1002/cam4.5421 PMid:36394085
PMCid:PMC10067127
- Montalban-Bravo, G.; Khanagal-Shamanna, R.; Class, C.A.; Sasaki, K.;
Ravandi, F.; Cortes, J.E.; Daver, N.; Takahashi, K.; Short, N.J.;
DiNardo, C.D.; et al. Outcomes of acute myeloid leukemia with
myelodysplasia related changes depend on diagnostic criteria and
therapy. Am J Hematol 2020, 95, 612-622.
https://doi.org/10.1002/ajh.25769 PMid:32112433
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.;
Kvasnicka, H.M.; et al. International consensus classification of
myeloid neoplasms and acute leukemia integrating morphological,
clinical, and genomic data. Blood 2022, 140, 1200-1228.
- Grob, T.; Al Hinai, A.; Sanders, M.A.; Kavelaars, F.G.; Rijken, M.;
Gradowska, P.L.; Biemond, B.J.; Breems, D.A.; Maertens, J.; van Marwijk
Kooy, M.; et al. Molecular characterization of mutant TP53 acute
myeloid leukemia and high-risk myelodysplastic syndrome. Blood 2022,
139, 2347-2354. https://doi.org/10.1182/blood.2021014472 PMid:35108372
- Zeidan, A.M.; Bewersdorf, J.P.; Hasle, V.; Shallis, R.M.; Thompson, E.;
Lopes de Meneres, D.; Rose, S.; Boss, I.; Helene, S.; Haferlach, T.; et
al. Prognostic implications of mono-hit and multi-hit TP53 alterations
in patients with acute myeloid leukemia and higher risk myelodysplastic
syndromes treated with azacitidine-based therapy. Leukemia 2023, 37,
240-243. https://doi.org/10.1038/s41375-022-01766-z
PMid:36437356
- Zeidan, A.M., Boss, I.; Beach, C.L.; Copeland, W.B.; Thompson, E.; Fox,
B.A.; Hasle, V.E.; Ogasawara, K.; Cavenagh, J.; Silverman, L.R.; et al.
A randomized phase 2 trial of azacitidine with or without durvalumab as
first-line therapy for higher-risk myelodysplastic syndromes. Blood Adv
2022, 6, 2207-2218. https://doi.org/10.1182/bloodadvances.2021005487
PMid:34972214 PMCid:PMC9006291
- Zeidan, A.M., Boss, I.; Beach, C.L.; Copeland, W.B.; Thompson, E.; Fox,
B.A.; Hasle, V.E.; Hellmann, A.; Taussig, D.C.; Tormo, M.; Voso, M.T.;
et al. A randomized phase 2 trial of azacitidine with or without
durvalumab as first-line therapy for older patients with AML. Blood Adv
2022, 6, 2219-2229. https://doi.org/10.1182/bloodadvances.2021006138
PMid:34933333 PMCid:PMC9006260
- Nibourel, O.; Guihard, S.; Roumier, C.; Pottier, N.; Terre, C.;
Paquet, A.; Peyrouze, P.; Geffroy, S.; Quentin, S.; Alberdi, A.;
et al. Copy-number analysis identified new prognostic marker in acute
myeloid elukemia. Leukemia 2017, 31, 555-564.
https://doi.org/10.1038/leu.2016.265 PMid:27686867
- Lee, W.Y.; Gutierrez-Lanz, E. ; Xiao, H. ; McClintock, D.; Chan, M.P.;
Bixby, D.L.; Shao, L. ERG amplification is a secondary recurrent driver
event in myeloid malignancy with complex karyotype and TP53 mutations.
Genes Chromos Cancer 2022, 61, 399-411.
https://doi.org/10.1002/gcc.23027 PMid:35083818
- Schandl, C.A.; Mazzoni, S.; Znoyko, I.; Nahhas, G.; Chung, D.; Ding,
Y.; Hess, B.; Wolff, D.J. Novel high-risk acute myeloid leukemia
subgoup with ERG amplification and biallelic loss of TP53. Cancer Genet
2023, in press. https://doi.org/10.1016/j.cancergen.2023.01.004
PMid:36657266
- Rucker, F.G.; Dolnik, A.; Blatte, T.J.; Teleanu, V.; Ernst, A.; Thol,
F.; Heuser, M.; Ganser, A.; Dohner, H.; Dohner, K.; et al.
Chromotripsis is linked to TP53 alterations, cell cycle impairment, and
dismal outcome in acute myeloid leukemia with complex karyotype.
Haematologica 2018, 103, e18.
https://doi.org/10.3324/haematol.2017.180497 PMid:29079594
PMCid:PMC5777208
- Bochtler, T.; Granzow, M.; Stolzel, F.; Kunz, C.; Mohr, B.;
Kartal-Kaess, M.; Hinderhofer, K.; Heilig, C.E.; Kramer, M.; Thiede,
C.; et al. Marker chromosomes may arise from chromotripsis and predict
adverse prognosis in acute myeloid leukemia. Blood 2017, 129,
1333-1342. https://doi.org/10.1182/blood-2016-09-738161
PMid:28119329
- Fontana, M.C.; Marconi, G.; Milosevic Feenstra, J.D.; Fonzi, E.;
Papayannidis, C.; Luserna di Rorà, A.G.; Padella, A.; Solli, V.;
Franchini, E.; Ottaviani, DE.; et al. Chomotripsis in acute myeloid
leukemia: biological featurers and impact on survival. Leukemia 2018,
32, 1609-1620. https://doi.org/10.1038/s41375-018-0035-y PMid:29472722
PMCid:PMC6035145
- Abel, H.J.; Oetjen, K.A.; Miller, C.A.; Ramakrishnan, S.M.; Dai, R.;
Helton, N.M.; FronicK, C.C.; Fulton, R.S.; Health, S.E.; Tarnawski,
S.P.; et al. Genomic landscape of TP53-mutated myeloid malignancies.
MedRxiv 2023, doi.org/10.1101/2023.01.
https://doi.org/10.1101/2023.01.10.23284322
- Nguyen, T.; Pepper, J.W. ; Nguyen, C. ; Fan, Y. ; Hu, Y. ; Chen, Q. ;
Yan, C. ; Meerzaman, D. Highest risk adult patients with acute myeloid
leukemia (AML) through multi-omics clustering. Front Genet 2021, 12,
777094. https://doi.org/10.3389/fgene.2021.777094 PMid:34777485
PMCid:PMC8585788
- Tsai, C.H.; Hou, H.A.; Tang, J.L:; Liu, C.Y.; Lin, C.C.; Chou, W.C.;
Tseng, M.H.; Chiang, Y.C.; Kuo, Y.Y.; Liu, M.C.; et al. Genetic
alterations and their clinical implications in older patients with
acute myeloid leukemia. Leukemia 2016, 30, 1485-1492.
https://doi.org/10.1038/leu.2016.65 PMid:27055875
- Silvranchez, J.; Francea, P.; Neumann, M.; Schroeder, M.P.; Vosberg,
S.; Schlee, V.; Isaakidis, K.; Ortiz-Tranchez, J.; Fransecky, L.R.;
Hatung, T.; Turemn, S.; et al. Acute myeloid leukemia in elderly is
characetrized by a distinct genetic and epigenetic landscape. Leukemia
2017, 31, 1640-1644. https://doi.org/10.1038/leu.2017.109
PMid:28366934
- Eisfeld, A.K.; Kohlschmidts, J.; Mrozek, K.; Blanchly, J.S.; Walker,
C.J.; Nicolet, D.; Orwick, S.; Maharry, S.E.; Caroll, A.J.; Stone,
R.M.; et al. Mutation patterns identify adult patients with de novo
acute myeloid leukemia aged 60 years or older who respond favorably to
standard chemotherapy: an analysis of Alliance studies. Leukemia 2018,
32, 1338-1348. https://doi.org/10.1038/s41375-018-0068-2 PMid:29563537
PMCid:PMC5992022
- Prassek, V.V.; Rothenberg-Thurley, M.; Suerland, M.C.; Herold, T.;
Janke, H.; Kaslezyh, B.; Kostandin, N.P.; Goerlich, D.; Krug, U.;
Faldum, A.; et al. Genetics of acute myeloid leukemia in elderly:
mutation spectrum and clinical impact in intensively treated patients
aged 75 years or older. Haematologica, 2018, 103, 1853-1861.
https://doi.org/10.3324/haematol.2018.191536 PMid:29903761
PMCid:PMC6278991
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley,
J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.; et al. The 5th
edition of the World Health Organization Classification of
haematolymphoid tumors myeloid and histiocytic/dendritic neoplasms.
Leukemia 2022, 36, 1703-1719.
https://doi.org/10.1038/s41375-022-01613-1 PMid:35732831
PMCid:PMC9252913
- Iacobucci, I.; Wen, J., Meggendorfer, M.; Choi, J.K.; Shi, L.; Pounds,
S.B.; Carmichael, C.L.; Masih, K.E.; Morris, S.M.; Lindsley, R.C.; et
al. Genomic subtyping and therapeutic targeting of acute
erythroleukemia. Nature Genet 2019, 51, 694-704.
https://doi.org/10.1038/s41588-019-0375-1 PMid:30926971
PMCid:PMC6828160
- Takeda, J.; Yoshida, K.; Nakagawa, M.M.; Nannya, Y.; Yoda, A.; Saiki,
R.; Ochi, Y.; Zhao, L.; Okuda, R.; Qi, X.; et al. Amplified EPOR/JAK2
genes define a unique subtype of acute erythroid leukemia. Blood Cancer
Discov 2022, 3, 410-427. https://doi.org/10.1158/2643-3230.BCD-21-0192
PMid:35839275 PMCid:PMC9894574
- Montalban-Bravo, G.; Benton, C.B.; Wang, S.A.; Ravandi, F.; Kadia, T.;
Cortes, J.; Daver, N.; Takahashi, K.; DiNardo, C.; Jabbour, E.; et al.
More than 1 TP53 abnormality is a dominant characteristic of pure
erythroid leukemia. Blood 2017, 129, 2584-2587.
https://doi.org/10.1182/blood-2016-11-749903 PMid:28246192
PMCid:PMC5418636
- Reichard, K.K.; Tefferi, A.; Abdelmagid, M.; Orazi, A.; Alexandres, C.;
Haack, J.; Greipp, P.T. Pure acute erythroid leukemia: morphology,
immunophenotype, cytogenetics, mutations, treatment details, and
survival data among 41 Mayo Clinic cases. Blood Cancer J 2022, 12, 147.
https://doi.org/10.1038/s41408-022-00746-x PMid:36323674
PMCid:PMC9630502
- Leone, G.; Franchi, L.; Pagano, L.; Voso, M.T. Incidence and
susceptibility to therapy-related myeloid neoplasms. Chimico-Biol
Interactions 2010, 184, 39-45.
https://doi.org/10.1016/j.cbi.2009.12.013 PMid:20026017
- Shih, A.H.; Chung, S.S.; Dolezal, E.K.; Zhang, S.T.; Abdal-Wehab, o.I.;
Park, C.Y.; Nimer, S.D.; Levine, R.L.; Klimek, V.M. Mutational analysis
of therapy-related myelodysplastic syndromes and acute myelogenous
leukemia. Haematologica 2013, 98, 908-913.
https://doi.org/10.3324/haematol.2012.076729 PMid:23349305
PMCid:PMC3669447
- Guerra, V.; Yuanquing,Y.; Hsu, J.;Wang, F.; Alfayez, M. ; Morita, K. ;
Song, X. ; DiNardo, C.D.; Konopleva, M.Y.; Borthakur, G.M.; et al.
Comprehensive analysis of genotype and prior exposures in
therapy-related myeloid neoplasms (t-MNs). Blood 2019, 134, suppl.1,
458. https://doi.org/10.1182/blood-2019-127498
- Lindsley, R.C.; Gibson, C.J.; Murdock, H.M.; Stone, R.M.; Cortes, J.E.;
Uy, G.L.; Lin, T.L.; Ritchie, E.K.; Prebet, T.; Ryan, R.J.; et al.
Genetic characteristics and outcomes by mutation status in a phase 3 of
CPX-351 versus 7+3 in older adults with newly diagnosed,
high-risk/secondary acute myeloid leukemia (AML). Blood 2019, 134,
suppl.1, 15. https://doi.org/10.1182/blood-2019-124500
- Ok, C.Y.; Patel, K.P.; Garcia-Manero, G.; Routbort, M.J.; Peng, J.;
Tang, G.; Goswami, M.; Young, K.H.; Singh, R.; Medeiros, L.J.; et al.
TP53 mutation characteristics in therapy-related myelodysplastic
syndromes and acute myeloid leukemia is similar to de novo diseases. J
Hematol Oncol 2015, 8, 45. https://doi.org/10.1186/s13045-015-0139-z
PMid:25952993 PMCid:PMC4431603
- Hiwase, D.; Hahn, C.; Tran, E.N.H.; Chhetri, R.; Baranwal, A.; Al-Kali,
A.; Sharplin, K.; Ladon, D.; Hollins, R.; Greipp, P.; et al. TP53
mutation in therapy-related myeloid neoplasm defines a distinct
molecular subtype. Blood 2023, in press.
https://doi.org/10.1182/blood.2022018236 PMid:36574363
- Weinberg, O.K.; Siddon, A.; Madanat, Y.F. ; Gagan, J.; Arber, D.A.; Dal
Cin, P.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjjian, R.P.
TP53 mutation defines a unique subgroup within complex karyotype de
novo and therapy-related MDS/AML. Blood Adv 2022, 6, 2847-2853.
https://doi.org/10.1182/bloodadvances.2021006239 PMid:35073573
PMCid:PMC9092405
- Tariq, H.; Slonim, L.B. ; Coty Fattal, Z.; Alikhan, M.; Segal, J.;
Gurbuxani, S.; Helenowski, I.B.; Zhang, H.; Sukhanova, M.; Lu, X.; et
al. Therapy-related myeloid neoplasms with normal karyotype show
distinct genomic and clinical characteristics compared to their
counterparts with abnormal karyotype. Brit J Haematol 2022, 197,
736-744. https://doi.org/10.1111/bjh.18154 PMid:35304738
- Cantu, M.D.; Kanagal-Shamanna, R.; Wang, S.; Kadia, T.; Bueso-Ramos,
C.E.; Patel, S.S.; Geyer, J.T.; Tam, W.; Madanat, Y.; Li, P.; et al.
Clinicopathologic and molecular analysis of normal karyotype
therapy-related and de novo acute myeloid leukemia: a
multi-institutional study by the bone marrow pahtology group. JCO
Precis Oncol 2023, 7, e2200400. https://doi.org/10.1200/PO.22.00400
PMid:36689697
- Schwartz, J.R.; Ma, J.; Kamens, J.; Westove r, T.; Walsh, M.P.; Brady,
S.W.; Michael, J.R.; Chen, X.; Montefiori, L.; Song, G.; et al. The
acquisition of molecular drivers in pediatric therapy-related myeloid
neoplasms. Nature Commun 2021, 12, 985.
https://doi.org/10.1038/s41467-021-21255-8 PMid:33579957
PMCid:PMC7880998
- Coorens, T.H.; Collord, G.; Lu, W.; Mitchell, E.; Ijaz, J.; Roberts,
T.; Oliver, T.; Burke, A.; Gattens, M.; Dickens, E.; et al. Clonal
hematopoiesis and therapy-related myeloid neoplasms following
neuroblastoma treatment. Blood 2021, 137, 2992-2996.
https://doi.org/10.1182/blood.2020010150 PMid:33598691
PMCid:PMC8160503
- Bertrum, E.; Rosendhal Huber, A.; de Kanter, J.K.; Brandsma, A.M.; van
Leeuwen, A.; Verheul, M.; van den Heuvel-Eibrink, M.; Oka, R.; van
Rosmalen, M.; de Groot-Kruseman, H.; et al.Elevated mutational age in
blood of children treated for concer contributes to therapy-related
myeloid neoplasms. Cancer Discovery 2022, 12, 1860-1872.
https://doi.org/10.1158/2159-8290.22541080 PMid:36815378
- Mitchell, E.; Chapman, M.S.; Williams, N.; Dawson, K.J.; Mende, N.;
Caiderbank, E.F.; Jung, H.; Mitchell, T.; Coorens, T.H.; Spencer, D.H.;
et al. Clonal dynamics of haematopoiesis across the human lifespan.
Nature 2022, 606, 349-357. https://doi.org/10.1038/s41586-022-04786-y
PMid:35650442 PMCid:PMC9177428
- Voso, M.T.; Falconi, G.; Fabiani, E. What's new in the pathogenesis and
treatment of therapy-related myeloid neoplasm. Blood 2021, 138,
749-757. https://doi.org/10.1182/blood.2021010764
PMid:33876223
- Gibson, G.J.; Lindsley, R.C.; Tcheknmedyian, V.; Mar, B.G.; Shi, J.;
Jaiswal, S.; Bosworth, A.; Francisco, L.; He, J.; Bansal, A.; et al.
Clonal hematopoiesis associated with adverse outcomes after autologous
stem cell transplantation for lymphoma. J Clin Oncol 2017, 35,
1598-1605. https://doi.org/10.1200/JCO.2016.71.6712 PMid:28068180
PMCid:PMC5455707
- Husby, S.; Favero, F.; Nielsen, C.; Sorensen, B.S.; Baech, J.; Grell,
K.; Hansen, J.W.; Rodriguez-Gonzalez, F.G.; Haastrup, E.K.;
Fischer-Nielsen, A.; et al. Clinical impact of clonal hemtaopoiesis in
patients with lymphoma undergoing ASCT: a national population-based
cohort study. Leukemia 2020, 34, 3256-3268.
https://doi.org/10.1038/s41375-020-0795-z PMid:32203146
- Liu, K.; Derkach, A.; Lewis, N.; Zhu, M.; Zhang, Y. ; Arcila, M.;
Salles, G.; Dogan, A.; Xiao, W. Clonal hematopoiesis in diffuse large
B-cell lymphoma: clinical impact and genetic relatedness to lymphoma
and lymphoma and therapy-related myeloid neoplasm. Haematologica 2022,
in press. https://doi.org/10.3324/haematol.2022.281724 PMid:36384248
PMCid:PMC9973483
- Sridharan, A.; Schinke, C.D.; Georgiev, G.; Da Silva Ferreira, M.;
Thiruthuvanathan, V.; MacArthur, I.; Bhagat, T.D.; Choudhary, G.S.;
Aluri, S.; Chen, J.; et al. Stem cell mutations can be detected in
myeloma patients years before onset of secondary leukemias. Blood Adv
2019, 3, 3962-3970. https://doi.org/10.1182/bloodadvances.2019000731
PMid:31805192 PMCid:PMC6963234
- Mouhieddine, T.H.; Sperling, A.S.; Redd, R.; Park, J.; Leventhal, M.;
Gibson, C.J.; Manier, S.; Nassar, A.H.; Capelletti, M.; Huynh, D.; et
al. Clonal hematopoiesis is associated with adverse outcomes in
multiple myeloma patients undergoing transplant. Nat Commun 2020, 11,
2996. https://doi.org/10.1038/s41467-020-16805-5 PMid:32533060
PMCid:PMC7293239
- Hazenberg, C.; de Graaf, A.O. ; Mulder, R. ; Bungener, L.; van Bergen,
M.; Mulder, A.B.; Choi, G.; Schuringa, J.J.; de Groot, M.; Vellenga,
E.; et al. Clonal hematopoiesis in patients with stem cell mobilization
failure: a nested case-control study. Blood Adv 2022, in press.
https://doi.org/10.1182/bloodadvances.2022007497 PMid:36219593
PMCid:PMC10119633
- Berger, G.; Kroeze, L.I.; Koorenhof-Scheele, T.N.; de Graaf, A.O.;
Yoshida, K., Ueno, H.; Shiaraishi, Y.; Miyano, S.; van den Berg, E.;
Schepers, H.; et al. Early detection and evolution of preleukemic
clones in therapy-related myeloid neoplasms following autologous SCT.
Blood 2018, 131, 1846-1857.
https://doi.org/10.1182/blood-2017-09-805879 PMid:29311096
- Weber-Lassalle, K.; Ernst, C.; Reuss, A.; Mollenhoff, K.; Baumann, K.;
Jackisch, C.; Hauke, J.; Dietrich, D.; Borde, J.; Park-Simon, T.W.; et
al. Clonal hematopoiesis-associated gene mutations in a clinical cohort
of 448 patients with ovarian cancer. J Natl Cancer Inst 2022, 114,
565-570. https://doi.org/10.1093/jnci/djab231 PMid:34963005
PMCid:PMC9002281
- Kwan, T.T.; Oza, A.M.; Tinker, A.V.; Ray-Coquard, I.; Oaknin, A.;
Aghajanian, C.; Lorusso, D.; Colombo, N.; Dean, A.; Weberplas, J.; et
al. Preexisting TP53-variant clonal hematopoiesis and risk of secondary
myeloid neoplasms in patients with high-grade ovarian cancer treated
with rucuparib. JAMA Oncol 2021, 7, 1-10.
https://doi.org/10.1001/jamaoncol.2021.4664 PMid:34647981
PMCid:PMC8517887
- Morice, P.M.; Leary, A.; Dolladille, C.; Chrétien, B.; Poulain, L.;
Gonzalez-Martin, A.; Moore, K.; O'Reilly, E.M.; Ray-Coquard, I.;
Alexandre, I. Myelodysplastic syndrome and acute myeloid leukaemia in
patients treated with PARP inhibitors: a safety meta-analysis of
randomised controlled trials and a retrospective study of the WHO
pharmacovigilance database. Lancet Oncol 2021, 8, e122-e134.
https://doi.org/10.1016/S2352-3026(20)30360-4 PMid:33347814
- Marmouset, V.; Decroocq, J.; Garciaz, S.; Etienne, G.; Belhabri, A.;
Bertoli, S.; Gastaud, L.; Simand, C.; Chantepie, S.; Uzunov, M.; et
al. Therapy-related myeloid neoplsms following PARP inhibitors:
real-life experience. Clin Cancer Res 2022, 28, 5211-5220.
-
Oliveira, J.L.; Greipp, P.T.; Rangun, A.; Jotoi, A.; Nguyen, P.L.
Myeloid malignancies in cancer patients treated with poly(ADP-ribose)
polymerase (PARP) inhibitors: a case series. Blood Cancer J 2022, 12,
11. https://doi.org/10.1038/s41408-022-00607-7 PMid:35078980
PMCid:PMC8789926
- Martin, J.E.; Khalife-Hachem, S.; Grinda, T.;Kfoury, M.; Garciaz, S.;
Pasquier, F.; Vargaftig, J.; Uzunov, M.; Belhabri, A.; Bertoli, S.; et
al. Therapy-related myeloid neoplasms following treatment with PARP
inhibitors: new molecular insights. Ann Oncol 2022, 32, 1046-1048.
https://doi.org/10.1016/j.annonc.2021.04.015 PMid:34107346
- Bolton, K.L., Moukarzel, L.A.; Ptashkin, R.; Gao, T.; Patel, M.;
Caltabellotta, N.; Braunstein, L.Z.; Aghajanian, C.; Hyman, D.M.;
Berger, M.F.; et al. The impact of poly ADP ribose polymerase (PARP)
inhibitors on clonal hematopoiesis. J Clin Oncol 2020, 38 (suppl. 15),
1513. https://doi.org/10.1200/JCO.2020.38.15_suppl.1513
- Baeten, J.T.; Chan, I.; Link, D.C.; Bolton, K.L. Effects of PARP
inhibitor therapy on p53-deficient hematopoietic stem and progenitor
cell fitness. Blood 2021, 138 (suppl. 1); 3275.
https://doi.org/10.1182/blood-2021-151373
- Khalife-Hachem, Saleh, K.; Pasquier, F.; Willekens, C.; Tarabay, A.;
Antoun, L.; Grinda, T.; Castilla-Llorente, C.; Duchmann, M.; Quivoron,
C.; et al. Molecular landscape of therapy-related myeloid neoplasms in
patients previously treated for gynecologic and breast cancers.
HemaSphere 2021, 5, 9(e632).
https://doi.org/10.1097/HS9.0000000000000632 PMid:34423258
PMCid:PMC8373540
- Sonbol, M.B.; Halfdanarson, T.R.; Hilal, T. Assessment of
therapy-related myeloid neoplasms in patients with neuroendocrine
tumors after peptide receptor radionuclide therapy: a systematic
review. JAMA Oncol 2020, 6, 1086-1092.
https://doi.org/10.1001/jamaoncol.2020.0078 PMid:32297906
- Singh, A.; Mencia-Trinchant, N.; Griffith, E.A.; Altahan, A.;
Swaminathan, M.; Gupta, M.; Gravina, M.; Tajammal, R.; Faber, M.G.;
Yan, L.; et al. Mutant PPMiD- and TP53-driven hematopoiesis populates
the hematopoietic compartment in response to peptide receptor
radionuclide therapy. JCO Precis Oncol 2022, 6, e100309.
https://doi.org/10.1200/PO.21.00309 PMid:35025619
PMCid:PMC8769150
- Miller, P.G.; Sperling, A.S.; Brea, E.J.; Leick, M.B.; Fell, G.G.; Jan,
M.; Gohil, S.H.; Tai, Y.T.; Munshi, N.C.; Wu, C.J.; et al. Clonal
hematopoiesis in patients receiving chimeric antigen receptor T-cell
therapy. Blood Adv 2021, 5, 2982-2986.
https://doi.org/10.1182/bloodadvances.2021004554 PMid:34342642
PMCid:PMC8361461
- Eder, L.N.; Martinovic, D.; Mazzeo, P.; Ganster, C.; Hasenkamp, J.;
Thomson, J.; Trummer, A.; Haase, D.; Wulf, G. Fatal progression of
mutated TP53-associated clonal hematopoiesis following anti-CD19 CAR-T
cell therapy. Curr Oncol 2023, 30,1146-1150.
https://doi.org/10.3390/curroncol30010087 PMid:36661736
PMCid:PMC9858310
- Pich, O.; Muinos, F.; Lolkema, M.P.; Steeghs, N.; Gonzalez-Perez, A.;
Lopez-Bigas, N. The mutational footprints of cancer therapies. Nat
Genet 2019, 51, 1732-1740. https://doi.org/10.1038/s41588-019-0525-5
PMid:31740835 PMCid:PMC6887544
- Landau, H.J.; Yellapantula, V.; Diamond, B.T.; Rustad, E.H.;
Maclachlan, K.H.; Gundem, G.; Medina-Martinez, J.; Ossa, J.A.; Levine,
M.F.; Zhou, Y.; et al. Accelerated single cell seeding in relapsed
myltiple myeloma. Nat Commun 2020, 11, 3617.
https://doi.org/10.1101/2020.02.25.963272
- Pinch, O.; Cortes-Bullich, A.; Muinos, F.; Pratcorona, M.;
Gonzalez-Perez, A.; Lopez-Bigas, N. The evolution of hematopoietic
cells under cancer therapy. Nat Commun 2021, 12, 4803.
https://doi.org/10.1038/s41467-021-24858-3 PMid:34376657
PMCid:PMC8355079
- Diamond, B.; Ziccheddu, B.; Maclachlan, K.; Taylor, J.; Boyle, E.;
Arrango Ossa, J.; Jahn, J.; Affer, M.; Totiger, T.M.; Coffey, D.; et
al. Tracking the evolution of therapy-related myeloid neoplasms using
chemotherapy signatures. Blood, 2023, in press.
https://doi.org/10.1101/2022.04.26.489507
- Sperling, A.S.; Guerra, V.A.; Kennedy, J.A.; Yan, Y.; Hsu, J.I.; Wang,
F.; Nguyen, A.T.; Miller, P.G.; McConkey, M.E.; Quevedo Barrios, V.A.;
et al. Lenalidomide promotes the development of TP53-mutated
therapy-related myeloid neoplasms. Blood 2022, 140, 1753-1763.
https://doi.org/10.1182/blood.2021014956 PMid:35512188 PMCid:PMC9837415
- Stratmann, S.; Yones, S.A.; Mayrhofer, M.; Norgren, N.; Skaftason, A.;
Sun, J.; Smolinska, K.; Komorowski, J.; Herlin, M.K.; Sundstrom, K.;
Genomic characterization of relapsed acute myeloid leukemia dreveals
novel putative therapeutic targets. Blood Adv 2021, 5, 900-909.
https://doi.org/10.1182/bloodadvances.2020003709 PMid:33560403
PMCid:PMC7876890
- Alwash, J.; Khoury, J.D.; Tashakori, M.; Kanagal-Shamanna, R.; Daver,
N.; Ravandi, F.; Kadia, T.M.; Konopleva, M.; Dinardo, C.D.; Issa, G.C.;
et al. Development of TP53 mutations over the course of therapy for
acute myeloid leukemia. Am J Hematol 2021, 96, 1420-1428.
https://doi.org/10.1002/ajh.26314 PMid:34351647 PMCid:PMC9167467
- Hasserjian, R.P.; Drazi, A.; Orfao, A.; Rozman, M.; Wang, S.A. The
international consensus classification of myelodysplastic syndromes and
related entities. Virchows Arch 2022, in press.
https://doi.org/10.1007/s00428-022-03417-1 PMid:36287260
- Neinberg, O.K.; Porwit, A.; Orazi, A.; Hasserjian, R.P.; Foucar, K.;
Duncavage, E.J.; Arber, D.A. The international consensus classification
of acute myeloid leukemia. Virchows Arch 2022, in press.
https://doi.org/10.1007/s00428-022-03430-4 PMid:36264379
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango
Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier,
L. Molecular international prognostic scoring system for
myelodysplastic syndromes. NEJM Evidence 2022, 1, EVIDoa2200008.
https://doi.org/10.1056/EVIDoa2200008
- Dutta, S.; Moritz, J.; Pregartmner, G.; Thallinger, G.G.; Bradstatter,
I.; Lind, K.; Renazia, S.; Lyssy, F.; Reinisch, A.; Zebisch, A.; et al.
Comparison of acute myeloid leukemia and myelodysplastic syndromes with
TP53 aberrations. Annals Hematol 2022, 101, 837-846.
https://doi.org/10.1007/s00277-022-04766-2 PMid:35083527
PMCid:PMC8913568
- Shallis, R.M.; Daver, N.G.; Altman, J.R.; Hasserjian, R.P.; Kantarjian,
H.M.; Platzbecker, U.; Santini, V.; Wei, A.H.; Sallman, D.A.; Zeidan,
A.M. TP53-altered acute myeloid leukemia and myelodysplastic syndrome
with excess blasts should be approached as a single entity. Cancer
2023, 129, 175-180. https://doi.org/10.1002/cncr.34535 PMid:36397669
- Stengel, A.; Haferlach, T.; Baer, C.; Hutter, S.; Meggendorfer, M.;
Kern, W.; Haferlach, C. Specific subtype distribution with impact on
prognosis of TP53 single hit and double hit events in AML and MDS.
Leukemia 2023, in press.
https://doi.org/10.1182/bloodadvances.2022009100 PMid:36735768
- Shah, M.V.; Hahn, C.N.; Hoa Tran, E.; Sharplin, K.M.; Chhetri, R.;
Baranwal, A.; Kutyna, M.M.; Wang , P.; Ladon, D.; Al_kali, A.; et al.
TP53 mutation status defines a distinct clinicopathological entity of
therapy-related myeloid neoplasm, characterized by genomic instability
and extremely poor outcome. Blood 2022, 140 (suppl.1), 4403.
https://doi.org/10.1182/blood-2022-165859
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.;
Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.;
et al. TP53 mutations in newly diagnosed acute myeloid leukemia:
clinicomolecular characteristics, response to therapy, and outcomes.
Cancer 2016, 122, 3484-3491. https://doi.org/10.1002/cncr.30203
PMid:27463065 PMCid:PMC5269552
- Lindsley, R.C.; Gibson, C.J.; Murdock, M.; Stone, R.M.; Cortes, J.E.;
Uy, G.L.; Ritchie, E.K.; Prebet, T.; Ryan, R.J.; Lancet, J.E. Genetic
characteristics and outcomes by mutation status in a phase 3 study of
CPX-351 versus 7+3 in older adults with newly diagnosed,
high-risk/secondary acute myeloid leukemia (AML). Blood 2019, 134
(suppl.1), 15. https://doi.org/10.1182/blood-2019-124500
- Goldberg, A.D.; Talati, C.; Desai, P.; Famulare, C.; Devlin, S.M.;
Farnoud, N.; Sallman, D.A.; Lancet, J.E.; Roboz, G.J.; Sweet, K.L., et
al. TP53 mutations predict poorer responses to CPX-351 in acute myeloid
leukemia. Blood 2018, 132 (suppl.1), 1433.
https://doi.org/10.1182/blood-2018-99-117772
- Badar, T.; Atallah, E.L.; Saliba, A.N.; Shallis, R.M.; Stahl, M.F.;
Bewersdof, J.P.; Sacchi de Camargo, N.; Patel, A.A.; Abaza, Y.; Murthy,
G.S.; et al. Clinical outcomes of patients with TP53-mutated AML after
first relapse or with primary refractory disease: results from
consortium on myeloid malignancies and neoplastic diseases (COMMAND).
Blood 2022, 140 (suppl 1), 2699.
https://doi.org/10.1182/blood-2022-163795
- Yanada, M.; Yamamoto, Y.; Iba, S.; Okamoto, A.; Inaguma, Y.; Tokuda,
M.; Morishima, S.; Kanie, T.; Mizuta, S.; Akatsuka, Y.; et al. TP53
mutations in older daults with acute myeloid leukemia. Int J Hematol
2016, 103, 429-435. https://doi.org/10.1007/s12185-016-1942-1
PMid:26781615
- Welch, J.S.; Petti, A.A.; Miller, C.A., Fronick, C.C.; O'Laughlin, M.;
Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et
al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic
syndromes. N Engl J Med 2016, 375, 2023-2036.
https://doi.org/10.1056/NEJMoa1605949 PMid:27959731 PMCid:PMC5217532
- Short, N. J.; Kantarjian, H.M.; Loghavi, S.; Huang, X.; Qiao, W.;
Borthakur, G.; Kadia, T.M.; Daver, N.; Ohanian, M.; Dinardo, C.D.; et
al. Treatment with a 5-day besus 10-day schedule of decitabine in older
patients with newly diagnosed acute myeloid leukemia: a randomised
phase 2 trial. Lancet Hematol 2019, 6, e29-e37.
https://doi.org/10.1016/S2352-3026(18)30182-0 PMid:30545576
- Ferraro, F.; Grusczczynska, A.; Ruzinova, M.B.; Miller, C.A.; Percival,
E.; Uy, G.L.; Pusik, I.; Jacoby, M.A.; Christopher, M.J.; Kim, M.Y., et
al. Decitabine salvage for TP53-mutated, relapsed/refractory acute
myeloid leukemia after cytotoxic induction therapy. Hematologica 2022,
107, 1709-1713. https://doi.org/10.3324/haematol.2021.280153
PMid:35236053 PMCid:PMC9244807
- Uy, G.L.; Duncavage, E.J.; Chang, G.S. ; Jacoby, M.A.; Miller, C.A.;
Shao, J.; Heath, S.; Elliott, K.; Reineck, T.; Fulton, R.S.; et al.
Dynamic changes in the clonal structure of MDS and AML in response to
epigenetic therapy. Leukemia 2017, 31, 872-881.
https://doi.org/10.1038/leu.2016.282 PMid:27740633
PMCid:PMC5382101
- Chang, C.K.; Zhan, Y.S.; Xu, F.; Guo, J.; Zhang, Z.; He, Q.; Wu, D.;
Wu, L.Y.; Su, J.Y.; Song, L.X.; et al. TP53 mutations predict
decitabine-induced complete responses in patients with myelodysplastic
syndromes. Brit J Haematol 2017, 176, 600-608.
https://doi.org/10.1111/bjh.14455 PMid:27984642
- Huls, G.; Chitu, D.A.; Pabst, T. ; Klein, S.K. ; Stussi, G. ;
Griskevicius, L. ; Valk, P. ; Cloos, J. ; van deLoosdrecht, A,A. ; van
Zeventer, I. ; et al. Ibrutinib added to 10-day decitabine for older
patients with AML and higher risk MDS. Blood Adv 2020, 4, 4267-4274.
https://doi.org/10.1182/bloodadvances.2020002846 PMid:32915972
PMCid:PMC7509861
- Falconi, G.; Fabiani, E.; Piciocchi, A.; Criscuolo, M.; Fianchi, L.;
Lindorfs Rossi, E.L.; Finelli, C.; Cerqui, E.; Ottone, T.; Molteni, A.;
et al. Somatic mutations as markerhs of outcome after azacitidine and
allogeneic stem cell transplantation in higher-risk myelodysplastic
syndromes. Leukemia 2019, 33, 785-790.
https://doi.org/10.1038/s41375-018-0284-9 PMid:30291338
PMCid:PMC6462855
- Hunter, A.M.; Komrokji, R.S.; Yun, S.; Al Ali, N.; Chan, O.; Song, J.;
Hussaini, M.; Talati, C.; Sweet, K.L., Lancet, J.E.; et al. Baseline
and serial molecular profiling predicts outcomes with hypomethylating
agents in myelodysplastic syndromes. Blood Adv 2021, 5, 1017-1028.
https://doi.org/10.1182/bloodadvances.2020003508 PMid:33591325
PMCid:PMC7903224
- Patel, A.A.; Cahill, K.; Sayugin, C.; Odenike, O.
Cedazuridine/decitabine: from preclinical to clinical development in
mhyeloid malignancies. 2021, 5, 2264-2270.
https://doi.org/10.1182/bloodadvances.2020002929 PMid:33904891
PMCid:PMC8095139
- Savona, M.R.; McCloskey, J.K.; Griffiths, E.A.; Yee, K.; Zeidan, A.M.;
Al-Kali, A.; Deeg, J.; Patel, P.; Sabloff, M.; Keating, M.M.; et al.
Prolonged survival in bi-allelic TP53-mutated (TP53mut) MDS subjects
treated with oral decitabine/cedazurudine in the Ascertain trial
(ASTX727-02). Blood 2022, 140 (suppl.1), 854.
https://doi.org/10.1182/blood-2022-163841
- DiNardo, C.D.; Pratz, K., Pullarkat, V.; Jonas, B.A.; Arellano, M.;
Becker,P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.;
et al. Venetoclax combined with decitabine or azacitidine in
treatment-naïve, elderly patients with acute myeloid leukemia. Blood
2019, 133, 7-17. https://doi.org/10.1182/blood-2018-08-868752
PMid:30361262 PMCid:PMC6318429
- Aldoss, I.; Zhang, J.; Pillai, R.; Shouse, G.; Sanchez, J.F.; Mei, M.;
Nakamura, R.; Stein, A.S.; Forman, S.J.; et al. Venetoclax and
hypomethylating agents in TP53-mutated acute myeloid leukemia. Brit J
Haematol 2019, 187, e33-e54. https://doi.org/10.1111/bjh.16166
PMid:31441045 PMCid:PMC6786919
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch,
C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al.
Outcomes of TP53-mutant acute myeloid leukemia with decitabine and
venetoclax. Cancer 2021, 127, 3772-3781.
https://doi.org/10.1002/cncr.33689 PMid:34255353
- Venugopal, S.; Shoukier, M.; Konopleva, M.; DiNardo, C.D.; Ravandi, F.;
Short, N.J.; Andreef, M.; Borthakur, G., Daver, N.; Pemmaraju, N., et
al. Outcomes in patients with newly diagnosed TP53-mutated acute
myeloid leukemia with or without venetoclx-based therapy. Cancer 2021,
127, 3541-3551. https://doi.org/10.1002/cncr.33675 PMid:34182597
- Pollyea, D.A.; Pratz, K.W. ; Wei, A.H.; Pullarkat, V.; Jonas, B.A.;
Recher, C.; Babu, S.; Schuh, A.C.; Dail, M.; Sun, Y.; et al. Outcomes
in patients with poor-risk cytogenetics with or without TP53 mutations
treated with venetoclax and azacitidine. Clin Cancer Res 2022, 28,
5272-5279. https://doi.org/10.1158/1078-0432.CCR-22-1183 PMid:36007102
PMCid:PMC9751752
- Kwag, D.; Cho, B.S.; Bang, S.Y.; Lee, J.H.; Min, G.J.; Park, S.S.;
Park, S.; Yoon, J.H.; Lee, S.E.; Eom, K.S.; et al. Venetoclax with
decitabine versus decitabine monotherapy in elderly acute myeloid
leukemia: a propensity score-matched analysis. Blood Cancer J 2022, 12,
169. https://doi.org/10.1038/s41408-022-00770-x PMid:36529771
PMCid:PMC9760636
- Othman, J.; Dillon, R.; Wilhelm-Benartzi, C.; Knapper, S.; Battern,
L.M.; Canharm, J.; Hinson, E.L.; Villiers, W.; Kleeman, M.; Gilkes, A.;
et al. Genomic correlates of outcome in a randomised comparison of
CPX-351 and FLAG-IDA in high-risk acute myeloid leukemia and
mylodysplastic syndrome: results from the UK NCRI AML19 trial. Blood
2022, 140 (suppl. 1), 1036-1038.
https://doi.org/10.1182/blood-2022-159433
- DiNardo, C.D.; Lachowiez, C.A.; Takashi, K.; Loghavi, S.; Xiao, L.;
Kadia, T.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al.
Venetoclax combined with FLAG-IDA induction and consolidation in newly
diagnosed and relapsed or refractory acute myeloid leukemia. J Clin
Oncol 2021, 39, 2768-2778. https://doi.org/10.1200/JCO.20.03736
PMid:34043428 PMCid:PMC8407653
- Desikan, S.P.; Konopleva, M.; Takashi, K., Lachowiz, C.A.; Loghavi, S.;
Xiao, L.C.; Kadia, T.M.; Daver, N.; Short, N.; Sasaki, K.; et al.
Updated phase II results of venetoclx with FLAG-IDA in relapsed or
refractory acute myeloid leukemia. Blood 2022, 140 (suppl.1), 221.
https://doi.org/10.1182/blood-2022-164922
- Daver, N.G.; Iqbal, S.; Renard, C.; Chan, R.J.; Hasegawa, K.; Hu, H.;
Tse, P.; Yan, J.; Zoratti, M.J.; Xie, F.; et al. Treatment outcomes for
newly diagnosed, untreated TP53-mutated acute myeloid leukemia: s
systematic review and meta-analysis. HemaSphere 2022, 6, 451-452.
https://doi.org/10.1097/01.HS9.0000845096.01949.78
- Daver, N.G.; Iqbal, S.; Huang, J.; Renard, C.; Lin, J.; Pan, H.; Xing,
G.; Pan, Y.; Ramsingh, G. Clinical characteristics and overall survival
among acute myeloid leukemia (AML) patients with TP53 gene mutations
(TP53m) or chromosme 17p deletion (17p del). Blood 2022, 140 (suppl.
1), 603. https://doi.org/10.1182/blood-2022-170298
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.;
Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al.
Molecular patterns of response and treatment failure after frontline
venetoclax combinations in older patients with AML. Blood 2020, 135,
791-803. https://doi.org/10.1182/blood.2019003988 PMid:31932844
PMCid:PMC7068032
- Nechiporuk, T.; Kurtz, S.E., Nikolova, O.; Liu, T.; Jones, C.L.;
D'Alessandro, A.; Culp-Hill, R.; d'Almeida, A.; Joshi, S.K., Rosenberg,
M.; et al. The TP53 apoptotic network is a primary mediator of
resistance to BCL2 inhibition in AML cells. Cancer Discov 2019, 9,
910-925. https://doi.org/10.1158/2159-8290.CD-19-0125 PMid:31048320
PMCid:PMC6606338
- Thijssen, R.; Diepstraten, S.T.; Moujalled, D.; Chew, E.; Flensburg,
C.; Shi, M.X.; Dengler, M.A.; Litalien, V.; MacRaild, S.; Chen, M.;
Anstee, N.S.; et al. Intact TP53 function is essnetial for sustaining
responses to BH3 mimetic drugs in leukemias. Blood 2021, 137,
2721-2735. https://doi.org/10.1182/blood.2020010167 PMid:33824975
PMCid:PMC8138548
- Barter, B.Z.; Mak, P.Y.; Tao, W.; Ruvolo, V.; Zhang, X.; Nishida, Y.;
Kornblau, S.M.; Hugehs, P.; Chen, X.; Morrow, P.K.; et al. Co-targeting
MCL-1 and BCL-2 in highly synergistic in BH3 mimetic- and
venetoclax/hypomethylating agent-resistant and TP53 mutated AML. Blood
2020, 136 (suppl.1), 7. https://doi.org/10.1182/blood-2020-141417
- Moujalled, D.M.; Pomilio, G.; Ghiurau, C.; Ivey, A.; Salmon, J.; Ryol,
S.; Macraild, S.; Zhang, L.; The, T.C.; Tiong, I.S.; et al. Combining
BH3 mimetics to target both BCL-2 and MCL1 has potent activity in
preclinical models of acute myeloid leukemia. Leukemia 2019, 33,
905-917. https://doi.org/10.1038/s41375-018-0261-3 PMid:30214012
PMCid:PMC6484700
- Chen, X.; Glitson, C.; Zhou, H.; Narang, S.; Reyna, D.E. ; Lopez, A. ;
Sakellaropoulos, T.; Gong, Y.; Kleotgen, A.; Sing Yip, Y.; et al.
Targeting mitochondrial structure sensitizes acute myeloid leukemia to
venetoclax treatment. Cancer Discov 2019, 9, 890-909.
https://doi.org/10.1158/2159-8290.CD-19-0117 PMid:31048321
PMCid:PMC6606342
- Schimmer, R.R.; Kovtnyuk, L.V.; Klemm, N.; Fullin, J.; Stolz, S.M.;
Mueller, J.; Caiado, F.; Kurppa, K.J.; Ebert, B.L.; Manz, M.G.; et al.
TP53 mutations confer resistance to hypomethylating agents and BCL-2
inhibition in myeloid neoplasms. Blood Adv 2022, 6, 3201-3205.
https://doi.org/10.1182/bloodadvances.2021005859 PMid:35026842
PMCid:PMC9198927
- Pan, R.; Ruvolo, V.; Mu, H.; Leverson, J.D.; Nichols, G.; Reed, J.C.;
Konopleva, M.; Andreef, M. Synthetic lethality of combined Bcl-2
inhibition and p53 activation in AML: mechanisms and superior
antileukemic efficacy. Cancer Cell 2017, 32, 748-760.
https://doi.org/10.1016/j.ccell.2017.11.003 PMid:29232553
PMCid:PMC5730338
- Shahzad, M.; Tariq, E.; Chaudhary, S.G.; Anwar, I.; Iqbal, Q.; Fatima,
H.; Abdelhakim, H.; Ahmed, N.; Balasu, R.; Hematti, P.; et al. Outcomes
with allogeneic hematopoietic stem cell trasnplantation in TP53-mutated
acute myeloid leukemia: a systematic review and meta-analysis. Leuk
Lymphoma 2022, 63, 3409-3417.
https://doi.org/10.1080/10428194.2022.2123228 PMid:36107118
- Vittayawacharin, P.; Kongtim, P.; Ciurea, S.O. Allogeneic stem cell
transplantation for patients with myelodysplastic syndroemes. Am J
Hematol 2023, 98, 322-337. https://doi.org/10.1002/ajh.26763
PMid:36251347
- Yoshizato, T.; Nannya, Y.; Atsuta, Y.; Shiozawa, Y.; Iijma-Yamashita,
Y.; Yoshida, K.; Shiraishi, Y.; Suzuki, H.; Nagata, Y.; Sato, Y.; et
al. Genetic abnormalities in myelodysplasia and secondary acute myeloid
leukemia: impact on outcome of stem cell transplantation. Blood 2017,
129, 2347-2358. https://doi.org/10.1182/blood-2016-12-754796
PMid:28223278 PMCid:PMC5409449
- Ciurea, S.O.; Chilkulwar, A.; Saliba, R.M.; Chen, J.; Rondon, G.;
Patel, K.P.; Khogeer, H.; Shah, A.R.; Randolph, B.V.; Ramos Perez,
J.M.; et al. Prognostic factors influencing suvrival after allogeneic
transplantation for AML/MDS patients with TP53 mutations Blood 2018,
131, 2989-2992. https://doi.org/10.1182/blood-2018-02-832360
PMid:29769261 PMCid:PMC7218750
- Daher-Reyes, G.; Kim, T.; Novitzky-Basso, I.; Kim, K.H.; Smith, A.;
Stockley, T.; Capochichi, J.M.; Al-Shaibani, Z.; Pasic, I.; Law, A.; et
al. Prognostic impact of the adverse molecular-genetic profile on
long-term outcomes following allogeneic stem cell trasnplantation in
acute myeloid leukemia. Bone Marrow Transplant 2021, 56, 1908-1918.
https://doi.org/10.1038/s41409-021-01255-4 PMid:33767401
- Badar, T.; Atallah, E.; Shallis, R.; Saliba, A.N.; Patel, A.;
Bewersdorf, J.P.; Grenet, J.; Stahl, M.; Duvall, A.; Burkart, M.; et
al. Survival of TP53-mutated acute myeloid leukemia patients receiving
allogeneic stem cell trasnplantation afetr first induction or salvage
therapy: results from the Consortium on myeloid malignancies and
neoplastic diseases (COMMAND). Leukemia 2023, in press.
https://doi.org/10.1038/s41375-023-01847-7 PMid:36807649
- Byrne MT, Kurian TJ, Patel DA, Tamari R, Hong S, Abdelhakim H, Klein V,
Rojas P, Madhavan R, Kent A, et al. Non-relapse mortality in
TP53-mutated MDS/AML - a multi-center collaborative study. Blood 2021,
138 (suppl 1), 2922-2924.
https://doi.org/10.1182/blood-2021-154151
- Loke, J.; Labopin, M.; Craddock, C.; Cornelissaen, J.J.;
Labussiere-Wallet, H.; Wagner-Drouet, E.M.; Van Gorkom, G.; Schaap, N.;
Kroger, N.M.; Veelken, J.H.; et al. Additional cytogenetic features
determine outcome in patients allografted for TP53 mutant acute myeloid
leukemia. Cancer 2022, 128, 2922-2931.
https://doi.org/10.1002/cncr.34268 PMid:35612815 PMCid:PMC9545190
- Hourigan, C.S.; Dillon, L.W.; Gui, G.; Logan, B.R. ; Fei, M.; Ghannam,
J.; Li, Y.; Licon, A.; Alyea, E.P.; Bashey, A.; et al. Impact of
conditioning intensity of allogeneic trasnplantation for acute myeloid
leukemia with genomic evidence of residual disease. J Clin Oncol 2019,
38, 1273-1283. https://doi.org/10.1200/JCO.19.03011 PMid:31860405
PMCid:PMC7164487
- Murdock, H.M; Kim, H.T.; Denlinger, N.; Vachhani, P.; Hambry, B.;
Manning, B.S.; Gier, S.; Cho, C.; Tsai, H.K.; McCurdy, S.; et al.
Impact of diagnostic genetics on remission MRD and transplantation
outcomes in older patients with AML. Blood 2022, 139, 3546-3557.
https://doi.org/10.1182/blood.2021014520 PMid:35286378
PMCid:PMC9203701
- Versluis, J.; Lindsley, R.C. Transplant for TP53-mutated MDS and AML:
because we can or because we should? Hematology 2022, ASH Educ Program,
522-527. https://doi.org/10.1182/hematology.2022000354 PMid:36485102
PMCid:PMC9820679
- Marvin-Peek, J.; Kishtagari, A.; Jayani, R.; Dholaria, B.; Kim, T.K.;
Mason, E.F.; Engelhardt, B.G.; Chen, H.; Kassim, A.A.; Savona, M.R.; et
al. TP53 mutations are associated with increased infections and reduced
hematopoietic cell transplantation rates in myelodysplastic syndrome
and acute myeloid leukemia. Blood 2022, 140 (suppl.1), 11638-11640.
https://doi.org/10.1182/blood-2022-164631
- Misra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen,
Y.B.; Deeg, H.J.; Sallman, D.; Gallagher, P.; Wennborg, A.; et al.
Eprenetapopt plus azacitidine after allogeneis stem-cell
transplantation for TP53-mutant acute myeloid leukemia and
myelodysplastic syndromes. J Clin Oncol 2022, 40, 3985-3993.
https://doi.org/10.1200/JCO.22.00181 PMid:35816664
- Zhao, H.; Song, S.; Ma, J.; Yan, Z.; Xie, H.; Feng, Y.; Che S. CD47 as
a promising therapeutic target in oncology. Front immunol 2022, 13,
757480. https://doi.org/10.3389/fimmu.2022.757480 PMid:36081498
PMCid:PMC9446754
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.;
Majeti, R.; Traver, D.; van Rouijen, N.; Weissman, I.L. CD47 is
upregulated on circulating hematopoietic stem cells and leukemia cells
to avoid phagocytosis. Cell 2009, 138, 271-285.
https://doi.org/10.1016/j.cell.2009.05.046 PMid:19632178
PMCid:PMC2775564
- Majeti, R.; Chiao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaswal, S.; Gibbs,
K.D.; van Roijen, N.; Weissman, I.L. CD47 is an adverse prognostic
factor and therapeutic antibody target on human acute myeloid leukemia
stem cells. Cell 2009, 138, 286-299.
https://doi.org/10.1016/j.cell.2009.05.045 PMid:19632179
PMCid:PMC2726837
- Yan, X.; Lai, B.; Zhou, X.; Yang, S.; Ge, Q. ; Zhou, M.; Shi, C.;
Xu, Z.; Ouyang, G. The differential expression of CD47 may be related
to the pathogenesis from myelodysplastic syndromes to acute myeloid
leukemia. Front Oncol 2022, 12, 872999.
https://doi.org/10.3389/fonc.2022.872999 PMid:35433462 PMCid:PMC9008711
- Sallman, D.; Asch, A.; Kambhampti, S.; Al Malki, M.; Zeidner, J.;
Donnellan, W.; Lee, D.; Vyas, P.; Jeyakumar, D.; Mannis, G.; et al.
AML-196: the first-in-class anti-CD47 antibody magrolimab in
combination with azacitidine is well tolerated and effective in AML
patients: phase 1b results. Clin Lymph Myeloma Leuk 2021, 21 (suppl.
1), S290. https://doi.org/10.1016/S2152-2650(21)01694-3
- Daver, N.; Konopleva, M.; Maiti, A.; Kadia, T.M.; DiNardo, C.D.;
Loghavi, S.; Pemmaraju, N.; Jabbour, E.J.; Montalban-Bravo, G.; Sasaki.
K.; et al. Phase I/II study of azacitidine (AZA) with venetoclax (VEN)
and magrolimab (Magro) in patients (pts) with newly diagnosed
older/unfit or high-risk acute myeloid leukemia (AML) and
relapsed/refractory (R/R) AML. Blood 2021, 138 (suppl.1), 371.
https://doi.org/10.1182/blood-2021-153638
- Daver, N.; Senapati, J.; Maiti, A.; Loghavi S.; Kadia, T.M.; DiNardo,
C.D.; Pemmaraju, N.; Jabbour, E.; Moltalban-Bravo, G.; Tang, G.; et al.
Phase I/II study of azacitidine (AZA) with venetoclax (VEN) and
magrolimab (Magro) in patients (pts) with newly diagnosed older/unfit
or high-risk acute myeloid leukemia (AML) and relapsed/refractory (R/R)
AML. Blood 2022, 140 (suppl.1), 141-144.
https://doi.org/10.1182/blood-2022-170188
- Daver, N.; Vyas, P.; Kambhampati, S.; Al Malki, M.; Larson, R.A.; Asch,
A.S.; Mannis, G.N.; Chai-Ho, W.; Tanaka, T.N.; Bradley, T.J.; et al.
Tolerability and efficacy of the first-in-class anti-CD47 antibody
magrolimab combined with azacitidine in frontline TP53m AML patients:
phase 1b results. J Clin Oncol 2022, 40 (suppl. 16), 7020.
https://doi.org/10.1200/JCO.2022.40.16_suppl.7020
- Johnson, L.; Zhang, Y.; Li, B.; Aviles, L.; Lal, I.; Ramsingh, G.;
Daver, N.; Vyas, P.; Sallman, D.A. Nature of clinical response and
depth of molecular response in patients with TP53 mutant
myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML)
treated with magrolimab and azacitidine. Blood 2022, 140 (suppl. 1),
3083. https://doi.org/10.1182/blood-2022-160337
- Garcia-Manero, G.; Przespolewski, A.; Abaza, Y.; Byrne, M.; Fong, A.P.;
Jin, F.;,Forgie, A.J.; Tsiatis, A.C.; Erba, H.P. Evorpacept (ALX148), a
CD47-blocking myeloid checkpoint inhibitor, in combination with
azacitidine ane venetoclx in patients with acute myeloid leukemia
(ASPEN-05): results from phase 1a dose escalation part. Blood 2022, 140
(suppl.1), 9046-9047. https://doi.org/10.1182/blood-2022-157606
- Qu, T.; Zhong, T.; Pang, X.; Huang, Z.; Jin, C.; Wang, Z.M.; Li, B.;
Xia, Y. Ligufalimab, a novel anti-CD47 antibody with no
hemagglutination demonstrates both monotherapy and combo antitumor
activity. J Immunother Cancer 2022, 10, e005517.
https://doi.org/10.1136/jitc-2022-005517 PMid:36450383 PMCid:PMC9717234
- Qi, J.; Li, J.; Jiang, B.; Liu, H.; Cao, X.; Zhang, M.; Meng, Y.; Ma,
X.; Jia, Y.; Guo, J.; Zhang, Y.; et al. A phase I/IIa of lemzoparlimab,
a monoclonal antibody targeting CD47, in patients with relapsed and/or
refractory acute myeloid leukemia (AML) and myelodysplastic syndrome
(MDS): initial phase I results. Blood 2020, 136 (suppl.1), 30-31.
https://doi.org/10.1182/blood-2020-134391
- Xiao, Z.; Chang, C.; Li, Q.; Yan, X.; Qin, T.; Tong, H.; Zhang, Y.;
Dai, M.; He, Y.; Weng, J.; et al. Molecular biomarker analyses for
exploring the therapeutic mechanism of lemzoparlimab and azacitidine
(AZA) in newly diagnosed higher risk myelodysplastic syndrome (HR-MDS).
Blood 2022, 140 (suppl.1), 3974.
https://doi.org/10.1182/blood-2022-163880
- Swords, R.T.; Coutre, S.; Maris, M.B.; Zeidner, J.F.; Foran, J.M.;
Cruz, J.; Erba, H.P.; Berdaja, J.G.; Tam, W.; Vardhanabhuti, S.; et
al. Pevenedistat, a first-in class NEDD8-activating enzyme inhibitor,
combined with azacitidine in patients with AML. Blood 2018, 131,
1415-1424. https://doi.org/10.1182/blood-2017-09-805895 PMid:29348128
PMCid:PMC5909884
- Saliba, A.N.; Kaufmann, S.H.; Stein, E.M.; Patel, P.A.; Baer, M.R.;
Stock, W.; Deininger, M.; Blum, W.; Schiller, G.J.; Olin, R.L.; et al.
Pevonedistat with azacitidine in older patients with TP53-mutated AML:
a phase 2 study with laboratory correlates. Blood Adv 2023, in press.
https://doi.org/10.1182/bloodadvances.2022008625 PMid:36315007
- Adès, L.; Gishova, L.; Doronin, V.A.; Diez-Campelo, M.; Valcarcel, D.;
Kambhapampti, S.; Viniou, N.A.; Woszczyk, D.; De Paz SArias, R.;
Simeonidis, A.; et al. Pevonedistat plus azacitidine vs azacitidine
alone in higher-risk MDS/chronic myelomonocytic leukemia or low-blast
percentage AML. Blood Adv 2022, 6, 5132-5145.
https://doi.org/10.1182/bloodadvances.2022007334 PMid:35728048
PMCid:PMC9631625
- Cojocari, D.; Smith, B.N.; Purkal, J.J.; Arrate, M.P.; Huska, J.D.;
Xiao, Y.; Gorska, A.; Hogdal, L.J.; Ramsey, H.E.; Boghaert. E.R.; et
al. Pevonedistat and azacitidine upregulate NOXA (PMAIP1) to increase
sensitivity to venetoclax in preclinical models of acute myeloid
leukemia. Haematologica 2022, 107, 825-835.
https://doi.org/10.3324/haematol.2020.272609 PMid:33853293
PMCid:PMC8968901
- Ong, F.; Garcia-Manero, G.; Montalban-Bravo, G.; Alvarado, Y.; Ohanina,
M.; Konopleva, M.; Jabbour, E.; Jain, N.; Borthakur, N.; DiNardo, C.D.;
et al. A phase I/II study of combination of azacitidine, venetoclax and
pevonedistat in patients with newly diagnosed secondary acute myeloid
leukemia (AML) and myelodysplastic syndrome (MDS) with hypomethylating
agent failure. Blood 2022, 140 (suppl.1), 2761.
https://doi.org/10.1182/blood-2022-165359
- Bykov, V.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.;
Chumakov, P.; Bergman, J.; Wiman, K.; Selivanova, G. Restoration of the
tumor suppressor function to mutant p53 by a low-molecular-weight
compound. Nat Med 2022, 8, 282-288. https://doi.org/10.1038/nm0302-282
PMid:11875500
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen,
B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor
growth delay in small cell lung cancer expressing mutant p53. Clin
Cancer Res 2011, 17, 2830-2841.
https://doi.org/10.1158/1078-0432.CCR-10-3168 PMid:21415220
- Fransson, A.; Glaessgen, D.; Alfredsson, J.; Wiman, K.G.;
Bajalica-Lagercrantz, S.; Mohell, N. Strong synergy with APR-246 and
DNA-damaging drugs in primary cancer cells from patients with TP53
mutant high-grade sreous ovarian cancer. J Ovarian Res 2016, 9, 27.
https://doi.org/10.1186/s13048-016-0239-6 PMid:27179933
PMCid:PMC4868029
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.B.; Soderqvist, J.M.,
Sagerback, D.; Bergam, I.; Feroht, A.R.; Heinaut, P.; Wiman, K.G.;
Bykov, V.I. PRIMA-1 reactivates mutant p53 by covalent binding to the
core domain. Cancer Cell 2009, 15, 376-388.
https://doi.org/10.1016/j.ccr.2009.03.003 PMid:19411067
- Birsen, R.; Larrue, C.; Decroocq, J.; Johnson, N.; Guraud, N.;
Gotanegre, M.; Cantero-Anguilar, L.; Grignao, E.; Huyhn, T.; Fontenay,
M.; et al. APR-246 induces early death by ferroptosis in acute myeloid
leukemia. Haematologica 2021, 107, 403.
https://doi.org/10.3324/haematol.2020.259531 PMid:33406814
PMCid:PMC8804578
- Maslah,
N.; Salomao, N.; Drevon, L.; Verger, E.; Partouche, N.; Ly, P.; Aubin,
P.; Naoui, N.; Schalageter, M.H.; Bally, C.; et al.
Synergistic effects of PRIMA-1Met (APR-246) and 5-azacitidine in
TP53-mutated myelodysplastic syndromes and acute myeloid leukemia.
Haematologica 2020, 105, 1539-1551.
https://doi.org/10.3324/haematol.2019.218453 PMid:31488557
PMCid:PMC7271596
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz,
G.J.; Sekeres, M.A.; Clezeau, T.; Sweet, K.L.; McLemore, A.; McGraw,
K.L.; et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant
myelodysplastic syndromes. J Clin Oncol 2021, 39, 1584-1594.
https://doi.org/10.1200/JCO.20.02341 PMid:33449813 PMCid:PMC8099410
- Cluzeau, T.; Sebert, M.; Rahmé, M.; Cuzzubbo, S.; Lehmann-Che, J.;
Madelaine, I.; Peterlin, P.; Bève, B.; Atallh, H.; Chermat, F.; et al.
Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes
and acute myeloid leukemia: a phase II study by the groupe Francophone
des myélosyplasies (GFM). J Clin Oncol 2021, 39, 1575-1583.
https://doi.org/10.1200/JCO.20.02342 PMid:33600210 PMCid:PMC8099409
- Slamman, D.A.; Komrokji, R.S.; DeZern, A.E.; Sebert, M.; Garcia-Manero,
G.; Rahmé, R.; Steensma, D.P.; Lehmann, J.; Roboz, G.J.; Madelaine, I.;
et al. Long term follow-up and combined phase 2 results of Eprenetapopt
(APR-246) and azacitidine (AZA) in patients with TP53 mutant
myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia
(AML). Blood 2021, 138 (suppl.1), 246.
https://doi.org/10.1182/blood-2021-153286
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi,
A.T.; Onenike, O.; Roboz, G.J.; Gallacher, P.; Wennborg, A.; Hickman,
D.K.; et al. Phase I and expansion study of Eprenetapopt (APR-246) in
combination with venetoclax (VEN) and azacitidine (AZA) in TP53-mutant
acute myeloid leukemia (AML). Blood 2021, 138 (suppl.1), 3409-3410.
https://doi.org/10.1182/blood-2021-148940
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen,
Y.B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al.
Eprenetapopt plus azacitidine after allogeneic hematopoietic stem-cell
transplantation for TP53-mutant acute myeloid leukemia and
myelodysplatic syndromes. J Clin Oncol 2022, 40, 3985-3993.
https://doi.org/10.1200/JCO.22.00181 PMid:35816664
- Kreur, T.L., Quintana, A.; Ferrall-Fairbanks, M.; Zhang, L.; Newmann,
H.; McLemore, AF, Bencrentsil, N.; Yang, Q.; Komrokji, R.S.; Chaudry,
S.; et al. XPO1 overexpression, a mechanism of resistance to
Eprenetapopt and 5-azacitidine therapy that can be therapeutically
exploited for the treatment of TP53 mutated myeloid malignancies. Blood
2022, 140 (suppl.1), 45.
https://doi.org/10.1182/blood-2022-169765
- Ghosh, A.; Michels, J., Mezzadra, R.; Venkatesh, D.; Dong, L.; Gomez,
R.; Samaan, F.; Ho, Y.J.; Campesato, L.F., Mangarin, L., et al.
Increased p53 expression induced by APR-246 reprograms tumor-associated
macrophages to augment immune checkpoint blockade. J Clin Invest 2022,
132, e148941. https://doi.org/10.1172/JCI148141 PMid:36106631
PMCid:PMC9479603
- Thorsson V.; Gibbs, D.L.; Bown, S.D. Cancer Genome Atlas Research
Network. The immune landscape of cancer. Immunity 2018, 48, 812-830.
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Redder, S.;
Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.;
et al. Immune landscapes predict chemotherapy resistance and
immunotherapy response in acute myeloid leukemia. Sci Transl Med 2020,
12, 10.1126. https://doi.org/10.1101/702001
- Vadakekolathu, J.; Lai, C.; Reeder, S.; Church, S.E.; Hood, T.;
Lourdusamy, A.; Rettig, M.P.; Aldoss, I.; Advani, A.S.; Godwin, J.; et
al. TP53 abnormalities correlate with immune infiltration and associate
with response to flotetuzumab immunotherapy in AML. Blood Adv 2020, 4,
5011-5024. https://doi.org/10.1182/bloodadvances.2020002512
PMid:33057635 PMCid:PMC7594389
- Pelosi, E.; Castelli, G.; Testa, U. CD123 a therapeutic target for
acute myeloid leukemia and blastic plamocytoid dendritic neoplasm. Int
J Mol Sci 2023, 24, 2718. https://doi.org/10.3390/ijms24032718
PMid:36769040 PMCid:PMC9917129
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduxilt, M.J.;
Advani, A.S.; Godwin, A.S.; Arellano, M.L.; Sweet, K.L.; Emodi, A.; et
al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid
leukemia. Blood 2021, 137, 751-762.
https://doi.org/10.1182/blood.2020007732 PMid:32929488 PMCid:PMC7885824
- Cataquiz Rimando, J.; Chend Amarai, E.; Rettig, M.P.; Jayasinghe, R.G.;
Christopher, M.; Richey, J.K.; Christ, S.; Kim, M.; Bonvini, E.;
DiPersio, J.F. Flotetuzumab and other T-cell imunotherapies upregulate
MHC class II expression on acute myeloid leukemia cells. Blood 2023, in
press. https://doi.org/10.1182/blood.2022017795 PMid:36563336
- Rutella, S.; Vadakekolathu, J.; Mazziotta, F.; Roder, S.; Yan, T.O.;
Mekhopadyay, R.; Dickins, B.; Altmann, H.; Kramer, M.; Knaus, H.A.; et
al. Immune dysfunction signatures predict outcomes and defines
checkpoint blockade-unresponsive meicroenvironments in acute myelois
leukemia. J Clin Invest 2022, 132, e159579.
https://doi.org/10.1101/2022.02.08.22270578
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.;
Montiel-Esparza, R.; Mukhopadhyay, R.; Vassura, K.; Blazar, B.R.; Karp,
J.E.; Luznik, L.; Signatures of CD8+ T cell dysfunction in AML patients
and their reversibility with response to chemotherapy. JCI Insight
2018, 3, e120974. https://doi.org/10.1172/jci.insight.120974
PMid:30385732 PMCid:PMC6238744
- Wen, X.; Xu, Z.; Jin, Y.; Xia, P.; Ma, J.; Qian, W.; Lin, J.; Qian, J.
Association analyses of TP53 mutation with prognosis, tumor mutational
buirden, and immunological features in acute myeloid leukemia. Fron
Immunol 2021, 12, 717527. https://doi.org/10.3389/fimmu.2021.717527
PMid:34745095 PMCid:PMC8566372
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw,
K.L.; Dhawan, A.; Geyer, S.; Hou, H.A.; Eksioglu, E.A.; et al. TP53
mutations in myelodysplastic syndromes and secondary AML confer an
immunosuppressive phenotype. Blood 2020, 136, 2812-2823.
https://doi.org/10.1182/blood.2020006158 PMid:32730593 PMCid:PMC7731792
- Kikushige, Y.; Miyamoto, T.; Yuda, J.; Jabbarzadeh-Tabrizi, S.; Shima,
T.; Takayanagi, S.I.; Niiro, H.; Yurino, A.; Miyawaki, K.; Takenaka,
K.; et al. TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal
of human myeloid leukemia stem cells and leukemia progression. Cell
Stem Cell 2015, 17, 341-352. https://doi.org/10.1016/j.stem.2015.07.011
PMid:26279267
- Schwartz, S.; Patel, N.; Longmire, T.; Jayaraman, P.; Jiang, X. ; Lu,
H. ; Baker, L. ; Velez, J. ; Ramesh, R. ; Wavreille, A.S.; et al.
Characterization of sabatolimab, a novel immunotherapy with
immuno-myeloid activity directed against TIM-3 receptor. Immunothrrapy
Adv 2022, 2, 1-14. https://doi.org/10.1093/immadv/ltac019 PMid:36196369
PMCid:PMC9525012
- Brunner, A.M.; Esteve, J.; Porkka, K.; Knapper, S.; Traer, E.; Scholl,
S.; Garcia-Manero, S.; Vey, N.; Werke, M.; Janssen, J.; et al. Efficacy
and safety of Sabatolimab (MBG453) in combination with hypomethylating
agents (HMA) in patients (Pts) with very high/high-risk myelodysplastic
syndrome (vHR-HR-MDS) and acute myeloid leukemia (AML): final analysis
from a phase Ib study. Blood 2021, 138 (suppl.1), 244. https://doi.org/10.1182/blood-2021-146039
- Zeidan, A.M.; Westermann, J.; Kovacsovics, T.; Assouline, S.; Schuh,
A.C.; Kim, H.J.; Macias, G.R.; Sanford, D.; Luskin, M.R.; Stein, E.M.;
et al. AML-484 first results of a phase II study (STIMULUS-AML1)
investigating Sabatolimab + Azacitidine + Venetoclax in patients with
newly diagnosed acute myeloid leukemia (AML). Clin Lymphoma Myeloma
Leuk 2022, 22 (suppl. S2), S255.
https://doi.org/10.1016/S2152-2650(22)01303-9
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.;
Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia,
T.; et al Efficacy, safety and biomarkers of response to azacitidine
and nivolumab in relapsed/refractory acute myeloid leukemia: a
non-randomized, open-label, phase II study. Cancer Discov 2019, 9,
370-383. https://doi.org/10.1158/2159-8290.CD-18-0774 PMid:30409776
PMCid:PMC6397669
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson,
B.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.; Konopleva, M.; et al
Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed
acute myeloid leukemia or high-risk myelodysplastic syndrome: a
single-arm, phase 2 study. Lancet Hematol 2019, 6, e480-e488.
https://doi.org/10.1016/S2352-3026(19)30114-0 PMid:31400961
- Reville, P.K.; Kantarjian, H.M.; TRavandi, F.; Jabbour, E.; DiNardo,
C.D.; Daver, N.; Pemmaraju, N.; Ohanian, M.; Alvarado, Y.; Xiao, L.; et
al. Nivolumab maintenance in high-risk acute myeloid leukemia patients:
a single-arm, open-label, phase II study. Blood Cancer J 2021, 11, 60.
https://doi.org/10.1038/s41408-021-00453-z PMid:33731681
PMCid:PMC7969746
- Liu, H.; Sharon, E.; Karrison, T.G.; Zha, Y.; Fulton, N.; Streicher,
H.; Sweet, K.; Yaghmour, G.; Liu, J.J.; Jonas, B.A.; et al. Randomized
phase II study to assess the role of nivolumab as single agent to
eliminate minimal residual disease and maintain remission in acute
myelogenous leukemia (AML) patients after chemotherapy (NCI9706
protocol; REMAIN trial). Blood 2022, 140 (suppl.1), 1716-1719.
https://doi.org/10.1182/blood-2022-157326
- Daver, N.; Garcia-Manero, G.; Konopleva, M.; Alfayez, M.; Pemmaraju,
N.; Kadia, T.; DiNardo, C.D.; Cortes, J.E.; Ravandi, F.; Abbas, H.; et
al. Azacitidine (AZA) with nivolumab (Nivo) and AZA with Nivo +
Ipilimumab (Ipi) in relapsed/refractory acute myeloid leukemia: a
non-randomized, prospective, phase 2 study. Blood 2019, 134 (suppl. 1),
830. https://doi.org/10.1182/blood-2019-131494
- Zeng, Z.; Maiti, A.; Herbrich, S.; Cai, T.; Cavazos, A.; Manzella, T.;
Ma, H.; Hayes, K.; Matthews, J.; DiNardo, C.D.; et al. Triple
combination targeting methyltrasnferase, BCL-2, and PD-1 facilitates
antileukemic responses in acute myeloid leukemia. Cancer 2023, 129,
531-540. https://doi.org/10.1002/cncr.34566 PMid:36477735
- Zeidner, J.F.; Vincent, B.G.; Ivanova, A.; Moore, D.; MkKinnon, K.P.;
Wilkinson, A.D.; Mukhopadhyay, R.; Mazziotta, F.; Knaus, H.A.; Foster,
M.C.; et al. Phase II trial of pembrolizumab after high-dose cytarabine
in relapsed/refractory acute myeloid leukemia. Blood Cancer Discover
2021, 2, 616-629. https://doi.org/10.1158/2643-3230.BCD-21-0070
PMid:34778801 PMCid:PMC8580622
- Gojo, I.; Stuart, R.K.; Webster, J.; Blackford, A.; Varela, J.C.;
Moreow, J.; DeZern, A.E.; Foster, M.C.; Lewis, M.J., Coombs, C.C.; et
al. Multi-center phase 2 study of pembrolizumab (Pembro) and
azacitidine (AZA) in patients with relapsed/refractory acute myeloid
leukemia (AML) and in newly diagnosed (≥65 years) AML patients. Blood
2019, 134, 832. https://doi.org/10.1182/blood-2019-127345
- Agrawal, V.; Croslin, C.; Beltran, A.L.; Zhang, J.; Palmer, J.;
Huyhn-Tran, Q.; Hernandez, R.; Thomas, S.; Robbins, M.; Aribi, A.; et
al. promising safety and efficacy results from an ongoing phase Ib
study of pembrolizumab combined with decitabine in patients with
relapsed/refractory (R/R) acute myeloid leukemia (AML). Blood 2022, 140
(suppl.1), 2774. https://doi.org/10.1182/blood-2022-165894
[TOP]