There are no pathognomonic diagnostic tests for AILDs and diagnosis is usually based on a combination of clinical, serological, biochemical, and histological findings.
Investigation of AILD usually involves a combination of auto-antibody measurement and histology evaluation. Other hepatitis causes including viral hepatitis should also be screened for using serological investigations as summarized below (Table 1). With regard to AIH, there are common antibodies which should be checked (Table 2) and The AIH Group’s criteria should be applied to calculate a score (Table 3).[32] A simplified score of >7 is 95% specific for AIH.[33]
 |
Table 1. Summary of investigations which we suggest considering using to evaluate for the presence of hepatitis viruses. |
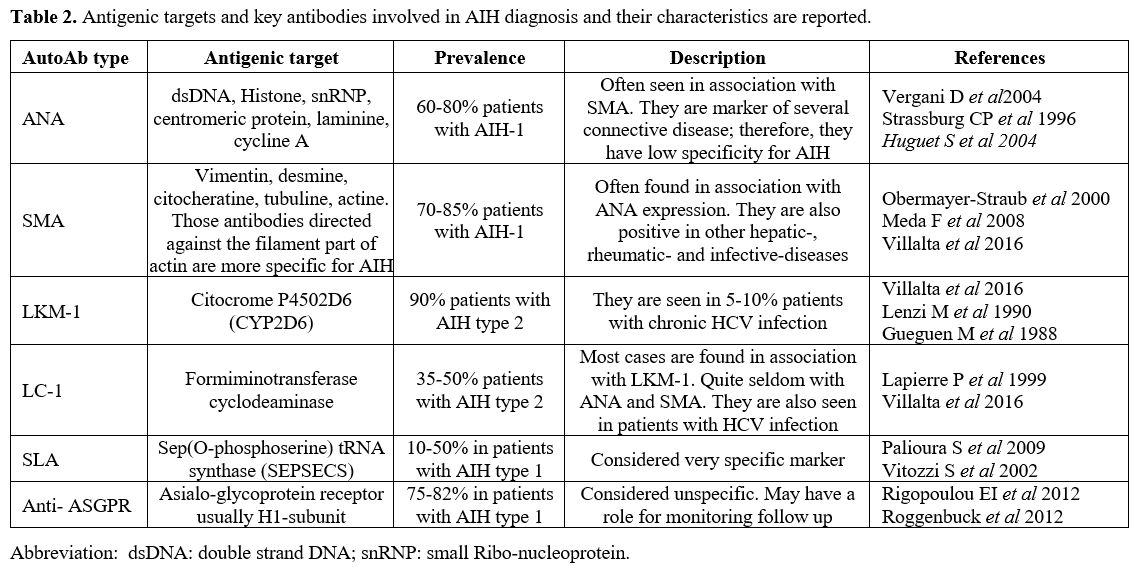 |
Table 2. Antigenic targets and key antibodies involved in AIH diagnosis and their characteristics are reported. |
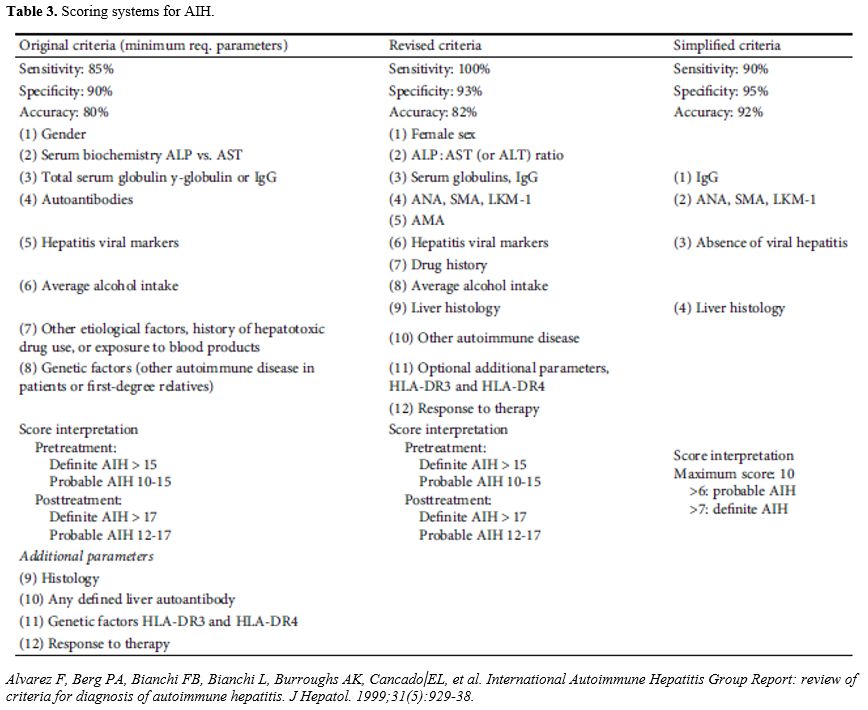 |
Table 3. Scoring systems for AIH. |
PBC and PSC similarly have a multifactorial approach to diagnosis. Radiological evaluation is often useful in addition to autoantibodies and biopsy.[34,35] Overlap syndromes of both AIH and either PBC or PSC (AIH/PBC or AIH/PSC) are more common than expected, if they were independent events. Overlap syndromes tend to be a challenging diagnosis.[36,37] There are special Paris criteria with 97% specificity for AIH/PBC.[38] Unfortunately, AIH/PSC doesn’t have a diagnostic system which is as established. Accurate diagnosis with overlap is important as without an AIH component corticosteroid is not likely to be as therapeutically beneficial and may precipitate vaso-occlusion in patients with co-existing SCD.[26,34,35]