Kun Yang*, Xiaodong Liu, Wei
Peng, Fang Hua, Lan Li, Kun Chen, Jin Zhang, Shan Luo, Wanting Li, Yuxi
Ding, Jie Chen and Jian Xiao*..
Department of Hematology, Zigong First People's Hospital, Zigong, China.
* The authors equally contributed to the work.
Correspondence to:
Kun Yang, Department of Hematology, Zigong First People's Hospital,
Zigong, 643000, China; E-mail:
1759874951@qq.com
Jian Xiao, Department of Hematology, Zigong First People's Hospital, Zigong, China, E-mail:
16188702@qq.com
Published: January 01, 2024
Received: July 05, 2023
Accepted: December 02, 2023
Mediterr J Hematol Infect Dis 2024, 16(1): e2024001 DOI
10.4084/MJHID.2024.001
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Thalidomide
is a therapeutic option for patients with βthalassemia by increasing
fetal hemoglobin and thereby reducing the requirement for blood
transfusions. However, information on changes in erythropoiesis and
iron homeostasis during thalidomide treatment is lacking. This study
investigated the effects of thalidomide treatment on hematologic,
erythropoietic, and iron-status parameters in 22 patients with
transfusion-dependent β-thalassemia (TDT). Thalidomide significantly
improved anemia endpoints, including increases in hemoglobin (p<0.001), red blood cells (p<0.001), and hematocrit (p<0.001),
as well as reducing erythropoietin levels (p=0.033) and ameliorating
erythropoiesis. Thalidomide treatment significantly reduced serum iron
levels (p=0.018) and transferrin saturation (p=0.039) and increased serum transferrin levels (p=0.030). Thalidomide had no observed effect on serum ferritin or hepcidin, but changes in hepcidin (r=0.439, p=0.041) and serum iron (r=−0.536, p=0.010)
were significantly correlated with hemoglobin increment. This
comprehensive study indicates that thalidomide treatment can ameliorate
erythropoiesis and iron homeostasis in patients with TDT, thus
supporting the effectiveness of this drug.
|
Introduction
Thalassemia is an inherited blood disorder affecting the synthesis of globin chains.[1]
The severity of anemia, need for transfusions, and clinical morbidity
of β-thalassemia are closely tied to the degree of imbalance between
α-globin and β-globin chains. Deficiency of β-globin chains leads to
the accumulation of excessive, unstable α-globin tetramers in
erythrocytes. Free α-globin is unstable, produces cytotoxic active
oxidants and cell pellets, impairs the maturation and viability of
erythrocyte precursors, and leads to ineffective erythropoiesis (IE)
and premature hemolysis of circulating erythrocytes, resulting in
anemia and decreased erythrocyte survival.[2]
Increased erythropoietin (EPO) due to chronic anemia further increases
IE, bone marrow dilation, and extramedullary hematopoiesis.[3]
Disordered iron homeostasis is a central feature of the pathophysiology
of thalassemia. In transfusion-dependent β-thalassemia (TDT) patients,
iron intake saturates serum transferrin, leading to
non-transferrin-bound iron species that accumulate in tissues and cause
damage to vital organs.[4]
Re-expression of
γ-globin and more efficient synthesis of fetal hemoglobin (HbF) can
reduce the imbalance between α-globin and β-globin chains, and
induction of HbF has been used as a treatment strategy for
β-thalassemia.[5] Thalidomide and its derivatives are
used to treat some malignant hematologic diseases because of their
anti-inflammatory, anti-tumor, anti-neovascularization, and
immunomodulatory properties.[6] Thalidomide can also induce expression of the γ-globin gene, which increases HbF levels.[7]
Previous studies demonstrated significant efficacy of thalidomide in
patients with TDT or non-transfusion-dependent β-thalassemia (NTDT);[8-10] however, information on changes in erythropoiesis and iron homeostasis during thalidomide treatment is lacking.
In
this study, we evaluated the effects of thalidomide on erythropoiesis
and iron homeostasis and analyzed the correlations between baseline
indicators and hemoglobin changes to explore the possible mechanisms of
thalidomide in the treatment of β-thalassemia and the possibility of
combining thalidomide with other agents.
Methods
Patients.
This study included TDT patients treated with thalidomide for >3
months in Zigong First People's Hospital. TDT was diagnosed according
to the Thalassemia International Federation guidelines.[4]
The inclusion criteria were: 1) age 14–18 years; 2) diagnosis of TDT
using accepted clinical and genetic methods; and 3) ECOG physical score
0–2 points. The exclusion criteria were: 1) therapy with drugs that
might affect Hb levels 3 months before enrolment; 2) other hemolytic
disorders; 3) cardiopulmonary, cerebrovascular, liver, kidney, or other
severe diseases; 4) allergy to thalidomide; and 5) currently
participating in any other clinical trial. Patients were informed of
the side effects and possible benefits of thalidomide. All patients
were warned against becoming pregnant or impregnating a woman while
taking the drug. The study protocol was approved by the ethics
committee of Zigong First People’s Hospital. The study adhered to the
principles of the Declaration of Helsinki, and written informed consent
was obtained from all participants and their guardians.
Treatment.
Thalidomide (Changzhou Pharmaceutical Factory, Changzhou, Jiangsu,
China) was administered at 100 mg/day. Transfusion was recommended to
maintain hemoglobin levels >9.0 g/dL during treatment, and regular
transfusion volumes were administered if hemoglobin fell below this
level. Aspirin was prescribed to patients with platelet counts
>500×109/L to prevent thrombosis.
These patients did not receive iron chelation therapy during the first
trimester of thalidomide treatment.
Laboratory examinations.
Venous blood samples were collected before thalidomide and after 3
months of treatment, respectively, and before transfusion. Complete
blood counts were analyzed using an XE 5000 automatic blood cell
analyzer (Sysmex Corporation, Kobe, Japan). Hb levels were quantified
by high-pressure liquid chromatography (Bio-Rad Variant II, Bio-Rad,
Hercules, CA, USA). Biochemical parameters were assessed using a
multichannel analyzer (Abbot Aeroset, Abbott Diagnostics, Bohemia, NY,
USA). Samples were tested for serum iron, total iron-binding capacity
(TIBC), unsaturated iron-binding capacity (UIBC), and transferrin
saturation by the colorimetric method (Pointe Scientific, Inc., Canton,
MI, USA), serum ferritin by immunoassay (Immulite 1000), and soluble
transferrin receptor (sTfR), EPO (R&D Systems), and hepcidin
(Intrinsic Life Sciences, La Jolla, CA, USA) by enzyme-linked
immunosorbent assay.
Statistical analysis.
Data were analyzed using SPSS Statistics 26.0 (SPSS Inc., Chicago, IL,
USA). Numerical data were presented as mean ± standard deviation or
median and interquartile range. Changes in continuous variables before
and after treatment were compared by paired t-test or Mann–Wilcoxon
rank-sum test. Correlations were analyzed by linear regression and
univariate analysis. A p-value <0.05 was considered significant in all analyses.
Results
Patient characteristics. Twenty-two
patients were included in this study between May 2021 and August 2022.
The patient cohort comprised 14 males and eight females, with a median
age of 15 years (range: 14–18 years). Splenectomy was performed in
18.2% (4/22) of the patients.
Effects of thalidomide treatment on hematologic and erythropoietic parameters in TDT patients.
Hematologic parameters improved after thalidomide treatment compared
with baseline. Specifically, thalidomide treatment significantly
improved anemia endpoints, including increased hemoglobin (p<0.001), red blood cells (RBCs) (p<0.001), and hematocrit (p<0.001) (Figure 1A-D). Thalidomide reduced the mean corpuscular volume (MCV) (p<0.001) and mean erythrocyte hemoglobin (MCH) (p=0.011) (Figure 1E-G), and reduced lactate dehydrogenase, suggesting that thalidomide treatment reduced hemolysis (Figure 1H-K). Thalidomide also significantly reduced EPO, further demonstrating an improvement in anemia after treatment (Figure 1L). The detailed data are shown in Supplementary Table S1.
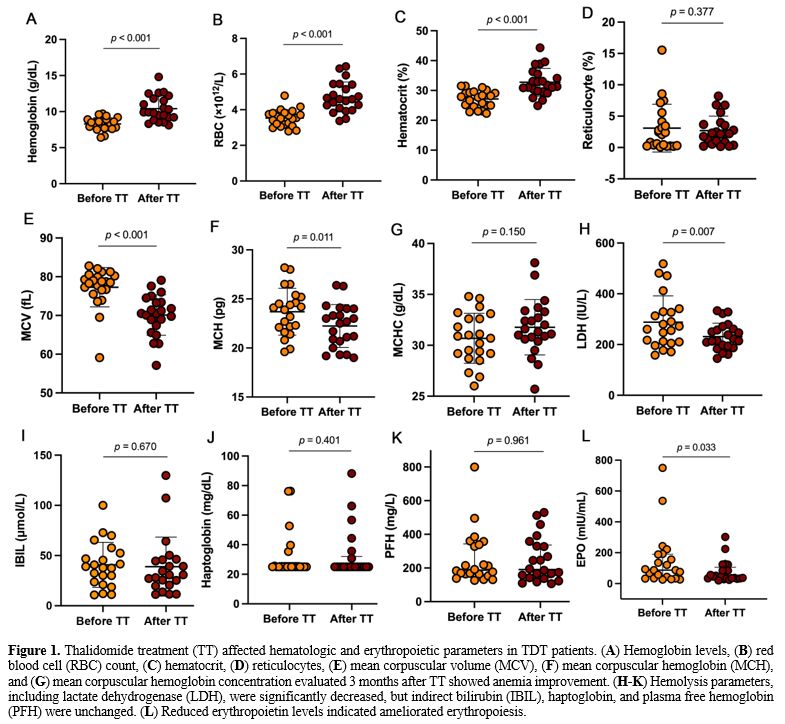 |
- Figure 1. Thalidomide treatment (TT) affected hematologic and erythropoietic parameters in TDT patients. (A) Hemoglobin levels, (B) red blood cell (RBC) count, (C) hematocrit, (D) reticulocyte (E) mean corpuscular volume (MCV), (F) mean corpuscular hemoglobin (MCH), and (G) mean corpuscular hemoglobin concentration evaluated 3 months after TT showed anemia improvement. (H-K)
Hemolysis parameters, including lactate dehydrogenase (LDH), were
significantly decreased, but indirect bilirubin (IBIL), haptoglobin,
and plasma free hemoglobin (PFH) were unchanged. (L) Reduced erythropoietin levels indicated ameliorated erythropoiesis.
|
Effects of thalidomide treatment on serum iron-status parameters in TDT patients. Thalidomide treatment significantly reduced serum iron levels (p=0.018) (Figure 2A) and transferrin saturation (p=0.039) (Figure 2B). Moreover, serum transferrin levels increased after thalidomide treatment (p=0.030) (Figure 2C). There was no significant change in serum ferritin, hepcidin, sTfR, TIBC, or UIBC (Figure 2D-H). The detailed data are shown in Supplementary Table S2.
 |
- Figure 2. Thalidomide
treatment (TT) affected serum iron-status parameters in TDT patients.
(A) Serum iron and (B) transferrin saturation were significantly
decreased, and (C) transferrin was significantly increased, (D-H) but
serum ferritin, hepcidin, soluble transferrin receptor (sTfR), total
iron-binding capacity (UIBC), and unsaturated iron-binding capacity
(UIBC) were unaffected.
|
Correlation between changes in erythropoiesis and iron-status parameters and hemoglobin increment.
We investigated the correlations between changes in erythropoiesis and
iron-status-related parameters and prolonged hemoglobin increment after
thalidomide treatment. Hemoglobin increment was significantly
correlated with changes in RBCs (r=0.839, p<0.001), hematocrit (r=0.813, p<0.001) and hepcidin (r=0.439, p=0.041) (Figure 3A-C). In addition, hemoglobin increment was negatively correlated with serum iron (r=−0.536, p=0.010) (Figure 3D). There were no correlations between changes in other parameters and hemoglobin increment after thalidomide treatment (Supplementary Table S3).
 |
- Figure 3. Plots of
prolonged hemoglobin increment after thalidomide treatment versus
changes in (A) red blood cells (RBCs), (B) hematocrit, (C) hepcidin,
and (D) serum iron.
|
Discussion
Thalidomide
has recently become a treatment option for patients with β-thalassemia,
under strict medical supervision or in clinical trials, especially for
patients who are unable to undergo hematopoietic stem cell
transplantation. Thalidomide has shown promise for increasing HbF and
reducing the need for transfusions in patients with β-thalassemia.[9-11]
In addition, thalidomide significantly reduced spleen size and may be
used to treat thrombocytopenia in patients with hypersplenism.[12,13] Chen et al.[13]
also observed that thalidomide improved organ iron deposition in
patients with TDT. The current study found that thalidomide increased
the concentration of circulating RBCs and Hb, reduced serum EPO levels,
reduced serum iron and transferrin saturation, and increased
transferrin levels in patients with TDT. To the best of our knowledge,
this study offers the first comprehensive analysis of erythropoiesis
and iron homeostasis in patients with thalassemia, providing evidence
to support the use of thalidomide for treating anemia in thalassemia.
After
thalidomide treatment, RBCs increased while EPO levels and
reticulocytes were reduced. Thalidomide thus appeared to increase RBC
production and maturation in patients with TDT, resulting in more
effective erythropoiesis. The decreased serum EPO concentration may be
due to feedback regulation by the increased number of circulating RBCs
and increased Hb concentration. In addition, improved erythropoiesis
may be associated with decreased formation of insoluble globins (α
chain/heme aggregates). Thalidomide acts as an HbF inducer to enhance
the expression of γ-globin, which binds to redundant α-globin chains
and reduces the deposition of α-globin chain tetramers, thereby
reducing their potential toxicity when adhering to erythrocyte
membranes and producing reactive oxygen species.[15]
The extramedullary hematopoiesis rate increases in patients with
β-thalassemia to compensate for anemia, resulting in increased
production and clearance of abnormal RBCs, with hypersplenism and
increased spleen size.[16] Chen et al.[13]
reported a progressive decrease in spleen length in thalassemia
patients treated with thalidomide at 12 months of follow-up, which may
also indicate improved erythropoiesis.
Blood transfusion can halt
disease progression by providing normal RBCs, inhibiting the production
of ineffective RBCs, and reducing extramedullary hematopoiesis.
However, repeated blood transfusions can lead to iron accumulation and
overload. Theoretically, thalidomide treatment will relieve the body's
iron burden by reducing blood transfusions in patients with severe
thalassemia. However, although serum ferritin levels decreased
following thalidomide treatment in the present study, the difference
was not significant. This may be because changes in hepatic iron
deposition after thalidomide treatment are inconsistent with changes in
serum ferritin levels over time. Chen et al.[14]
observed a significant decrease in serum ferritin levels up to 12
months after thalidomide treatment but significant reductions in
hepatic iron deposition at 3 and 12 months of treatment, suggesting
that hepatic iron deposits may be reduced during thalidomide therapy
even when serum ferritin levels are not significantly changed.
Thalidomide tended to reduce transferrin saturation and serum iron
levels after treatment, possibly due to increased iron consumption due
to enhanced erythropoiesis. When transferrin saturation is reduced, the
predominant form of transferrin in circulation is
monoferric-transferrin, whose each molecule delivers less iron to
erythroid precursors than holo-transferrin.[17] This
enables more erythroid precursors to receive a smaller portion of the
iron pool to offset developing anemia and is consistent with a low MCV
and MCH. Therefore, Thalidomide treatment results in a state of
iron-restricted-like erythropoiesis. Typically, as in iron-deficiency
anemia, iron-restricted-like erythropoiesis is associated with low MCV
and MCH values, where the amounts of heme and Hb per cell are lower due
to the delivery of less iron to each RBC precursor and the production
of fewer cells, resulting in low MCV anemia. Iron is a rate-limiting
factor for heme synthesis, a transcriptional regulator of globin
synthesis; decreased iron concentration may thus reduce heme synthesis,
leading to decreased α-globin precipitation on erythrocyte membranes.[18]
In addition, transferrin levels are markedly elevated after thalidomide
treatment, and additional transferrin may have the inherent ability to
distribute small doses of iron to a large number of RBCs in
thalassemia.[18] Patients with TDT may thus benefit from the reductions in MCV and MCH caused by thalidomide treatment.
The
mechanism by which thalidomide benefits patients with thalassemia is
currently unclear. On the one hand, thalidomide effectively enhanced
the expression of GATA-1 and EKLF in erythroid progenitor cells and induced the expression of the γ-globin gene.[20]
On the other hand, thalidomide induced γ-globin gene expression and
increased HbF synthesis through reactive oxygen species-dependent
activation of the p38 mitogen-activated protein kinase signaling
pathway and histone H4 acetylation.[7] Thalidomide can
also increase the number of hematopoietic colonies, including
erythrocyte colonies, and increase demethylation of H3 histone and
acetylation of H4 histones in erythroid precursor cells, making it more
effective in upregulating HbF.[21,22] In addition, thalidomide promotes erythropoiesis by inducing STAT5 and GATA-1 transcription factors.[23]
The current results showed that thalidomide improved erythropoiesis and
iron homeostasis to some extent. The effects of thalidomide on
thalassemia are thus multifaceted, and more comprehensive research is
needed to elucidate the key targets and pathways of thalidomide in
treating thalassemia, including a long-term study in a larger cohort of
TDT patients.
Conclusions
In
summary, our findings demonstrate that thalidomide improves TDT,
possibly via improvements in erythropoiesis and iron homeostasis. This
study expands our understanding of the effects of thalidomide in
thalassemia and provides evidence to support its use in treating this
disease.
Aknowledgment
We
want to thank the participating families for their continuous support
and participation in this study. This study was financially supported
by Zigong's Key Science and Technology Project (grant no. 2020YXY04).
References
- Kattamis A, Kwiatkowski JL, Aydinok Y. Thalassaemia. Lancet. 2022;399:2310-24. https://doi.org/10.1016/S0140-6736(22)00536-0 PMid:35691301
- Taher AT, Musallam KM, Cappellini MD. beta-Thalassemias. N Engl J Med. 2021;384:727-43. https://doi.org/10.1056/NEJMra2021838 PMid:33626255
- Casu
C, Pettinato M, Liu A, Aghajan M, Lo Presti V, Lidonnici MR, Munoz KA,
O'Hara E, Olivari V, Di Modica SM, Booten S, Guo S, Neil G, Miari R,
Shapir N, Zafir-Lavie I, Domev H, Ferrari G, Sitara D, Nai A, Rivella
S. Correcting beta-thalassemia by combined therapies that restrict iron
and modulate erythropoietin activity. Blood. 2020;136:1968-79. https://doi.org/10.1182/blood.2019004719 PMid:32556142 PMCid:PMC8209564
- Farmakis
D, Porter J, Taher A, Domenica Cappellini M, Angastiniotis M,
Eleftheriou A. 2021 Thalassaemia International Federation Guidelines
for the Management of Transfusion-dependent Thalassemia. Hemasphere.
2022;6:e732. https://doi.org/10.1097/HS9.0000000000000732 PMid:35928543 PMCid:PMC9345633
- Musallam
KM, Taher AT, Cappellini MD, Sankaran VG. Clinical experience with
fetal hemoglobin induction therapy in patients with beta-thalassemia.
Blood. 2013;121:2199-212; quiz 372. https://doi.org/10.1182/blood-2012-10-408021 PMid:23315167
- Stewart AK. Medicine. How thalidomide works against cancer. Science. 2014;343:256-7. https://doi.org/10.1126/science.1249543 PMid:24436409 PMCid:PMC4084783
- Aerbajinai
W, Zhu J, Gao Z, Chin K, Rodgers GP. Thalidomide induces gamma-globin
gene expression through increased reactive oxygen species-mediated p38
MAPK signaling and histone H4 acetylation in adult erythropoiesis.
Blood. 2007;110:2864-71. https://doi.org/10.1182/blood-2007-01-065201 PMid:17620452 PMCid:PMC2018668
- Ren
Q, Zhou YL, Wang L, Chen YS, Ma YN, Li PP, Yin XL. Clinical trial on
the effects of thalidomide on hemoglobin synthesis in patients with
moderate thalassemia intermedia. Ann Hematol. 2018;97:1933-9. https://doi.org/10.1007/s00277-018-3395-5 PMid:29931453
- Yang
K., Wu Y., Zhou Y., Long B., Lu Q., Zhou T., Wang L., Geng Z., Yin X.
Thalidomide for patients with β-thalassemia: A multicenter experience.
Mediterr J Hematol Infect Dis 2020, 12(1): e2020021 https://doi.org/10.4084/mjhid.2020.021 PMid:32395210 PMCid:PMC7202343
- Chen
JM, Zhu WJ, Liu J, Wang GZ, Chen XQ, Tan Y, Xu WW, Qu LW, Li JY, Yang
HJ, Huang L, Cai N, Wang WD, Huang K, Xu JQ, Li GH, He S, Luo TY, Huang
Y, Liu SH, Wu WQ, Lu QY, Zhou MG, Chen SY, Li RL, Hu ML, Huang Y, Wei
JH, Li JM, Chen SJ, Zhou GB. Safety and efficacy of thalidomide in
patients with transfusion-dependent beta-thalassemia: a randomized
clinical trial. Signal Transduct Target Ther. 2021;6:405. https://doi.org/10.1038/s41392-021-00811-0 PMid:34795208 PMCid:PMC8602273
- Li
X, Hu S, Liu Y, Huang J, Hong W, Xu L, Xu H, Fang J. Efficacy of
Thalidomide Treatment in Children With Transfusion Dependent
beta-Thalassemia: A Retrospective Clinical Study. Front Pharmacol.
2021;12:722502. https://doi.org/10.3389/fphar.2021.722502 PMid:34456732 PMCid:PMC8397440
- Chen
J, Zhu W, Cai N, Bu S, Li J, Huang L. Thalidomide induces haematologic
responses in patients with beta-thalassaemia. Eur J Haematol.
2017;99:437-41. https://doi.org/10.1111/ejh.12955 PMid:28850716
- Chen
Y, Cai N, Lai Y, Xu W, Li J, Huang L, Huang Y, Hu M, Yang H, Chen J.
Thalidomide for the Treatment of Thrombocytopenia and Hypersplenism in
Patients With Cirrhosis or Thalassemia. Front Pharmacol. 2020;11:1137. https://doi.org/10.3389/fphar.2020.01137 PMid:32792958 PMCid:PMC7394185
- Che
J, Luo T, Huang L, Lu Q, Yan D, Meng Y, Xie J, Chen W, Chen J, Long L.
Magnetic Resonance Imaging Quantification of the Liver Iron Burden and
Volume Changes Following Treatment With Thalidomide in Patients With
Transfusion-Dependent ss-Thalassemia. Front Pharmacol. 2022;13:810668. https://doi.org/10.3389/fphar.2022.810668 PMid:35250561 PMCid:PMC8894715
- Gardenghi
S, Ramos P, Marongiu MF, Melchiori L, Breda L, Guy E, Muirhead K, Rao
N, Roy CN, Andrews NC, Nemeth E, Follenzi A, An X, Mohandas N, Ginzburg
Y, Rachmilewitz EA, Giardina PJ, Grady RW, Rivella S. Hepcidin as a
therapeutic tool to limit iron overload and improve anemia in
beta-thalassemic mice. J Clin Invest. 2010;120:4466-77. https://doi.org/10.1172/JCI41717 PMid:21099112 PMCid:PMC2993583
- Kolnagou
A, Michaelides Y, Kontoghiorghe CN, Kontoghiorghes GJ. The importance
of spleen, spleen iron, and splenectomy for determining total body iron
load, ferrikinetics, and iron toxicity in thalassemia major patients.
Toxicol Mech Methods. 2013;23:34-41. https://doi.org/10.3109/15376516.2012.735278 PMid:23039902
- Ginzburg
Y, Rivella S. beta-thalassemia: a model for elucidating the dynamic
regulation of ineffective erythropoiesis and iron metabolism. Blood.
2011;118:4321-30. https://doi.org/10.1182/blood-2011-03-283614 PMid:21768301 PMCid:PMC3204905
- Chen JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109:2693-9. https://doi.org/10.1182/blood-2006-08-041830 PMid:17110456 PMCid:PMC1852217
- Li
H, Rybicki AC, Suzuka SM, von Bonsdorff L, Breuer W, Hall CB,
Cabantchik ZI, Bouhassira EE, Fabry ME, Ginzburg YZ. Transferrin
therapy ameliorates disease in beta-thalassemic mice. Nat Med.
2010;16:177-82. https://doi.org/10.1038/nm.2073 PMid:20098432
- Jalali
Far MA, Dehghani Fard A, Hajizamani S, Mossahebi-Mohammadi M, Yaghooti
H, Saki N. Thalidomide is more efficient than sodium butyrate in
enhancing GATA-1 and EKLF gene expression in erythroid progenitors
derived from HSCs with beta-globin gene mutation. Int J Hematol Oncol
Stem Cell Res. 2016;10:37-41.
- Fard
AD, Kaviani S, Noruzinia M, Soleimani M, Abroun S, Chegeni R,
Hajifathali A, Zonoubi Z, Ahmadvand M, Mohammadi MM, Saki N. Evaluation
of H3 histone methylation and colony formation in erythroid progenitors
treated with thalidomide and sodium butyrate. Lab Hematol. 2013;19:1-5.
https://doi.org/10.1532/LH96.12003 PMid:23538327
- Fard
AD, Hosseini SA, Shahjahani M, Salari F, Jaseb K. Evaluation of Novel
Fetal Hemoglobin Inducer Drugs in Treatment of beta-Hemoglobinopathy
Disorders. Int J Hematol Oncol Stem Cell Res. 2013;7:47-54.
- Grzasko
N, Chocholska S, Goracy A, Hus M, Dmoszynska A. Thalidomide can promote
erythropoiesis by induction of STAT5 and repression of external pathway
of apoptosis resulting in increased expression of GATA-1 transcription
factor. Pharmacol Rep. 2015;67:1193-200. https://doi.org/10.1016/j.pharep.2015.05.011 PMid:26481541
Supplementary files
 |
Table S1. Erythropoiesis-related parameters before and after thalidomide treatments.
|
 |
Table S2. Serum iron parameters before and after thalidomide treatments.
|
 |
Table S3. Correlation between changes in erythropoiesis and iron-status parameters and hemoglobin increment.
|