Justification of Universal Iron Supplementation for Infants 6-12 months in Regions with a High Prevalence of Thalassemia
Phakatip Sinlapamongkolkul1, Pacharapan Surapolchai1* and Vip Viprakasit2*.
1 Department of Pediatrics, Faculty of Medicine, Thammasat University, Pathumthani, Thailand.
2 Department of Pediatrics and Thalassemia Center, Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand.
* Both authors equally contributed to the work.
Published: September 1, 2023
Received: July 11, 2023
Accepted: August 17, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023056 DOI
10.4084/MJHID.2023.056
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Introduction:
Many clinicians hesitate to adopt a universal infant iron
supplementation program due to the risk of increased iron absorption
for those with thalassemia. We aimed to determine thalassemia
prevalence in 6- to 12-month-old infants, along with the iron status of
those with and without thalassemia.
Methods: We performed a cross-sectional descriptive study of
infants attending the Well Baby Clinic at Thammasat University Hospital
for routine checkups. Complete blood count, hemoglobin electrophoresis,
iron parameters, and molecular genetics for common α- and β-thalassemia
were evaluated.
Results: Overall, 97 of 206 (47%) participants had thalassemia
minor, the majority having Hb E traits. None had thalassemia intermedia
or major. Familial history of anemia or thalassemia presented an
increased risk of detecting thalassemia minor in offspring (OR 5.18;
95% CI 2.60-10.33, p=0.001).
There were no statistical differences in transferrin saturation, serum
ferritin and hepcidin between iron-replete infants with thalassemia
minor and those without. However, one-third of infants with thalassemia
minor (31/97) also had iron deficiency anemia (IDA), with a similar
risk of having iron deficiency to infants without thalassemia. There
was no hepcidin suppression in our infants with thalassemia minor as
compared to controls.
Conclusions: Both thalassemia and IDA are endemic to Southeast
Asia. Infants with thalassemia minor, particularly with Hb E and
α-thalassemia traits, are at risk of IDA. Our short-term universal iron
supplementation program for 6- to 12-month-old infants does not appear
to increase the risk of those with thalassemia minor developing iron
overload in the future.
|
Introduction
An
estimated 300-million children worldwide had anemia in 2011,[1] and
iron deficiency anemia (IDA) remains the most common cause of this to
date. The World Health Organization (WHO) publishes an international
anemia control guideline that states all children and women living in
settings where the prevalence of anemia exceeds 20% should receive
supplemental iron.[2] In Thailand, this recommendation has been adopted
by the Department of Health, Ministry of Public Health, which
recommends a universal iron supplement for Thai infants over six months
old to prevent IDA when these babies come for routine vaccination. Iron
supplementation continues until 24 months of age with 12.5 mg of
elemental iron weekly, according to WHO Guideline (2011).[3,4]
Compared to placebos or no intervention, intermittent iron
supplementation is considered effective in reducing the risk of anemia
or iron deficiency (ID) in children younger than 12 years old.[5] This
is because infants older than six months have a high prevalence of IDA,
which can impair physical, behavioral, and cognitive functions and
result in persistent neurocognitive defects, despite receiving iron
therapy later.[6] However, local Thai practitioners, particularly
pediatricians, have concerns about this policy since there is a high
prevalence of thalassemia and hemoglobin disorders in the country.[7]
It is widely accepted that thalassemia disease could significantly
increase the risk of iron overload, leading to iron toxicity later in
life.[8-9]
Thalassemia is characterized by inherited mutations of α and β
globin genes causing decreased globin synthesis. At least 5.2% of the
world population carries one allele of globin gene variants (carrier or
trait).[10] For α-thalassemia, there are two types based on molecular
defects: α0-thalalassemia caused by deletions of two linked α-globin genes in cis (--/αα) and α+-thalassemia caused by deletions of one α-globin gene (-α/αα) or nucleotide mutations (αTα/αα or αα/ααT).
Coinheritance of two affected alleles in autosomal recessive mode leads
to chronic hemolytic anemia and ineffective erythropoiesis, known as
thalassemia disease.[11] On the other hand, hemoglobinopathy is mainly
caused by mutations of coding sequences and produces qualitative
defects. Several hemoglobinopathies are innocuous and do not lead to
any clinical consequences.[12] However, some mutations such as
hemoglobin E (Hb E) at codon 26 of the β-globin genes (GAG>AAG) also have quantitative effects, and an interaction of Hb E with β-thalassemia mutations results in Hb E/β-thalassemia syndrome with heterogeneous clinical severity.
Around 30 to 40 percent of Thais are thalassemia carriers, including α-thalassemia, β-thalassemia, and Hb E. Due to a high prevalence of all genotypes, it is not uncommon to find individuals with combined α and β-globin
abnormalities.[13-16] Collectively, these thalassemia traits, simple or
in combination, are asymptomatic and do not require specific treatment;
these are classified as “thalassemia minor”. Individuals with
homozygous Hb E (Hb E/E) carrying two defective β-globin genes also present with milder forms of anemia without hepatosplenomegaly or blood transfusion being required.[17]
Several previous studies have examined iron status in patients with
thalassemia,[18-21] but little is known about thalassemia minor in
comparison to normal populations,[22] particularly in infants. Iron
overload is one of the most common complications in those with
thalassemia due to blood transfusions and increased iron
absorption.[8-9] Intestinal iron intake in thalassemia is usually
enhanced due to hepcidin suppression by the upregulation of
erythropoietic markers such as GDF-11, GDF-15 and Erfe in response to
chronic anemia and erythropoietin drive.[23-25] Hepcidin controls iron
intake through duodenal enterocytes by limiting the expression of
ferroportin: an intestinal iron gateway into circulation. Research
consistently shows hepcidin suppression in thalassemia patients.[26,27]
Recently, a Sri Lankan study demonstrated that β-thalassemia carriers
had mildly suppressed hepcidin concentrations out of proportion to
their iron stores. It has been suggested that a widespread distribution
of iron supplementation could possibly increase the risk of harmful
iron overload in β-thalassemia carriers.[28] In Thailand, there has
been no data on iron status and hepcidin levels in the pediatric
population with our common thalassemia traits of α-thalassemia and Hb E and homozygous HbE, especially in infants who receive supplements through our national program.
Our main objective was to determine the iron status in infants aged six
to 12 months from our Well Baby Clinic and identify the prevalence of
ID and IDA among those with or without thalassemia. In addition, we
evaluated clinical and laboratory characteristics of both groups to
identify which factors, including hepcidin levels, would significantly
influence iron status. We aimed to illustrate whether infants with
thalassemia are at similar or lower risk of ID or IDA as compared to
the similarly-aged general population and, thus, supply evidence
regarding safe universal iron supplementation for Thai infants.
Materials and Methods
Study population.
This is a cross-sectional descriptive study approved by the Human
Research Ethics Committee of Thammasat University (Medicine)
(MTU-EC-PE-2-006/59). From June 2016 to June 2017, six- to 12-month-old
infants attending the Well Baby Clinic at Thammasat University Hospital
for vaccinations and scheduled checkups were randomly recruited.
Written informed consent was given by their parents or legal guardians.
Our inclusion criteria were term newborns (38-42 weeks gestation) with
a birthweight between 2,500-4,000 grams having no prenatal and
perinatal complications such as severe birth asphyxia, severe
respiratory distress, or neonatal intensive care unit admission.
Infants with chromosome abnormalities/syndromes,
infectious/inflammatory diseases and any acute health problems were
excluded. All clinical samples were collected before routine universal
iron supplementation. We obtained demographic/clinical data through
direct interviews with two investigators (PSi and PSu). Weight and
length of participants were measured and evaluated by Z-score,
according to WHO guidelines.[29] The Z-scores of weight-for-lengths
below or above two standard deviations (SD) are categorized as
underweight and overweight, respectively.
Hematological and biochemical evaluation.
All participants underwent complete blood count evaluation using
automated cell count (UniCel®DxH 800, Beckman Coulter, Brea, USA),
hemoglobin typing by automated capillary electrophoresis analyzer
(MINICAP, Sebia, Lisses, France), iron parameters including serum iron
(SI), total iron binding capacity (TIBC) using a fully automated
quantitative assay, and serum ferritin by electrochemiluminescence
immunoassay (ECLIA or Elecsys® technology, Roche Diagnostics, Penzberg,
Germany). SI, TIBC and serum ferritin assays were performed using a
ROCHE COBAS BIO centrifugal analyzer according to manufacturer’s
instructions.[30]
Hepcidin, a cysteine-rich 25-amino acid peptide hormone, is produced
from 84-amino acid pro-hepcidin in the liver and secreted into blood
circulation, functioning iron homeostasis and anti-microbial activity.
Accordingly, methods have been developed for identification and
quantitation of pro-hepcidin and hepcidin in blood and urine specimens,
including high-performance liquid chromatography/electrospray
ionization-mass spectrometry (HPLC/ESI-MS), enzyme-linked immunosorbent
assay (ELISA) methods. The HPLC/ESI-MS method is sensitive and
selective; nonetheless, it requires a very expensive sophisticated
instrument system and a highly experienced analyst.[31,32] In
comparison, the ELISA, including competitive- and sandwich types,
require specific antibody/antibodies against hepcidin epitopes,
sensitive colorimetric /chemiluminescent reactions (rank of ng/mL and
pg/mL), and commercially available kits by many manufacturers. In this
study, we used a competitive ELISA kit (Product number CEB979Hu,
Cloud-Clone Corporation, Wuhan, PR China), with afternoon blood
sampling to prevent diurnal variations,[33,34] for determination of
serum hepcidin, of which the quality control shows very good
sensitivity (low limit of detection <11 pg/mL), specificity (no
significant cross reactivity between hepcidin and analogues), recovery
(82-90%), precision (coefficient of variation <10% for intra-assay
and <12% for inter-assay) and linearity (86-102% for diluted serum
samples).
Molecular analysis.
Genomic DNA was extracted from peripheral blood leukocytes using a
standard protocol of phenol-chloroform extraction. Alpha-globin
genotyping was performed by a single-tube multiplex gap polymerase
chain reaction (Gap-PCR) for detecting seven common α-globin deletions (--SEA, --THAI, -(α) 20.5, --FIL, -- MED-α3.7, -α4.2) and a single-tube multiplex amplification refractory mutation system (ARMS-PCR) for screening six common non-deletional α-globin
mutations in Thailand: initiation codon (ATG➝A-G), codon 30 (ΔGAG),
codon 59 (GGC➝GAC), codon 125 (CTG➝CCG) or Hb QuangSze, termination
codon (TAA➝CAA) or Hb Constant Spring, and termination codon (TAA➝TAT)
or Hb Paksé.[35] Beta-globin genotyping was performed by ARMS-PCR for
detecting 16 common beta-globin mutations (-28, CD8/9, CD17, CD19, CD26
(Hb E), CD26 G>T (stop codon), CD27/28, IVSI-I, IVSI-5, CD35, CD41,
CD41/42, CD43, CD71/72, CD95 and IVSII-654).[36] A single-tube
multiplex Gap-PCR and enzymatic amplification was used for common
beta-globin gene deletions (3.48 kb, 619 bp, Filipino (β)°, SEA HPFH (β)°, Chinese Gγ( Aγδβ)°, Thai (δβ)°,Hb Lepore, HPFH-6 Gγ( Aγδβ)°, Siriraj-thal Gγ( Aγδβ)°,
Asian Indian type A, and Asian Indian type B).[37,38] Hemoglobin E
testing was studied by restriction fragment length polymorphism
(RFLP)-PCR utilizing MnlI restriction enzyme.[39]
Definitions.
Hemoglobin (Hb) <11 g/dL was used to define anemia, in line with WHO
criteria.[40] Participants were classified as having ID if their serum
ferritin (SF) was <30 ng/mL or transferrin saturation (TS) was
<16% (TS = SI/TIBC x100).[41] IDA was diagnosed if there was
compatibility with either laboratory criteria or a therapeutic response
to oral iron therapy (syrup of ferric hydroxide polymaltose complex 3-6
mg/kg/day for 8-12 weeks, as prescribed) and followed up by
hematologists. The coexisting causes and possible risk factors of ID
need to be further identified, especially unresponsiveness to oral iron
therapy. The diagnosis of β-thalassemia
trait was based on Hb A2 level >3.5%. Infants with Hb F >10% were
investigated for common beta-globin gene deletions to diagnose δβ-thalassemia traits and hereditary persistence of fetal hemoglobin (HPFH), as described in previous studies.[37,42]
Statistical analysis. The sample size was calculated from this formula: N = Z2pq/d2,
Z = 1.96, p = 0.4, q = 0.6, d = 0.07. The study population, according
to the prevalence of thalassemia in Thailand (30-40%),[13,15] was 188;
we approximated needing 200 participants, reflecting a 10% dropout.
Demographic data was summarized as frequency and percentage for
qualitative data and as mean and SD for quantitative data. Student’s
t-test or Mann-Whitney U test was used to compare continuous variables;
the chi-square or Fisher’s exact test was used for categorical
variables, as appropriate. Univariate and multivariate logistic
regression analyses were performed to identify risk factors: P of < 0.05 was considered statistically significant.
Results
Clinical characteristics.
A total of 206 infants, with 114 males (55.3%) and a mean age of 8.2
months (SD 2.0, range 6-12 months), were randomly enrolled.
Interestingly, we found 97 individuals (47%) with some form of
thalassemia minor, 39 with α-thalassemia (18.9% of total population), 45 with β-globin mutation mainly Hb E (21.8%), and 13 with combined α and β
globin abnormalities (6.3%). None of these individuals had genotypes
found in thalassemia diseases such as Hb H disease or Hb E/β-thalassemia;
therefore, they were classified as thalassemia minor and subsequently
used for further analysis. Details of all comprehensive genotype data
are shown in Table 1. Infants with thalassemia minor had no history of blood transfusion and no hepatosplenomegaly.
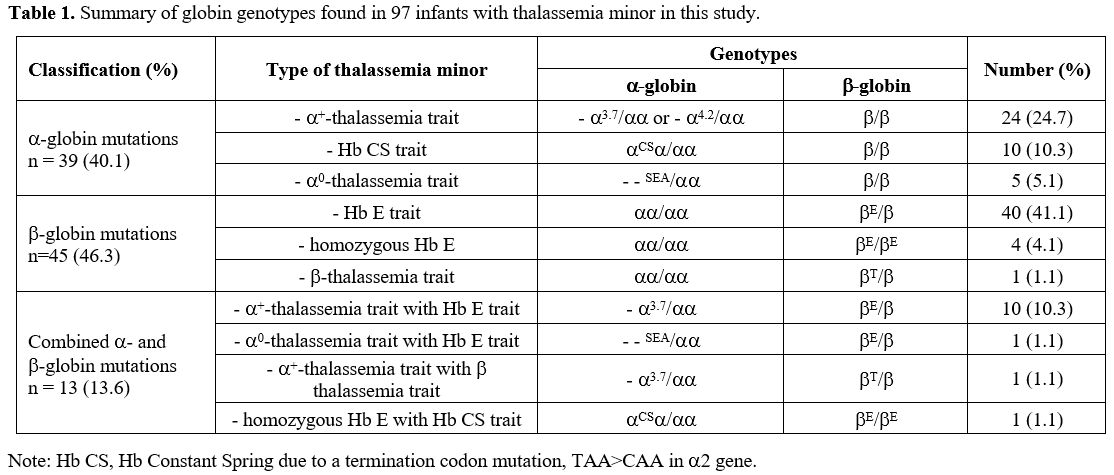 |
- Table
1. Summary of globin genotypes found in 97 infants with thalassemia minor in this study.
|
We found no significant differences in all clinical characteristics:
age, gender, growth and nutrition normal iron and those with IDA to see
the effects of parameters, and iron markers such as SF, TS, and
hepcidin levels. The exception was having a family history of anemia or
thalassemia being more common in infants with thalassemia minor (43.3%)
versus those without thalassemia (12.8%): Table 2.
In our logistic regression analysis, having a history of anemia or
thalassemia showed an increased risk of thalassemia minor in infants
with an odds ratio of 5.18 (95% CI: 2.60-10.33), p
= 0.001. Notably, the number of infants with both thalassemia minor and
IDA at first diagnosis was higher than those with normal globin
genotypes (32.0% vs. 20.2%); moreover, the number of infants with
normal iron status and ID was significantly different among infants
with and without thalassemia minor (p = 0.037): Table 2.
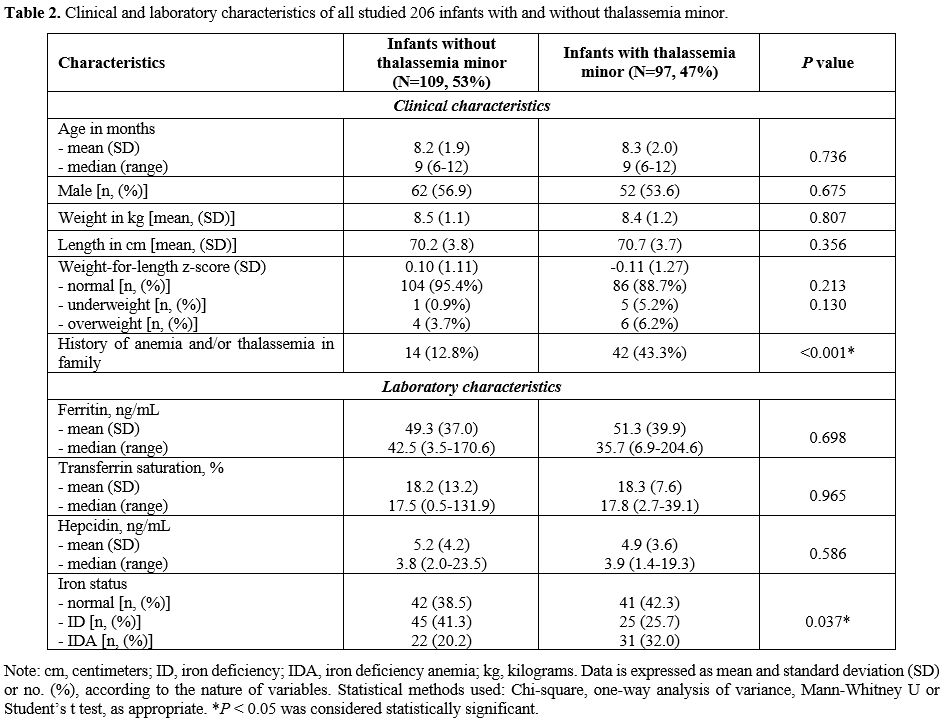 |
- Table
2. Clinical and laboratory characteristics of all studied 206 infants with and without thalassemia minor.
|
The primary diagnosis of infants with IDA using Hb levels with SF and
TS values was further confirmed when the majority responded to iron
therapy. Thirty-four out of 53 IDA infants with (n = 31) and without
thalassemia minor (n = 22) have received iron therapy, and all showed
therapeutic response displayed as a significant increase across all red
blood cell (RBC) parameters, compared to the baseline within their
groups (p
< 0.05). Of note, IDA infants without thalassemia minor (n = 18) had
slightly greater increments in Hb and mean corpuscular volume (MCV)
after iron therapy than IDA infants with some type of thalassemia minor
(n = 16); however, there were no statistically significant differences
between groups: Supplementary Table 1.
Iron status in infants with or without thalassemia minor.
We performed subgrouping analysis within the groups of infants with and
without thalassemia minor to determine the effects of normal iron
status and IDA on hematological parameters (Table 3).
There were significant differences in Hb, hematocrit (Hct), MCV, mean
corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration
(MCHC), and red blood cell distribution width (RDW) within both groups
suggesting iron played a significant role in determining Hb, Hct, and
red blood cell indices. With the exception of increased RBC counts and
RDW, the other RBC parameters were lower in those with IDA versus
normal iron. Interestingly, infants with thalassemia minor who had IDA
(n = 31) displayed statistically lower values in MCV, MCH, with the
higher RDW: Table 3.
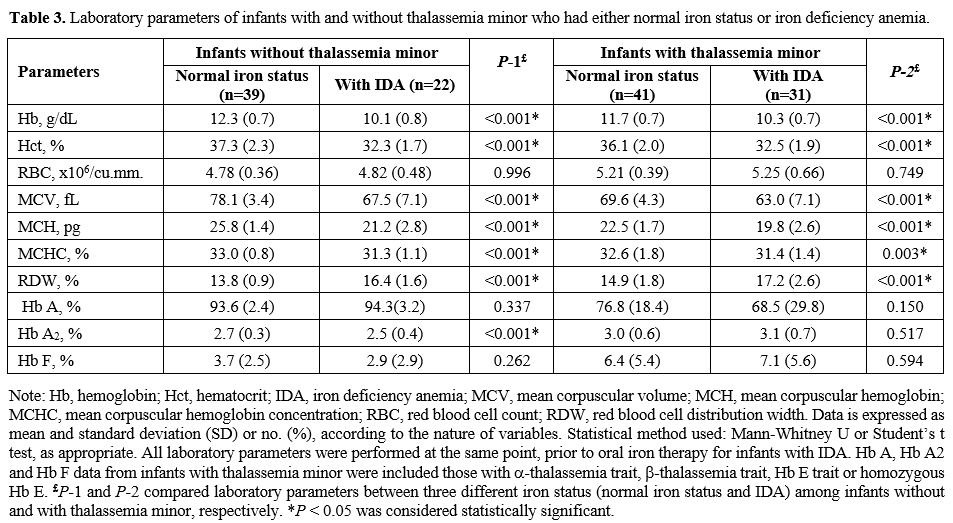 |
- Table
3. Laboratory parameters of infants with and without thalassemia minor
who had either normal iron status or iron deficiency anemia.
|
This suggests that the coinheritance of globin mutations can have
epistatic hematological effects on top of any primary effects on iron
status.
Effects of thalassemia minor on hematologic and iron parameters.
We further compared those infants with thalassemia minor. Coinheritance
of thalassemia had significant effects on Hb, Hct, RBC, MCV, MCH, RDW,
but not MCHC, solely in those with normal iron status: Supplementary Table 2.
However, we found only RBC and MCV to be significantly different in
infants who already had IDA, suggesting this epistatic effect took
place only within those two parameters. In addition, we found an effect
of iron on significantly decreased levels of Hb A2 in infants without
thalassemia minor but not in those with thalassemia. On the other hand,
the basal Hb F in infants with thalassemia minor was generally higher
than those without, suggesting a delay in globin switching, one of the
consequences of globin abnormality.[43,44] Iron status does not appear
to be significantly associated with the levels of persistent Hb F
expression within the groups of both infants with and without
thalassemia minor (Table 3).
Effects of thalassemia minor on hepcidin expression.
We compared serum hepcidin, serum ferritin, and TS in infants without
thalassemia having normal iron status to those having different types
of thalassemia minor: Figure 1. Measurements for each group are in Supplementary Table 3A (iron replete) and Supplementary Table 3B
(iron deplete). Levels of hepcidin appeared to be slightly lower in
those with thalassemia minor and lowest in those with combined α and β
globin mutations. This was consistent with the slightly higher serum
ferritin and TS seen in those with combined thalassemia minors,
although without statistically significant differences. No differences
within these parameters were found between groups or by gender (data
not shown).
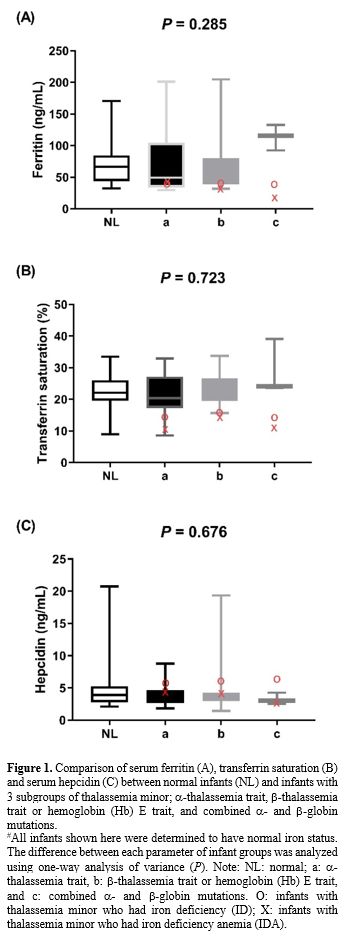 |
- Figure
1. Comparison of serum ferritin (A), transferrin saturation (B) and
serum hepcidin (C) between normal infants (NL) and infants with 3
subgroups of thalassemia minor; α-thalassemia trait, β-thalassemia trait or hemoglobin (Hb) E trait, and combined α- and β-globin mutations.
#All
infants shown here were determined to have normal iron status. The
difference between each parameter of infant groups was analyzed using
one-way analysis of variance (P). Note: NL: normal; a: α-thalassemia trait, b: β-thalassemia trait or hemoglobin (Hb) E trait, and c: combined α- and β-globin
mutations. O: infants with thalassemia minor who had iron deficiency
(ID); X: infants with thalassemia minor who had iron deficiency anemia
(IDA).
|
Discussion
We
found the prevalence of thalassemia minor (carrier) in nearly half of
the infants, at 47%. It was higher than previous reports of
30-40%.[13,15] One of the main reasons was the DNA testing used in our
current study was far more comprehensive than the approaches such as
cord blood hemoglobin studies, hemoglobin typing, etc. used 20 years
ago. This is consistent with a report on thalassemia prevalence by
Viprakasit V et al. in 2009,[45] stating that Hb E trait was the most
common type of thalassemia minor[14,15,45] in Thailand, with a
frequency up to 50-60% in Southeast Asia.[14] We found no individuals
with thalassemia disease. This may point to the effectiveness of
Thailand’s prevention and control program that screens for thalassemia
carriers in pregnant women and their partners in order to identify the
genetic risk of severe thalassemia syndromes.[16] Therefore, our
studied population is likely to represent relatively “healthy” infants
receiving routine health care and are a primary target for the iron
supplementation program.
A previous study of β-thalassemia
traits noted the presence of mildly increased erythropoiesis, as seen
through elevated erythropoietin levels.[46] It has also been observed
that adults with α- or β-thalassemia
traits have shown increases in soluble transferrin receptors or
erythropoietin concentrations, indicating ineffective erythropoiesis
and increased erythropoietic drive leading to hepcidin suppression and
upregulated iron absorption. Prior studies in India and Iran examining
the iron status of adults with β-thalassemia traits concluded that β-thalassemia
traits had higher serum ferritin than the controls, representing an
advantage in iron balance.[47,48] These particular findings did not
concur with others, which had stated that ID might commonly coexist
with thalassemia traits.[20,49,50] These conflicting results have
caused uncertainty in iron supplementation strategies for areas with a
high prevalence of hemoglobinopathy. Of note, a universal iron
supplementation program for infants might not be directly applicable to
countries where differences in the genetic background of thalassemia,
especially Hb E and α-thalassemia traits are uncommon and β-thalassemia
traits are more frequent.[51,52] With a potential increase in the risk
of iron overload for individuals with thalassemia minors, universal
iron supplementation programs remain a point of contention.
A recent community study of 1821 Sri Lankan schoolchildren aged 8-18
years (48.3% males) from the Oxford group has shown that this might be
the case for those with β-thalassemia traits.[28] Eighty-two β-thalassemia
carriers with iron-replete had evidence of increased erythropoiesis, a
slight but significant reduction in hepcidin, and suppression of
hepcidin out of proportion to their iron stores: lower
hepcidin-ferritin ratio compared with non-carrier controls (n = 176
with normal MCV and MCH). Another Sri Lankan cross-sectional study of
2273 children (aged 12–19 years) from a total of 7526 students,
reported the same effect in iron-replete α‐thalassemia carriers as compared to the non‐iron deficient controls without thalassemia minor (4.8 ng/mL vs 5.3 ng/mL, p = 0.02).[53]
However, this was not observed in those with Hb E traits from both
cohorts.[28,53] Based on these results, it has been proposed that a
hepcidin cutoff of < 3.2 ng/mL could be used to select cases for
iron supplementation in countries with high rates of thalassemia
carriers.[53] Both studies were conducted in primary and secondary
school students as this is the age group at which iron supplementation
is given in Sri Lanka. However, the effects of being a thalassemia
carrier on hepcidin suppression, as well as the risk of iron
accumulation in younger cases with thalassemia minor, remain unclear.
Our study determined this iron supplement issue in infants with
thalassemia minor. While we could not find significant hepcidin
suppression in our infants with thalassemia minors as compared to
previous studies, our results were somewhat in line with such findings.
Most of our thalassemia minors were Hb E traits, and this condition did
not show a significant enough globin imbalance leading to ineffective
erythropoiesis and subsequent hepcidin suppression. Moreover, even for
individuals with homozygous Hb E, we found no evidence of this effect.
Our infants with α-thalassemia
carriers also demonstrated no effects of hepcidin suppression,
differing from the previous study.[53] This may be because our
population was younger with remaining Hb F expression (Table 1 and 3) and had less globin imbalance and ineffective erythropoiesis per se. It is, therefore, possible the erythropoietic drive that suppresses hepcidin was not fully operative yet.
In addition, the normal physiology of hepcidin expression, especially
within the first year of life, might be more dynamic. A recent study in
late preterm infants (32-36 weeks gestation) described a physiologic
decrease of hepcidin levels during the first four months of life to
increase iron availability.[54] Another longitudinal study that
followed 140 Spanish healthy and full-term infants found hepcidin
levels increased from six to 12 months of age with hepcidin levels
positively correlated with iron status.[55] These results suggested
that, in normal babies, a regulation of hepcidin production is under
development during the first year of life; this may also be true for
infants with thalassemia. Therefore, the effects of ineffective
erythropoiesis on hepcidin suppression in thalassemia traits are likely
not fully apparent during the first year of their life. This warrants
further study to define at what age this effect would first be
identified.
We still found our infants with thalassemia minor having a high
proportion of iron depletion (57.7%), similar to infants without
thalassemia (61.5%); the number of infants with both thalassemia and
IDA was even significantly higher than infants without thalassemia
minor (32 vs 20.2%) (Table 2).
Comparing with the two previous studies at well baby and well child
clinics of different university hospitals, we reported markedly higher
prevalence of IDA (25.7% VS 19% by Linpisarn et al and 14.3% by
Tantracheewathorn et al).[56,57] Nevertheless, our prevalence was in
disagreement with the Fifth National Nutritional Survey of Thailand in
2003 by Department of Health, Ministry of Public Health of Thailand
which reported prevalence of anemia in 6–11-month-old infants up to
56.3%.[58] These discordances might be due to differences in geographic
settings, methodology and socioeconomic level of the populations. Since
Thailand is an endemic area of thalassemia and malaria, we urge
exploration and explanation of all causes of anemia, including
inherited and acquired, and the application of proper management.
Additionally, the long-term irreversible neurodevelopmental
consequences of ID,[59,60] as well as the extremely critical diagnosis
and prevention of ID and IDA in infants, are well-known and published,
including preventive strategies of iron deficiency anemia and other
micronutrient deficiencies in reproductive-aged and pregnant women
which might be affecting their child health and developmental
outcomes.[61] Universal screening for anemia by Hb concentrations at
approximately 12 months of age, as recommended by the American Academy
of Pediatrics (AAP)[62] and the Royal College of Pediatricians of
Thailand & Pediatric Society of Thailand, might not be suitable for
Thailand. We would, instead, strongly recommend screening and early
detection of ID and anemia in infants aged 6-12 months in
well-childcare settings by CBC, RBC morphology and the proper iron
parameters. Thus, the coexisting causes (such as chronic hemolysis and
inflammation) and possible risk factors of ID need to be further
identified, especially unresponsiveness to oral iron therapy, including
inadequate iron in complementary feeding (maternal preference for
staple foods), micronutrient deficiency, intestinal infection,
parasitic infestation, and malabsorption.[63] Nevertheless, infants
with thalassemia minor who have IDA or ID would benefit from proper
iron supplementation.
Interestingly, infants with coexisting thalassemia minor and IDA had
significantly reduced Hb, MCV, MCH, and MCHC with increased RDW versus
those having thalassemia minor with normal iron (Table 3).
These findings were consistent with previous studies in India where MCV
and MCH were significantly lower in adults with combined thalassemia
traits and IDA than with either of these conditions.[49] Moreover,
discordance between RBC count and RDW was also observed in infants with
coexisting thalassemia minor and IDA as described in recent
studies.[64,65] In addition, diminished HbA2 levels in patients with
concomitant β-thalassemia
traits and iron deficiency have been observed.[51,66] We believe our
RBC indices to present a comprehensive analysis of thalassemia carriers
in this age group. Our findings could be useful as references.
Among 36 thalassemia minor infants with anemia, we found five cases who did not have coexisting IDA, including infants with two α-thalassemia traits (-α3.7/αα and --SEA/αα), one β-thalassemia trait, one Hb E trait and one homozygous Hb E. This suggested that α- and β-thalassemia
traits may be the cause of mild anemia in some infants. Accordingly,
anemic infants unresponsive to oral iron therapy should be investigated
for thalassemia rather than continuously undergoing long-term iron
therapy by default, as toxicity or other side effects may develop. A
familial history of anemia or thalassemia, as shown herein, was found
to be strongly associated with thalassemia minor in offspring and could
be used to diagnose future cases early.
Conclusions
Our study showed that infants in Thailand, from 6 to 12 months old, with thalassemia minor, in which the majority had Hb E and α-thalassemia
traits, are at similar risk of developing IDA as the general
population. This may partially be due to a lack of hepcidin suppression
at this age or the type of mutations we found. Therefore, a universal
short-term period of iron supplementation in infants would likely not
be harmful. More than half of this population could benefit from this
strategy. Beyond this age group, however, particularly for school
children, a proper measurement of serum hepcidin along with using a
cutoff as described earlier would be an alternative approach to select
those who should genuinely receive iron supplementation. This would
minimize the chance of overtreating individuals with thalassemia minor
in areas of high prevalence of thalassemia and hemoglobinopathies.[53]
Acknowledgments
The
authors would like to express our gratitude to all infants and their
parents/guardians for their participation, Miss Pornpen Gamnarai
(Faculty of Medicine, Thammasat University) for her technical
assistance, and Prof Somdet Srichairatanakool (Faculty of Medicine,
Chiang Mai University) for his contribution on the analysis of
iron-related parameters. The English language editing was done by Debra
Kim Liwiski and Sam Ormond, international instructors, Clinical
Research Center, Faculty of Medicine, Thammasat University. The authors
gratefully acknowledge the financial support provided by Thammasat
University Research Fund under TU Research Scholar, contract no
2/67/2560. VV was supported by a Chalermprakiat Grant, Faculty of
Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand.
Author Contributions
PSi
and PSu were the co-principal investigators of the project, evaluated
all study participants, collected data, performed analysis, and drafted
the manuscript. VV, as senior author, developed the concept, analysis
plan, overall interpretation of results, and revised the manuscript.
All authors read and approved the final version of the manuscript.
References
- Guideline: Daily iron supplementation in infants and children. Geneva: World Health Organization; 2016.
- Essential
Nutrition Actions: Improving maternal, newborn, infant and young child
health and nutrition. Geneva: World Health Organization; 2013.
- Department
of Health: Ministry of Public Health of Thailand. Guidelines for
controlling and preventing anemia from iron deficiency. Available at:
URL: http://hpc.go.th/director/data/mch/IDAControl.pdf. Accessed Mar 27, 2019.
- Guideline: Intermittent iron supplementation in preschool and school-age children Geneva: World Health Organization; 2011.
- De‐Regil
LM, Jefferds MED, Sylvetsky AC, Dowswell T. Intermittent iron
supplementation for improving nutrition and development in children
under 12 years of age. Cochrane Database Syst Rev.
2011;2011(12):CD009085. https://doi.org/10.1002/14651858.CD009085.pub2 PMID: 22161444.
- Lukowski
AF, Koss M, Burden MJ, Jonides J, Nelson CA, Kaciroti N,et al. Iron
deficiency in infancy and neurocognitive functioning at 19 years:
evidence of long-term deficits in executive function and recognition
memory. Nutritional Neuroscience. 2010;13:54-70. https://doi.org/10.1179/147683010X12611460763689 PMID: 20406573.
- Surapolchai
P, Sinlapamongkolkul P, Thaweekul P, Viprakasit V. Prevalence of iron
deficiency in 6-12 month-old infants at the Well Baby Clinic. In:
Navarawong W, editor. Proceedings of the 52th TSH Annual meeting; 2018 Mar 4-7; Bangkok, Thailand. Bangkok: TSH; 2018. p. 212.
- Porter
J, Viprakasit V. Iron overload and chelation. In: Cappellini MD, Cohen
A, Porter J, Taher A, Viprakasit V, eds. Guidelines for the management
of transfusion dependent thalassaemia, 3rd ed. Thalassaemia international federation; 2014. p. 42-97.
- Taher
A, Vichinsky E, Musallam K, Cappellini MD, Viprakasit V. Iron overload
and chelation. In: Taher A, Vichinsky E, Musallam K, Cappellini MD,
Viprakasit V, editors. Guidelines for the management of non transfusion
dependent thalassaemia, 1st ed. Thalassaemia international federation;
2013. p. 35-50.
- Modell
B, Darlison M. Global epidemiology of haemoglobin disorders and derived
service indicators. Bull World Health Organ. 2008;86:480–487. https://doi.org/10.2471/blt.06.036673 PMID: 18568278.
- Weatherall DJ, Clegg JB, eds. The Thalassaemia Syndromes. 4th ed. Oxford: Blackwell Science; 2001.
- Globin gene server home page. Available at: URL: http://globin.cse.psu.edu/. Accessed Mar 1, 2019.
- Panich
V, Pornpatkul M, Sriroongrueng W. The problem of thalassemia in
Thailand. Southeast Asian J Trop Med Public Health 1992;23 Suppl 2:1-6.
PMID: 1298980.
- Fucharoen
S, Winichagoon P. Hemoglobinopathies in Southeast Asia: molecular
biology and clinical medicine. Hemoglobin. 1997;21:299-319. https://doi.org/10.3109/03630269709000664 PMID: 9255610.
- Fucharoen
S, Winichagoon P, Wisedpanichkij R, Sae-Ngow B, Sriphanich R, Oncoung
W, et al. Prenatal and postnatal diagnoses of thalassemias and
hemoglobinopathies by HPLC. Clin Chem. 1998;44:740–748. PMID: 9554484.
- Viprakasit
V, Limwongse C, Sukpanichnant S, Ruangvutilert P, Kanjanakorn C,
Glomglao W, et al. Problems in determining thalassemia carrier status
in a program for prevention and control of severe thalassemia
syndromes: a lesson from Thailand. Clin Chem Lab Med. 2013;
51:1605–1614. https://doi.org/10.1515/cclm-2013-0098 PMID: 23525874.
- Tachavanich
K, Viprakasit V, Chinchang W, Glomglao W, Pung-Amritt P, Tanphaichitr
VS. Clinical and hematological phenotype of homozygous hemoglobin E:
revisit of a benign condition with hidden reproductive risk. Southeast
Asian J Trop Med Public Health. 2009;40:306-316. PMID: 19323016.
- Taher
A, Hershko C, Cappellini MD. Iron overload in thalassaemia intermedia:
reassessment of iron chelation strategies. Br J Haematol.
2009;147:634-640. https://doi.org/10.1111/j.1365-2141.2009.07848.x PMID: 19681884.
- Winichakoon
P, Tantiworawit A, Rattanathammethee T, Hantrakool S, Chai-adisaksopha
C, Rattarittamrong E, et al. Prevalence and risk factors for
complications in patients with nontransfusion dependent alpha- and
beta-thalassemia. Anemia. 2015;2015:1-7. https://doi.org/10.1155/2015/793025 PMID: 26664743
- Aydinok
Y, Porter JB, Piga A, Elalfy M, Beshlawy AE, Kilinc Y, et al.
Prevalence and distribution of iron overload in patients with
transfusion-dependent anemias differs across geographic regions:
results from the CORDELIA study. Eur J Haematol. 2015;95:244-253. https://doi.org/10.1111/ejh.12487 PMID: 25418187.
- Krittayaphonga
R, Viprakasit V, Saiviroonpornc P, Siritanaratkuld N, Siripornpitake S,
Meekaewkunchornf A, et al. Prevalence and predictors of cardiac and
liver iron overload in patients with thalassemia: A multicenter study
based on real-world data. Blood Cells Mol Dis. 2017;66:24–30. https://doi.org/10.1016/j.bcmd.2017.08.002 PMID: 28806577.
- Zimmermann
MB, Fucharoen S, Winichagoon P, Sirankapracha P, Zeder C, Gowachirapant
S, et al. Iron metabolism in heterozygotes for hemoglobin E (HbE),
α-thalassemia 1, or β-thalassemia and in compound heterozygotes for
HbE/ β –thalassemia. Am J Clin Nutr. 2008;88:1026–1031. https://doi.org/10.1093/ajcn/88.4.1026 PMID: 18842790.
- Tanno
T, Bhanu NV, Oneal PA, Goh SH, Staker P, Lee YT, et al .High levels of
GDF15 in thalassemia suppress expression of the iron regulatory protein
hepcidin. Nat Med. 2007;13:1096–1101. https://doi.org/10.1038/nm1629 PMID: 17721544.
- Kim A, Nemeth E. New insights into iron regulation and erythropoiesis. Curr Opin Hematol. 2015;22:199–205. https://doi.org/10.1097/MOH.0000000000000132 PMID: 25710710.
- Gupta
R, Musallam KM, Taher AT, Rivella S. Ineffective erythropoiesis: anemia
and iron overload. Hematol Oncol Clin N Am. 2018;32:213–21. https://doi.org/10.1016/j.hoc.2017.11.009 PMID: 29458727.
- Gardenghi
S, Marongiu MF, Ramos P, Guy E, Breda L, Chadburn A, et al. Ineffective
erythropoiesis in beta-thalassemia is characterized by increased iron
absorption mediated by down-regulation of hepcidin and up-regulation of
ferroportin. Blood. 2007;109:5027-5035. https://doi.org/10.1182/blood-2006-09-048868 PMID: 17299088.
- Camberlein
E, Zanninelli G, Détivaud L, Lizzi AR, Sorrentino F, Vacquer S, et al.
Anemia in β-thalassemia patients targets hepatic hepcidin transcript
levels independently of iron metabolism genes controlling hepcidin
expression. Haematologica. 2008;93:111-115. https://doi.org/10.3324/haematol.11656 PMID: 18166793.
- Jones
E, Pasricha SR, Allen A, Evans P, Fisher CA, Wray K. Hepcidin is
suppressed by erythropoiesis in hemoglobin E β-thalassemia and
β-thalassemia trait. Blood. 2015;125:873-880. https://doi.org/10.1182/blood-2014-10-606491 PMID: 25519750.
- WHO
Anthro for personal computers, version 3.2.2, 2011: Software for
assessing growth and development of the world's children. Geneva: WHO,
2010. (http://www.who.int/childgrowth/software/en/)
- Schlosnagle
DC, Hutton PS, Conn RB. Ferrozine assay of serum iron and total
iron-binding capacity adapted to the COBAS BIO centrifugal analyzer.
Clin Chem. 1982;28:1730-1732. PMID: 7083590.
- Toldi
G, Stenczer B, Molvarec A, Takats Z, Beko G, Rigo J, Jr., et al.
Hepcidin concentrations and iron homeostasis in preeclampsia. Clin Chem
Lab Med. 2010;48:1423-1426. https://doi.org/10.1515/CCLM.2010.290
- Murphy
AT, Witcher DR, Luan P, Wroblewski VJ. Quantitation of hepcidin from
human and mouse serum using liquid chromatography tandem mass
spectrometry. Blood. 2007;110:1048-1054. https://doi.org/10.1182/blood-2006-11-057471
- Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood. 2008;112:4292-4297. https://doi.org/10.1182/blood-2008-02-139915 PMID: 18689548.
- Troutt
JS, Rudling M, Persson L, Ståhle L, Angelin B, Butterfield AM, et al.
Circulating human hepcidin-25 concentrations display a diurnal rhythm,
increase with prolonged fasting, and are reduced by growth hormone
administration. Clin Chem. 2012;58:1225-1232. https://doi.org/10.1373/clinchem.2012.186866 PMID: 22679180.
- Eng
B PM, Walker L, Chui DHK, Waye JS. Detection of severe nondeletional
α-thalassemia mutations using a single-tube multiplex ARMS assay. Genet
Test. 2001;5:327-329. https://doi.org/10.1089/109065701753617471 PMID: 11960579.
- Newton
CR, Graham A, Heptinstall LE, Powell J, Summers C, Kalsheker N, et al.
Analysis of any point mutation in DNA. The amplification refractory
mutation system (ARMS). Nucleic Acids Research. 1989;17:2503–2516. https://doi.org/10.1093/nar/17.7.2503 PMID: 2785681.
- Tritipsombut
J, Phylipsen M, Viprakasit V, Chalaow N, Fucharoen S, Harteveld CL, et
al. A single-tube multiplex gap-polymerase chain reaction for the
detection of eight beta-globin gene cluster deletions common in
Southeast Asia. Hemoglobin. 2012;3:571-580. https://doi.org/10.3109/03630269.2012.747441 PMID: 23181748.
- Craig
JE, Barnetson RA, Prior J, Raven JL, Thein SL. Rapid detection of
deletions caused β-thalassemia and hereditary persistence of fetal
hemoglobin by enzymatic amplification. Blood. 1994;83:1673-1682. PMID:
7510147.
- Ekwattanakit
S MY, Riolueang S, Tachavanich K, Viprakasit V. Association of XmnI
polymorphism and hemoglobin E haplotypes on postnatal gamma globin gene
expression in homozygous hemoglobin E. Adv Hematol. 2012;2012:1-5. https://doi.org/10.1155/2012/528075 PMID: 23049556.
- Iron
deficiency anemia: assessment, prevention and control. A guide for
programme managers. Geneva, World Health Organization, 2001
(WHO/NHD/01.3).
- Camaschella
C. Iron deficiency: new insights into diagnosis and treatment.
Hematology Am Soc Hematol Educ Program. 2015;2015:8-13. https://doi.org/10.1182/asheducation-2015.1.8 PMID: 26637694.
- Galanello
R. Screening and diagnosis for haemoglobin disorders. In: Old J,
editor. Prevention of thalassaemias and other haemoglobin disorders:
volume 1, 2nd ed. Nicosia, Cyprus: Thalassaemia international federation; 2013.
- Tachavanich
K, Viprakasit V, Chinchang W, Glomglao W, Pung-Amritt P, Tanphaichitr
VS. Clinical and hematological phenotype of homozygous hemoglobin E:
revisit of a benign condition with hidden reproductive risk. Southeast
Asian J Trop Med Public Health. 2009;40:306-316. PMID: 19323016.
- Vrettou
C, Kanavakis E, Traeger-Synodinos J, Metaxotou-Mavrommati A, Basiakos
I, Maragoudaki E, et al. Molecular studies of beta-thalassemia
heterozygotes with raised Hb F levels. Hemoglobin. 2000;24:203-220. https://doi.org/10.1016/j.bcmd.2017.06.001 PMID: 28651846.
- Viprakasit
V, Lee-Lee C, Chong QT, Lin KH, Khuhapinant A. Iron chelation therapy
in the management of thalassemia: the Asian perspectives. Int J
Hematol. 2009;90:435-445. https://doi.org/10.1007/s12185-009-0432-0
- Tassiopoulos
T, Konstantopoulos K, Tassiopoulos S, Rombos Y, Alevizou-Terzaki V,
Kyriaki P, et al. Erythropoietin levels and microcytosis in
heterozygous beta-thalassaemia. Acta Haematol. 1997;98:147-149. https://doi.org/10.1159/000203609 PMID: 9352745.
- Mehta BC, Pandya BG. Iron status of beta thalassemia carriers. Am J Hematol. 1987;24:137-141. https://doi.org/10.1002/ajh.2830240204 PMID: 3812467.
- Hoorfar
H, Sadrarhami S, Keshteli AH, Ardestani SK, Ataei M, Moafi A.
Evaluation of iron status by serum ferritin level in Iranian carriers
of beta thalassemia minor. Int J Vitam Nutr Res. 2008;78:204-207. https://doi.org/10.1024/0300-9831.78.45.204 PMID: 19326343.
- Dolai
TK, Nataraj KS, Sinha N, Mishra S, Bhattacharya M, Ghosh MK. Prevalance
of iron deficiency in thalassemia minor: a study from tertiary
hospital. Indian J Hematol Blood Transfus. 2012;28:7–9. https://doi.org/10.1007/s12288-011-0088-9 PMID: 23449336.
- Hinchliffe
RF, Lilleyman JS. Frequency of coincident iron deficiency and
beta-thalassaemia trait in British Asian children. J Clin Pathol.
1995;48:594-595. https://doi.org/10.1136/jcp.48.6.594. PMID: 7665713.
- Yousafzai
YM, Wahid QUA, Khan S, Mir A, Khan A, Raziq F. Co-existing iron
deficiency/overload in beta-thalassemia trait. J Pak Med Assoc.
2019;69(6):806-810. PMID: 31189286.
- Wang
M, Zhang X, Zhao Y, Zhang Y, Lin Y, Xiao M, Li L. Prevalence of
iron-deficiency anemia in pregnant women with various thalassemia
genotypes: Thoughts on iron supplementation in pregnant women with
thalassemia genes. Front Nutr. 2022 17;9:1005951 https://doi.org/10.3389/fnut.2022.1005951 PMID: 36466428.
- Wray
K, Allen A, Evans E, Fisher C, Premawardhena A, Perera L, et al.
Hepcidin detects iron deficiency in Sri Lankan adolescents witha high
burden of hemoglobinopathy: A diagnostic test accuracy study. Am J
Hematol. 2017;92:196–203. https://doi.org/10.1002/ajh.24617 PMID: 27883199.
- Uijterschout
L, Domellöf M, Berglund SK, Abbink M, Vos P, Rövekamp L, et al. Serum
hepcidin in infants born after 32 to 37 wk of gestational age. Pediatr
Res. 2016;79:608-613. https://doi.org/10.1038/pr.2015.258 PMID: 26672736.
- Aranda
N, Bedmar C, Arija V, Jardí C, Jimenez-Feijoo R, Ferré N, et al. Serum
hepcidin levels, iron status, and HFE gene alterations during the first
year of life in healthy Spanish infants. Ann Hematol.
2018;97:1071-1080. https://doi.org/10.1007/s00277-018-3256-2 PMID: 29404719.
- Linpisarn
S, Tienboon P, Promtet N, Putsyainunt P, Santawanpat S, Fuchs GJ. Iron
deficiency and anaemia in children with a high prevalence of
haemoglobinopathies: implications for screening. Int J Epidemiol.
1996;25(6):1262-1266. https://doi.org/10.1093/ije/25.6.1262 PMID: 9027533.
- Tantracheewathorn
S, Lohajaroensub S. Incidence and risk factors of iron deficiency
anemia in term infants. J Med Assoc Thai. 2005;88(1):45-51. PMID:
15960216.
- Department
of Health, Ministry of Public Health of Thailand. The Fifth National
Nutritional survey of Thailand. Bangkok: Department of Health, Ministry
of Public Health of Thailand; 2003.
- Lozoff B, Georgieff MK. Iron deficiency and brain development. Semin Pediatr Neurol. 2006;13:158-165. https://doi.org/10.1016/j.spen.2006.08.004 PMID: 17101454.
- Lukowski
AF, Koss M, Burden MJ, Jonides J, Nelson CA, Kaciroti N, et al. Iron
deficiency in infancy and neurocognitive functioning at 19 years:
evidence of long-term deficits in executive function and recognition
memory. Nutritional Neuroscience. 2010;13:54-70. https://doi.org/10.1179/147683010X12611460763689 PMID: 20406573.
- Oh
C, Keats EC, Bhutta ZA. Vitamin and Mineral Supplementation During
Pregnancy on Maternal, Birth, Child Health and Development Outcomes in
Low- and Middle-Income Countries: A Systematic Review and
Meta-Analysis. Nutrients. 2020;12(2):491. https://doi.org/10.3390/nu12020491 PMID: 32075071.
- Baker
RD, Greer FR; Committee on Nutrition American Academy of Pediatrics.
Diagnosis and prevention of iron deficiency and iron-deficiency anemia
in infants and young children (0-3 years of age). Pediatrics.
2010;126(5):1040-1050. https://doi.org/10.1542/peds.2010-2576 PMID: 20923825.
- Mantadakis
E, Chatzimichael E, Zikidou P. Iron Deficiency Anemia in Children
Residing in High and Low-Income Countries: Risk Factors, Prevention,
Diagnosis and Therapy. Mediterr J Hematol Infect Dis. 2020 Jul
1;12(1):e2020041. https://doi.org/10.4084/MJHID.2020.041 PMID: 32670519.
- Burdick C. Combined iron deficiency and thalassemia minor. Am J Clin Pathol. 2013;139(2):260. https://doi.org/10.1309/AJCPCZU11FHTLCST PMID: 23355213.
- Schoorl M. The authors' reply. Am J Clin Pathol. 2013;139(2):261. https://doi.org/10.1093/ajcp/139.2.261 PMID: 23355214.
- Verma
S, Gupta R, Kudesia M, Mathur A, Krishan G, Singh S. Coexisting iron
deficiency anemia and Beta thalassemia trait: effect of iron therapy on
red cell parameters and hemoglobin subtypes. ISRN Hematol.
2014;2014:293216. https://doi.org/10.1155/2014/293216 PMID: 25006473; PMCID: PMC3972954.
Supplementary files
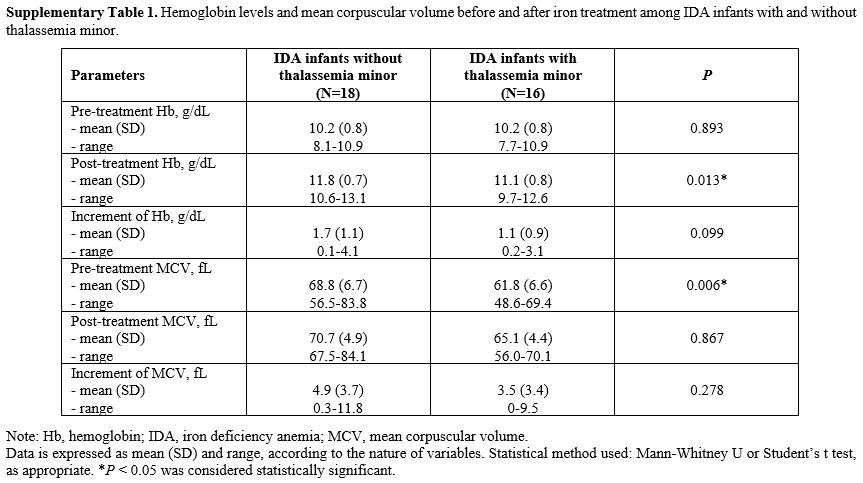 |
Supplementary Table 1.
Hemoglobin levels and mean corpuscular volume before and after
iron treatment among IDA infants with and without thalassemia minor. |
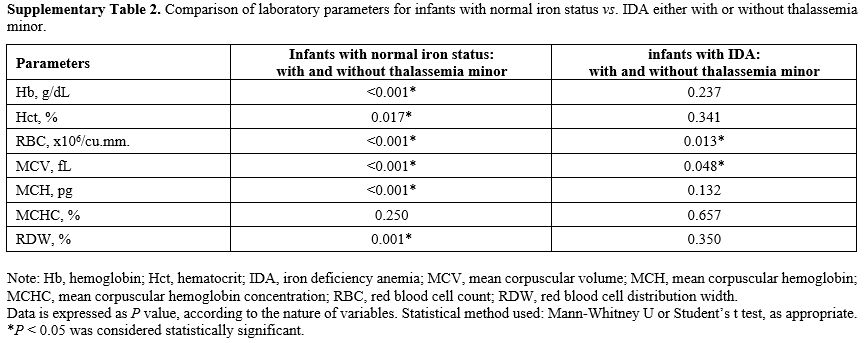 |
Supplementary Table
2. Comparison of laboratory parameters for infants with normal iron
status vs. IDA either with or without thalassemia minor. |
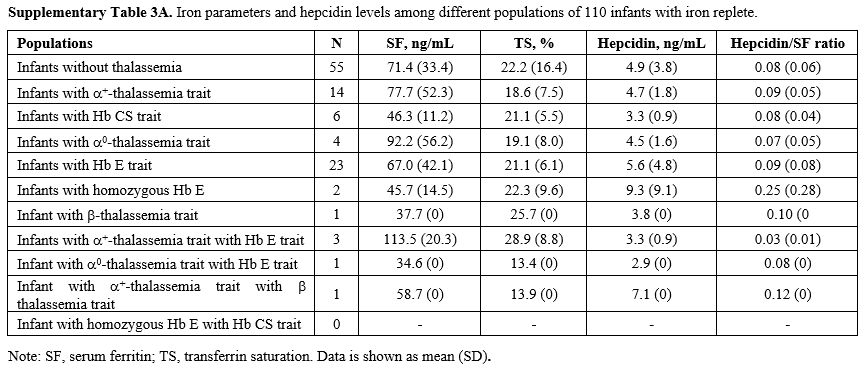 |
Supplementary Table 3A. Iron parameters and hepcidin levels among different populations of 110 infants with iron replete. |
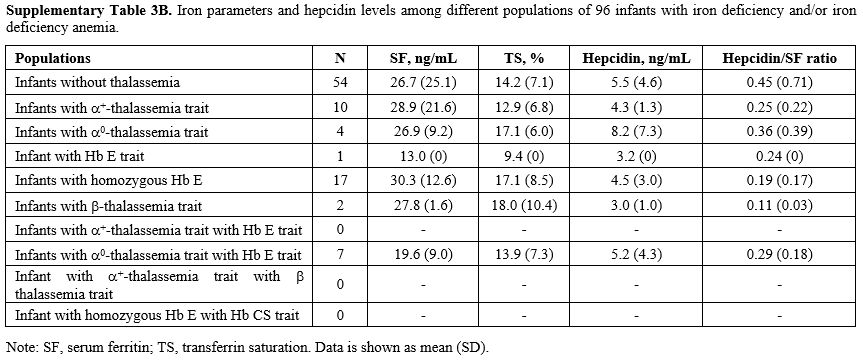 |
Supplementary Table
3B, Iron parameters and hepcidin levels among different populations of
96 infants with iron deficiency and/or iron deficiency anemia. |
[TOP]