Yali Zhou1, Jing Li2, Manlv Wei1, Linan Lu1, Jingting Luo3*, Xiuren Jin4, Shijie Yang4, Lei Yang5, Guiping Liao1, Tianhong Zhou1, Jie Huang2, Yaopeng Chen1 and Xiaolin Yin1*.
1 Department
of Hematology, The 923rd Hospital of the Joint Logistics Support Force
of the People's Liberation Army, Nanning, China.
2
Department of Blood Transfusion, The 923rd Hospital of the Joint
Logistics Support Force of the People's Liberation Army, Nanning, China.
3
Key Laboratory of Optoelectronic Devices and Systems of Education
Ministry and Guangdong Province, College of Physics and Optoelectronic
Engineering, Shenzhen University, Shenzhen, China.
4 WellYearn Technology (Shenzhen) Company Limited, Shenzhen, China.
5 Shenzhen Institute for Technology Innovation, Shenzhen, China.
Correspondence to:
Jingting Luo, Key Laboratory of Optoelectronic Devices and Systems of
Education Ministry and Guangdong Province, College of Physics and
Optoelectronic Engineering, Shenzhen University, Shenzhen, China;
E-mail:
luojt@szu.edu.cn.
Xiaolin
Yin, Department of Hematology, The 923rd Hospital of the Joint
Logistics Support Force of the People's Liberation Army, Nanning,
Guangxi, China; E-mail:
yin-xl@163.com.
Published: September 1, 2023
Received: July 14, 2023
Accepted: August 11, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023050 DOI
10.4084/MJHID.2023.050
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
To the editor
Thalassemia
comprises a diverse group of genetic disorders that affects the
synthesis of globin chains, with a global distribution.[1]
The type of thalassemia depends on the defective globin chain, with
α-thalassemia and β-thalassemia being the most clinically important
forms.[2] From a clinical perspective, thalassemia can
be classified according to the severity of the anemia and the need for
regular red blood cell (RBC) transfusions. Mild thalassemia is caused
by the heterozygous inheritance of one thalassemia mutation and
presents clinically as minimal, microcytic, and hypochromic anemia.
Patients with severe thalassemia require regular lifelong transfusions
of RBCs from early childhood, while moderate thalassemia is associated
with less severe anemia that does not require blood transfusions or
sporadic transfusions. However, some patients with thalassemia
intermedia may eventually benefit from regular blood transfusions,
while patients with thalassemia major may discontinue transfusions
after splenectomy.[1,3]
An imbalance in globin
chain production in thalassemia leads to erythrocyte skeleton damage
and cell dysfunction, resulting in a shortened lifespan. Erythrocyte
lifespan (ELS) indicates the survival time of RBCs in the blood
circulation and is the most direct and reliable indicator of the degree
of RBC destruction.
Methods for determining ELS fall into two
general categories, namely radioactive or nonradioactive labeling and
CO breath tests. Conventional methods of ELS detection have
disadvantages such as radiation hazards, allergy risk, and cumbersome
operation, which make them unsuitable for largescale detection and
application in clinical practice. The 15N-glycine
labeling technique is the gold standard method for determining ELS;
however, although this is an accurate technique for measuring ELS that
avoids safety issues related to radioactivity or possible allergic
reactions, its usefulness in clinical settings is seriously hindered by
the fact that it takes several weeks to complete the analyses.[4,5]
In contrast, Levitt’s CO breath test also provides a reliable technique
for determining ELS and has a simpler protocol with faster results,
making it more useful in clinical applications.[6] Furthermore, the CO breath test performs as well as the 15N-glycine labelling technique for distinguishing hemolysis.[7]
The
current study aimed to investigate the use of Levitt’s CO breath test
as a quantitative measure of ELS, to explore the feasibility of using
ELS as an indicator for determining the severity of different types of
thalassemia, and to assess the impact of treatment.
Materials and Methods
A total of 209 patients with thalassemia were enrolled from the 923rd
Hospital of the Joint Logistics Support Force of the People’s
Liberation Army between March 2022 and May 2023. Thalassemia syndromes
can be classified phenotypically into non-transfusion-dependent
thalassemia (NTDT) (n=70), or transfusion-dependent thalassemia (TDT)
(n=139) based on whether blood transfusions are required for long-term
survival. Additionally, patients with thalassemia were subsequently
classified into four types based on their clinical manifestations and
genotype: thalassemia major (TM, n=83), thalassemia intermedia (TI,
n=46), hemoglobin H (HbH, n=63), and α-thalassemia co-inherited with
β-thalassemia (Mix, n=17). HbH disease was further subcategorized into
deletional HbH (del-HbH, n=12) and non-deletional types (all with HbH
Constant Spring, HbH-CS, n=51).
This study was approved by the
ethics committee of the 923rd Hospital of the Joint Logistics Support
Force of the People’s Liberation Army, and all participants provided
written informed consent. For participants under 14 years of age, their
guardians signed the informed consent form on their behalf.
Hemoglobin
levels were analyzed using a Bio-Rad Variant II high-performance liquid
chromatography system (Hercules, CA, USA). ELS was determined using an
ELS Tester (Seekya Biotec Co., Ltd., Shenzhen, China), which measured
endogenous CO by nondispersive infrared comparison of the CO content
within an alveolar air sample versus that in the accompanying
atmospheric air sample according to Levitt’s formula.[6,8]
Statistical analysis.
Statistical analysis was performed using SPSS 26.0, and data were
presented as median and range. Numerical variables were compared
between groups using Student’s t-test or the Mann-Whitney rank-sum test. Correlation analysis was carried out using Spearman’s rank correlation coefficient. P values <0.05 were considered statistically significant.
Results
CO concentrations and ELS values in different types of thalassemia.
The endogenous alveolar CO concentration and ELS were compared between
patients with different types of thalassemia. Patients with TI had the
highest CO concentration [7.2 ppm (3.9–22)] compared with patients with
TM [5.7 ppm (2.1–18.1)] (P<0.001), HbH-CS [6.0 ppm (2.2–12.7)] (P=0.023), del-HbH [4.05 ppm (1.4–13.6)] (P<0.001), and Mix [5.1 ppm (2.2–11.7)] (P=0.004) (Figure 1A). The CO concentration was significantly higher in patients with HbH-CS compared with patients with del-HbH (P=0.011) (Figure 1A).
In contrast, the ELS was significantly lower in patients with TI [16.5
ppm (6–30)] compared with patients with TM [22 days (7–68)] (P<0.001), HbH-CS [22 days (6–72)] (P=0.025), del-HbH [32.5 days (11–72)] (P<0.001), and Mix [23 days (6–61)] (P=0.002) (Figure 1B). In addition, the ELS was significantly higher in patients with del-HbH compared with patients with HbH-CS (P=0.030) (Figure 1B). However, there was no difference in CO concentration [6.05 ppm (1.4–16.8) vs. 5.8 ppm (2.1–22.0), P=0.208] and ELS [19 days (6–72) vs. 20 days (6–68), P=0.250] between NTDT and TDT.
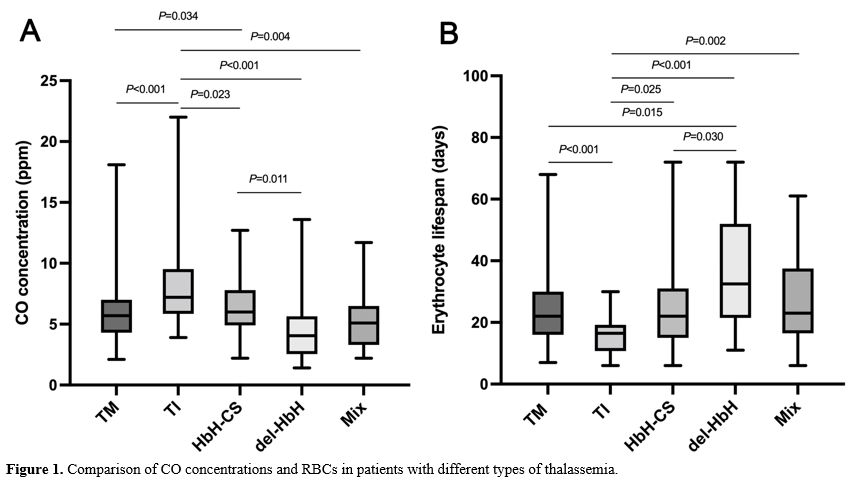 |
- Figure 1. Comparison of CO concentrations and RBCs in patients with different types of thalassemia.
|
Effects of splenectomy on different types of thalassemia.
Few patients had del-HbH or Mix, and we therefore compared the effects
of splenectomy among patients with TM, TI, and HbH-CS. There was no
significant difference in CO concentration or ELS between patients with
TM and TI who underwent splenectomy compared to those who did not (Figure 2A-D). However, patients with HbH-CS who underwent splenectomy had significantly lower CO levels (P=0.01) (Figure 2E) and higher ELS than patients with HbH-CS without splenectomy (P=0.02) (Figure 2F).
Furthermore, there was no significant difference in CO concentration or
ELS between patients with NTDT and TDT who underwent splenectomy
compared to those who did not (P>0.05).
 |
- Figure 2. Effects of splenectomy in patients with different types of thalassemia.
|
Effects of hemoglobin levels and transfusion in patients with different types of thalassemia. Spearman’s correlation analysis showed that hemoglobin levels were negatively correlated with CO concentration (r=-0.158, P=0.023) (Figure 3A) and positively correlated with ELS (r=0.535, P<0.001) (Figure 3B).
To investigate the association between the number of transfusions or
transfusion interval and CO concentration or ELS, separate studies were
conducted in patients with TDT. As a result, patients with more than 10
transfusions in the last year had a lower CO concentration [4.9 ppm
(2.1–12.9) vs. 7.05 ppm (3.3–22.0), P<0.001] and higher ELS [27 days (8–68) vs. 17 days (6–28), P<0.001]
than patients with fewer than 10 transfusions. In addition, the length
of time between blowing and the last transfusion in patients with TDT
was positively correlated with CO concentration (r=0.209, P=0.014) (Figure 3C) and negatively correlated with ELS (r=-0.267, P=0.001) (Figure 3D). However, no association was found in patients with NTDT.
 |
- Figure
3. Plots of hemoglobin levels or transfusion intervals versus CO concentration or ELS.
|
Discussion
For
simpler and better management, thalassemia is currently divided into
NTDT and TDT based on whether patients rely on blood transfusions for
long-term survival. Traditionally, thalassemia phenotypes can be
divided into carrier, mild, intermediate, and severe. Carriers and
patients with mild disease are usually asymptomatic and do not require
treatment. In contrast, patients with the most severe type of
α-thalassemia, Hb Bart’s hydrops fetalis syndrome, usually die very
early. The main types of thalassemia that require clinical attention
are thus TM, TI, and HbH disease.[9] The current study compared ELS values among different types of thalassemia and used them to validate the role of splenectomy.
In
this study, hemoglobin levels were similar in patients with various
types of thalassemia, but the expiratory CO concentration was highest
in patients with TI, followed by HbH-CS, TM, and Mix, and lowest in
del-HbH disease. ELS showed the opposite pattern, being shortest in
patients with TI, followed by HbH-CS, TM, and MIX, and longest in
del-HbH disease. Li et al.[10] reported that the ELS
values in patients with mild β-thalassemia and severe α/β thalassemia
were 67.5 days and 30.2 days, respectively, but did not provide
definitions of severe and mild β-thalassemia or clarify the type of
α-thalassemia. HbH disease can be classified as deletional or
non-deletional based on genotype. HbH-CS accounts for most cases of
non-deleted HbH in Guangxi, China, and its clinical manifestations are
more severe than del-HbH.[11] Among patients with
HbH, ELS was shorter in patients with HbH-CS than in patients with
del-HbH, although the severity of anemia was similar in both groups.
This also reflected the more severe hemolysis in patients with
non-deletional compared with deletional HbH disease.
Patients
with TI had a lower ELS than patients with TM, possibly because
patients with TM had more blood transfusions. Patients with TM had a
shorter interval from the last transfusion to insufflation and a higher
frequency of transfusions in the last year compared with patients with
TI. The average lifespan of transfused RBCs is higher than that in
patients with thalassemia.[12] In addition, blood
transfusions can also inhibit hematopoiesis in thalassemia patients,
reduce ineffective hematopoiesis, and reduce the corresponding RBC
destruction.[13,14] In the present study, transfusion
intervals were positively correlated with ELS in patients with TDT.
Previous studies also reported a higher incidence of patients with
erythroferrone, an indicator of ineffective hematopoiesis, among TI
compared with TM patients.[15,16] This is consistent
with clinical observations, in that patients with TM with standardized
blood transfusion have fewer complications than patients with TI.[17]
In addition, patients with TI can be inadequately treated if hemoglobin
values alone are used to guide blood transfusion. All the patients with
Mix included in this study were β-globin gene double heterozygotes with
one or two α-globin gene mutations, largely associated with TI. In
patients with β-thalassemia, co-inheritance of α-globin gene variants,
leading to absence or reduction of α-globin synthesis, were associated
with a milder clinical course.[18] In our cohort, ELS
was significantly higher in patients with Mix and significantly higher
than that in patients with TI, which further confirmed that
co-inheritance of α-thalassemia alleviated hemolysis in patients with
β-thalassemia. However, the CO concentration and ELS in patients with
Mix were more similar to those in patients with TM. Overall, ELS or CO
concentration thus better reflect the severity of different types of
thalassemia compared with hemoglobin.
Splenectomy is a therapeutic
option for thalassemia. The current study found no significant
difference in CO concentrations or ELS in relation to splenectomy in
patients with either TM or TI. However, splenectomy reduced the CO
concentration and prolonged ELS in patients with HbH-CS, suggesting
that splenectomy reduced RBC destruction in patients with α-thalassemia
rather than β-thalassemia. We also previously observed that splenectomy
reduced blood transfusions and increased hemoglobin in patients with
HbH.[19] This is mainly due to the different sites of
RBC destruction, the former being mainly in the spleen and the latter
mainly in the bone marrow.[20] There was no
significant difference in hemoglobin levels between HbH-CS patients
with and without splenectomy, but the difference in ELS was
significant, suggesting that ELS was a more sensitive indicator than
hemoglobin, as confirmed in clinical trials of other drugs.[21]
This
study had certain limitations. First, the sample sizes were small.
Although there was a total of 209 cases of thalassemia, after dividing
the patients into five groups, the size of each group was relatively
small, especially in the case of del-HbH disease, which only included
12 cases. Second, the sample was not representative. All the data were
from patients presenting at outpatient or inpatient visits, there was
selection bias, and only patients with relatively severe disease
generally came to hospital, and the proportions of patients with
different types of thalassemia were therefore not consistent with the
distribution of large sample surveys. The results of this study should
thus be interpreted with caution. Notably, however, the findings
reflected the actual situation of clinical patients.
Conclusions
In
conclusion, our observations suggested that there were large
differences in CO concentrations and ELS values among patients with
different types of thalassemia. Measuring ELS will provide more
information for assessing the severity of thalassemia and judging the
effects of blood transfusions and treatments, especially in clinical
drug trials.
Acknowledgments
We
wish to thank all of the study participants. This study was supported
by the Scientific Research Project of Guangxi Zhuang Autonomous Region
Health Committee (Z20211228, Z-A20231086).
References
- Kattamis A, Kwiatkowski JL, Aydinok Y. Thalassaemia. Lancet. 2022;399:2310-24. https://doi.org/10.1016/S0140-6736(22)00536-0 PMid:35691301
- Tesio N, Bauer DE. Molecular basis and genetic modifiers of thalassemia. Hematol Oncol Clin North Am. 2023;37:273-99. https://doi.org/10.1016/j.hoc.2022.12.001 PMid:36907603
- Osataphan
N, Dumnil S, Tantiworawit A, Punnachet T, Hantrakun N, Piriyakhuntorn
P, Rattanathammethee T, Hantrakool S, Chai-Adisaksopha C,
Rattarittamrong E, Norasetthada L, Fanhchaksai K, Charoenkwan P. The
long-term efficacy in blood transfusions, hematologic parameter
changes, and complications after splenectomy in patients with
transfusion-dependent thalassemia. Transfus Apher Sci. 2023;62:103620. https://doi.org/10.1016/j.transci.2022.103620 PMid:36509632
- Franco RS. Measurement of red cell lifespan and aging. Transfus Med Hemother. 2012;39:302-7. https://doi.org/10.1159/000342232 PMid:23801920 PMCid:PMC3678251
- Khera
PK, Smith EP, Lindsell CJ, Rogge MC, Haggerty S, Wagner DA, Palascak
MB, Mehta S, Hibbert JM, Joiner CH, Franco RS, Cohen RM. Use of an oral
stable isotope label to confirm variation in red blood cell mean age
that influences HbA1c interpretation. Am J Hematol. 2015;90:50-5. https://doi.org/10.1002/ajh.23866 PMid:25293624 PMCid:PMC4276493
- Zhang
HD, Ma YJ, Liu QF, Ye TZ, Meng FY, Zhou YW, Yu GP, Yang JP, Jiang H,
Wang QS, Li GP, Ji YQ, Zhu GL, Du LT, Ji KM. Human erythrocyte lifespan
measured by Levitt's CO breath test with newly developed automatic
instrument. J Breath Res. 2018;12:036003. https://doi.org/10.1088/1752-7163/aaacf1 PMid:29400658
- Ye
L, Ji Y, Zhou C, Luo J, Zhang L, Jing L, Zhao X, Guo J, Gao Q, Peng G,
Li Y, Li Y, Li J, Fan H, Yang W, Yang Y, Ma Y, Zhang F. Comparison of
Levitt's CO breath test and the (15) N-glycine labeling technique for
measuring the lifespan of human red blood cells. Am J Hematol.
2021;96:1232-40. https://doi.org/10.1002/ajh.26290 PMid:34265098
- Strocchi
A, Schwartz S, Ellefson M, Engel RR, Medina A, Levitt MD. A simple
carbon monoxide breath test to estimate erythrocyte turnover. J Lab
Clin Med. 1992;120:392-9.
- Lee JS, Cho SI, Park SS, Seong MW. Molecular basis and diagnosis of thalassemia. Blood Res. 2021;56:S39-S43. https://doi.org/10.5045/br.2021.2020332 PMid:33935034 PMCid:PMC8093999
- Li
LQ, Deng HM, Ma W, Zhou YW. Diagnosis of microcytic hypochromic anemia
with red blood cell survival via carbon monoxide breath-red blood cell
survival. Food Sci Technol, 2022;42:e53121. https://doi.org/10.1590/fst.53121
- Yin
XL, Zhang XH, Zhou TH, Zhang TL, Luo RG, Wang L, Zhou YL, Chen YS, Kong
XJ, Liang B, He YY, Peng L, Lu LB, Fang SP, Wu ZK. Hemoglobin H disease
in Guangxi province, Southern China: Clinical review of 357 patients.
Acta Haematol. 2010;124:86-91. https://doi.org/10.1159/000314058 PMid:20639625
- Luten
M, Roerdinkholder-Stoelwinder B, Bost HJ, Bosman GJ. Survival of the
fittest?--survival of stored red blood cells after transfusion. Cell
Mol Biol (Noisy-le-grand). 2004;50:197-203.
- Pasricha
SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses
erythropoiesis and increases hepcidin in adult patients with
beta-thalassemia major: a longitudinal study. Blood. 2013;122:124-33. https://doi.org/10.1182/blood-2012-12-471441 PMid:23656728
- James
EB, Vreman HJ, Wong RJ, Stevenson DK, Vichinsky E, Schumacher L, Hall
JY, Simon J, Golden DW, Harmatz P. Elevated exhaled carbon monoxide
concentration in hemoglobinopathies and its relation to red blood cell
transfusion therapy. Pediatr Hematol Oncol. 2010;27:112-21. https://doi.org/10.3109/08880010903536227 PMid:20201692
- Srole
DN, Ganz T. Erythroferrone structure, function, and physiology: Iron
homeostasis and beyond. J Cell Physiol. 2021;236:4888-901. https://doi.org/10.1002/jcp.30247 PMid:33372284 PMCid:PMC8026552
- Ozturk
Z, Gumuslu S, Yalcin K, Kupesiz A. Erythropoiesis and iron parameters
in transfusion-dependent and nontransfusion-dependent thalassemias. J
Pediatr Hematol Oncol. 2021;43:186-92. https://doi.org/10.1097/MPH.0000000000002046 PMid:34157011
- Meloni
A, Detterich J, Pepe A, Harmatz P, Coates TD, Wood JC. Pulmonary
hypertension in well-transfused thalassemia major patients. Blood Cells
Mol Dis. 2015;54:189-94. https://doi.org/10.1016/j.bcmd.2014.11.003 PMid:25488617 PMCid:PMC4297514
- Diamantidis
MD, Karanikola RA, Polyzoudi C, Delicou S, Manafas A, Savera H, Xydaki
A, Kotsiafti A, Tsangalas E, Ikonomou G, Mani E, Ntoulas K, Alexiou E,
Argyrakouli I, Koskinas J, Fotiou P. Clinical significance of
mutational variants in beta and alpha genes in patients with
hemoglobinopathies from two large Greek centers: a complex interplay
between genotype and phenotype. J Mol Med (Berl). 2023. https://doi.org/10.1007/s00109-023-02342-3 PMid:37420139
- Zhou
YL, Zhang XH, Liu TN, Wang L, Yin XL. Splenectomy improves anaemia but
does not reduce iron burden in patients with haemoglobin H Constant
Spring disease. Blood Transfus. 2014;12:471-8.
- Rigas
DA, Koler RD. Decreased erythrocyte survival in hemoglobin H disease as
a result of the abnormal properties of hemoglobin H: The benefit of
splenectomy. Blood. 1961;18:1-17. https://doi.org/10.1182/blood.V18.1.1.1
- Chen
JM, Zhu WJ, Liu J, Wang GZ, Chen XQ, Tan Y, Xu WW, Qu LW, Li JY, Yang
HJ, Huang L, Cai N, Wang WD, Huang K, Xu JQ, Li GH, He S, Luo TY, Huang
Y, Liu SH, Wu WQ, Lu QY, Zhou MG, Chen SY, Li RL, Hu ML, Huang Y, Wei
JH, Li JM, Chen SJ, Zhou GB. Safety and efficacy of thalidomide in
patients with transfusion-dependent beta-thalassemia: a randomized
clinical trial. Signal Transduct Target Ther. 2021;6:405. https://doi.org/10.1038/s41392-021-00811-0 PMid:34795208 PMCid:PMC8602273
[TOP]