The proband was a 32-year-old female who was the family's only child. The family history was unremarkable, and the parents were non-consanguineous. At age 27, the proband required blood transfusions during her first pregnancy due to severe anemia. At that time, due to the low level of vitamin B12, the proband was incorrectly diagnosed with megaloblastic anemia.
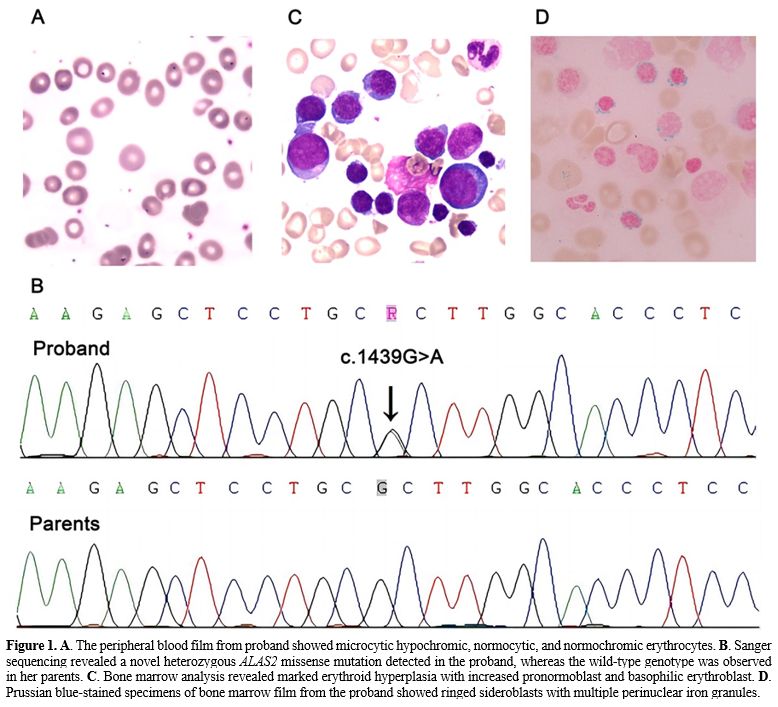
At age 32, the proband again developed severe anemia during her second pregnancy and required a blood transfusion. Physical examination revealed that the spleen of the proband was enlarged to 5 cm below the costal margin. A hemogram at presentation to our center revealed a hemoglobin level of 5.9 g/dL (reference value: 12.0–15.0 g/dL), a red blood cell count of 1.41×1012/L (reference value: 3.8–5.1×1012/L), a mean corpuscular volume of 127.7 fL (reference value: 80–100 fL), mean corpuscular hemoglobin of 41.8 pg (reference value: 27–34 pg), a mean corpuscular hemoglobin concentration of 32.8 g/dL (reference value: 31.6–35.4 g/dL), a reticulocyte percentage of 2.35% (reference value: 0.5–1.5%), a white blood cell count of 3.95×109/L (reference value: 3.5–9.5×109/L), and a platelet count of 132×109/L (reference value: 100–300×109/L). A peripheral blood smear was dimorphic, including microcytic hypochromic, normocytic, and normochromic erythrocytes (Figure 1A). Serum biochemical tests revealed a total bilirubin level of 15 μmol/L (reference value: 0–23 μmol/L), an indirect bilirubin level of 8.7 μmol/L (normal range: 5–18 μmol/L), and a lactate dehydrogenase level of 117 U/L (reference value: 120–250 U/L). Iron metabolism testing revealed that serum ferritin, serum iron, and transferrin were 751.4 ng/mL (reference value: 4.63–204 ng/mL), 37.18 μmol/L (reference value: 7–30 μmol/L), and 1.57 g/L (reference value: 1.7–3.4 g/L), respectively. Levitt’s CO breath test showed that the erythrocyte life span of the proband was 24 days (normal range: 70–140 days). Laboratory tests for megaloblastic anemia, autoimmune hemolytic anemia, paroxysmal nocturnal hemoglobinuria, glucose-6-phosphate dehydrogenase deficiency, thalassemia, and hepatopathy were negative.
Next-generation sequencing of the proband and her family was carried out to investigate underlying variants associated with anemia in the proband. A de novo heterozygous missense mutation of ALAS2: c.1439G>A was found and further confirmed by Sanger sequencing (Figure 1B). The c.1439G>A mutation led to a substitution of a conserved arginine to histidine at residue 480 (p.Arg480His) in exon 9 of the ALAS2 protein. Given the ALAS2 mutation, a diagnosis of CSA was suspected.
Bone marrow analysis revealed marked erythroid hyperplasia with increased pronormoblast and basophilic erythroblast (Figure 1C). In Prussian blue-stained specimens, a bone marrow smear showed erythroid hyperplasia with a 66% proportion of total sideroblasts, and the proportion of ring sideroblasts was 16% (Figure 1D). Chromosome analysis revealed a normal karyotype (46, XY). A bone marrow biopsy showed hyperactive hyperplasia and no increase in reticulin fibers (MF0). Following the diagnosis of SA in the bone marrow, further investigations were conducted, including tests for pyridoxine, zinc, lead, and copper levels and chromosomal microarray analysis for myelodysplastic syndromes. All tests came back normal. The proband was thus diagnosed with CSA caused by this mutation of the ALAS2 gene. The proband had a good response to pyridoxine treatment, but her ferritin level gradually rose to 925.85 ng/mL.
 |
|
XLSA typically affects younger males due to an X-linked recessive pattern of inheritance. However, as a result of familial-skewed inactivation of the normal X chromosome, females with ALAS2 mutations may have a late-onset clinical phenotype, as with the proband in this study.[3] The management of CSA remains primarily supportive rather than definitive. ALAS2 catalyzes the first step of the heme biosynthetic pathway by condensing glycine and succinyl-CoA to form delta-aminolevulinic acid in the presence of pyridoxal 5’-phosphate, which is the metabolite of vitamin B6.[4] Pyridoxine can enhance the activity of the ALAS2 enzyme, and more than half of XLSA patients are responsive to supplementation with pyridoxine.[5] More than 100 distinct mutations in the ALAS2 gene have been reported. Most disease-associated variants occur in exons 5 and 9; the latter contains the pyridoxal-binding amino acid.[2] This finding was confirmed by the good response to pyridoxine treatment observed in the proband.
Patients with CSA are prone to iron overload, whether pyridoxine-responsive or not, regardless of red blood cell transfusions. Iron overload is partly attributed to reduced hepcidin level secondary to ineffective erythropoiesis, which promotes intestinal iron absorption.[6] The ferritin level of our reported XLSA patient increased gradually. It is therefore necessary to monitor regularly patients' clinical, laboratory, and radiological parameters to detect these long-term complications. Anecdotal reports of hematopoietic stem cell transplantation in CSA describe effective remission.[7] However, early diagnosis and management of CSA remain fundamental, especially as iron overload should be kept at a minimum to ensure a better outcome of a potential future transplantation. Additionally, developing definitive treatments for CSA is an area of need. Preclinical studies and clinical trials are essential to determine whether novel agents such as luspatercept or approaches involving gene therapy for CSA would benefit its treatment.[8]
In conclusion, we identified a novel ALAS2 missense mutation causing CSA in the Chinese population. Our findings will provide valuable insights to broaden the clinical phenotypic spectrum of CSA and improve understanding of ALAS2 gene variants.