Emiliano Fabiani1,2, A. Cristiano1, H. Hajrullaj1, G. Falconi1, G. Leone3 and MT. Voso1.
1 Department of Biomedicine and Prevention, University of Tor Vergata, Rome, Italy.
2 UniCamillus-Saint Camillus International University of Health Sciences, Rome, Italy.
3 Università Cattolica del Sacro Cuore, Roma, Italy.
Correspondence to:
Emiliano Fabiani, PhD. University of Rome "Tor Vergata", Department of Biomedicine and Prevention. E-mail:
emiliano.fabiani@uniroma2.it; ORCID ID: 0000-0002-6209-8934.
Published: November 01, 2023
Received: September 19, 2023
Accepted: October 16, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023064 DOI
10.4084/MJHID.2023.064
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Therapy-related Myeloid Neoplasm
(t-MN) represents one of the worst long-term consequences of cytotoxic
therapy for primary tumors and autoimmune disease. Poor survival and
refractoriness to current treatment strategies characterize affected
patients from a clinical point of view. In our aging societies, where
newer therapies and ameliorated cancer management protocols are
improving the life expectancy of cancer patients, therapy-related
Myeloid Neoplasms are an emerging problem. Although several research
groups have contributed to characterizing the main risk factors in t-MN
development, the multiplicity of primary tumors, in association with
the different therapeutic strategies available and the new drugs in
development, make interpreting the current data still complex. The main
risk factors involved in t-MN pathogenesis can be subgrouped into
patient-specific, inherited, and acquired predispositions.
Although
t-MN can occur at any age, the risk tends to increase with advancing
age, and older patients, characterized by a higher number of
comorbidities, are more likely to develop the disease. Thanks to the
availability of deep sequencing techniques, germline variants have been
reported in 15-20% of t-MN patients, highlighting their role in cancer
predisposition.
It is becoming increasingly evident that t-MN
with driver gene mutations may arise in the background of Clonal
Hematopoiesis of Indeterminate Potential (CHIP) under the positive
selective pressure of chemo and/or radiation therapies. Although CHIP
is generally considered benign, it has been associated with an
increased risk of t-MN. In this context, the phenomenon of clonal
evolution may be described as a dynamic process of expansion of
preexisting clones, with or without acquisition of additional genetic
alterations, that, by favoring the proliferation of more aggressive
and/or resistant clones, may play a crucial role in the progression
from preleukemic states to t-MN.
|
Introduction
Therapy-related
Myeloid Neoplasms (t-MN) include Acute Myeloid Leukemia (AML),
MyeloDysplastic Syndromes (MDS), and MyeloDysplastic/MyeloProliferative
Neoplasm (MDS/MPN) arising in patients treated with chemo and/or
radiation therapy for a previous cancer or an autoimmune disease.[1,2]
In
the 5th edition of the WHO classification of hematolymphoid tumors,
t-MN have been included in a new segregated category of secondary
myeloid neoplasm, encompassing diseases that arise in the setting of
specific known predisposing factors such as Myeloid Neoplasms post
Cytotoxic Therapy (MN-pCT). This term formally replaces
“therapy-related”.[3]
On the other hand, the
term therapy-related has been retained by the “International Consensus
Classification of myeloid neoplasms and acute leukemia” (ICC), but
without differences in the concept that now both terms, “MN-pCT” and
“therapy-related", are considered diagnostic qualifiers that should be
added following the specific MDS or AML diagnosis.[4]
The
10-year cumulative incidence of t-MN ranges from 1-10% according to
different cancers and different chemo and/or radiation regimens.[5,6]
However, due to the steady improvement in the overall survival (OS) of
cancer patients, an increase in the percentage of t-MN diagnoses has
been shown in recent years.[7] Nowadays, t-MN represents one of the worst long-term side effects of cytotoxic therapy, since compared to de novo
myeloid neoplasms, t-MN patients are characterized by a poor prognosis
and refractoriness to current treatment strategies (about 5-year
survival rate of 10%).[3,6]
In
this line, in our aging societies, where newer therapies and
ameliorated cancer management protocols are improving the life
expectancy of cancer patients, t-MNs are an emerging problem.
Over
the last few decades, according to the available screening
technologies, factors predisposing the development of t-MN have been
investigated in different directions.
Several Authors have
studied the impact of the host's genetic background on cancer
predisposition. Polymorphisms in genes involved in detoxification, DNA
repair, and apoptosis may modify the individual risk of developing a
t-MN. In particular, when detoxification and/or DNA repair are
ineffective, the DNA damage induced by therapy can cause chromosomal
instability, leading to severe failure of cell functions and/or
apoptosis. Moreover, these polymorphisms have been shown to influence
the individual response to cancer treatment by increasing the
concentration of active drug metabolites or impairing enzymatic
pathways that rescue cancer cells from genotoxic damage and apoptosis.[8-13]
In
the same line, germline variants typical of familial predisposition
syndromes like Fanconi Anemia and Li-Fraumeni have been reported at
higher frequencies in t-MN patients, and TP53 uncommon germline variants may play a key role in t-MN pathogenesis.[14-16]
More
recently, Clonal Hematopoiesis of Indeterminate Potential (CHIP) has
been considered one of the main risk factors and has been identified at
the time of the primary cancer diagnosis in 30%-70% of patients
developing a t-MN, representing a pre-malignant state, which the
exposure to cytotoxic agents[6,17,18] can further trigger.
Despite
several research groups that have contributed to characterize the main
risk factors in t-MN development, the multiplicity of primary tumors,
in association with the different therapeutic strategies available and
the new drugs in development, make interpreting the current data still
complex. In addition, we also need to keep in mind that in 10-15% of
cases, myeloid neoplasm occurs as a second neoplasm in patients who
underwent surgery alone to treat the primary tumor, and a familial
and/or personal history of multiple neoplasms is present in 5-10% of
patients.[19]
Therefore, in order to better
characterize the genotypic and phenotypic profiles of t-MN, the aim to
be pursued in the early future should be to select a homogeneous study
population consisting of patients affected by the same primary tumor,
treated with similar treatment protocols before t-MN development, in
order to limit the biases involved in the study of heterogeneous
populations and treatments.
Risk Factors in t-MN Pathogenesis: Patient-Specific Predisposition
Due
to the limited incidence of t-MN in treated patients, during past
years, many researchers have tried to identify the main potential
contributors involved in t-MN onset. Understanding the risk factors
associated with t-MN development is crucial for identifying high-risk
individuals and implementing preventive strategies to improve patient
outcomes. To date, three main categories of risk factors have been
identified: patient-specific, inherited, and acquired predisposition.
In
the patient category, specific risk factors are included: age, previous
cancer, autoimmune diseases (AD), and environmental exposure (smoking,
benzene, irradiation, etc.).
Age is one of the most significant
risk factors for t-MN development. Although t-MN can occur at any age,
the risk tends to increase with advancing age. Older patients,
characterized by a higher number of comorbidities and frequency of
Clonal Hematopoiesis (CH), are more likely to develop the disease.[20,21]
Although
the frequency of t-MN can be considered very low, some primary tumors
have been associated with a higher risk of t-MN development. In
particular, the most common primary malignancies are breast cancer and
lymphoproliferative disorders such as Hodgkin's and non-Hodgkin's
lymphoma, Multiple Myeloma (MM), and Chronic Lymphocytic Leukemia
(CLL).[22-25]
The direct correlation between
the type of primary tumor and the risk of developing a t-MN can be
related to the specific type of treatment to which patients are
subjected and to their survival duration.
Some chemotherapy drugs,
alkylating agents, and topoisomerase II inhibitors (cyclophosphamide,
busulfan, and melphalan, as well as etoposide and doxorubicin), have
been associated with an increased risk of t-MN. Alkylating agents
damage DNA by adding alkyl groups to its structure. At the same time,
topoisomerase II inhibitors interfere with the topoisomerase II
enzyme's function, which helps manage DNA structure during cell
division. As a result, DNA sequence and chromosomal structure would be
altered, increasing the risk of t-MN.[26] Recently, PARP1 inhibitor therapy has been added in the 5th edition of WHO 2022 as a qualifying criterion for t-MN, while the treatment with methotrexate has been excluded.[3]
The time of insurgence of t-MN is generally earlier (1-3 years) in
patients treated with Topoisomerase inhibitors than Alkylating
agents and/or radiation (7-10 years), even if the frequent
contemporaneous administration of these drugs makes this difference not
significant.[25]
In 10-15% of cases, myeloid
neoplasms may occur as a second neoplasm in patients who underwent
surgery alone to treat the primary tumor. Surgery is not typically
associated with an increased risk of myeloid neoplasms; however,
surgery can include adjuvant and neo-adjuvant therapies, such as
chemotherapy or radiation, recommended to reduce the initial tumor mass
or eradicate residual cancer cells to reduce the risk of new
occurrence. The administration of these adjuvant treatments may also
play a role in t-MN pathogenesis.[19]
Autoimmune
diseases, such as Systemic Lupus Erythematosus (SLE), Rheumatoid
Arthritis (RA), Multiple Sclerosis (MS), and Inflammatory Bowel Disease
(IBD) have also been considered potential risk factors in t-MN
development.[27] The involvement of the immune system
and inflammation has been indicated as a possible driving factor
contributing to myeloid neoplasm development and progression.
Systemic-Inflammatory-Autoimmune-Diseases (SIAD) are increasingly
considered in the hematological context.[28]
However,
myeloid neoplasm development depends on several factors not yet fully
elucidated, including the specific subtype of AD, the chronic immune
stimulation, the duration and anti-rheumatic/anti-inflammatory
treatment, and the genetic predisposition. The most well-documented
leukemogenic potential is related to drugs such as azathioprine,
cyclophosphamide, and mitoxantrone, which can impair the hematopoietic
processes.[29-31]
Finally, environmental
exposure to cigarette smoking, benzene, pesticides, chemicals including
Formaldehyde, and ionizing radiation has been associated with myeloid
neoplasm pathogenesis. So, it should be included in the category of
patient-specific risk factors.[32,33]
Mutations
in ASXL1 have been significantly associated with smoking history. Of
note, current smokers showed a higher rate of ASXL1 mutations than
former smokers.[33]
The higher incidence of
myeloid neoplasms in survivors of the Nagasaki and Hiroshima atomic
bombs reinforces the causal relationship between ionizing radiation and
hematological disorders.[34,35]
In this
context, the exposure of cells to ionizing radiation results in the
increased formation of Reactive Oxygen Species (ROS), such as hydrogen
peroxide, superoxide, and hydroxyl radicals. These molecules can
oxidize and deaminate the nitrogenous bases of DNA, triggering damage
to DNA structure. Cells with DNA damage are genomically unstable,
cumulating somatic mutations and cytogenetic alterations, which are the
basis for developing myeloid neoplasms.[36,37]
Similarly,
Benzene exposure is now considered casually related to myeloid
neoplasms. Benzene and its metabolites are found to be harmful to
Hematopoietic Stem Cells (HSC), giving rise to a reduction in the
number of HSC and impairing their maturation and differentiation in
myeloid and lymphoid lineages. Although the majority of evidence comes
from case-control studies and occupational studies with a relatively
small number of cases, genotoxicity, immunotoxicity, altered gene
expression, chronic inflammation, and induction of immunodepression are
described as the main causes of benzene-induced damage.[38,39]
Risk Factors in t-MN Pathogenesis: Inherited Predisposition
Germline
variants (mutations and polymorphisms) have also been reported as risk
factors in t-MN development. Thanks to the availability of deep
sequencing techniques, a germline cancer predisposition has been
confirmed in 15-20% of t-MN patients.[14,40]
These germline variants can affect genes involved in DNA repair, cell
cycle regulation, genotoxic metabolism, and other biological pathways
related to cancer development.[41-43]
Moreover,
germline variants can make individuals more vulnerable to the harmful
effects of chemo and/or radiation therapy. In this line, polymorphisms
in genes belonging to the xenobiotic detoxification pathway, such as
cytochrome p450, NADPH-quinone oxidoreductase 1 (NQO1), and glutathione
S-transferase (GST), and DNA repair pathways like RAD51, XRCC1, XRCC2,
XRCC3 and XPD, were among the first candidates to be studied for their
possible involvement in t-MN development, since the ineffective repair
of damaged cells, that survive to genotoxic stress, may be crucial for
cancer genesis. Similarly, polymorphisms in apoptotic modulators could
deregulate the apoptotic pathway, rescuing damaged cells from apoptosis
and modifying the risk of t-MN (Figure 1).
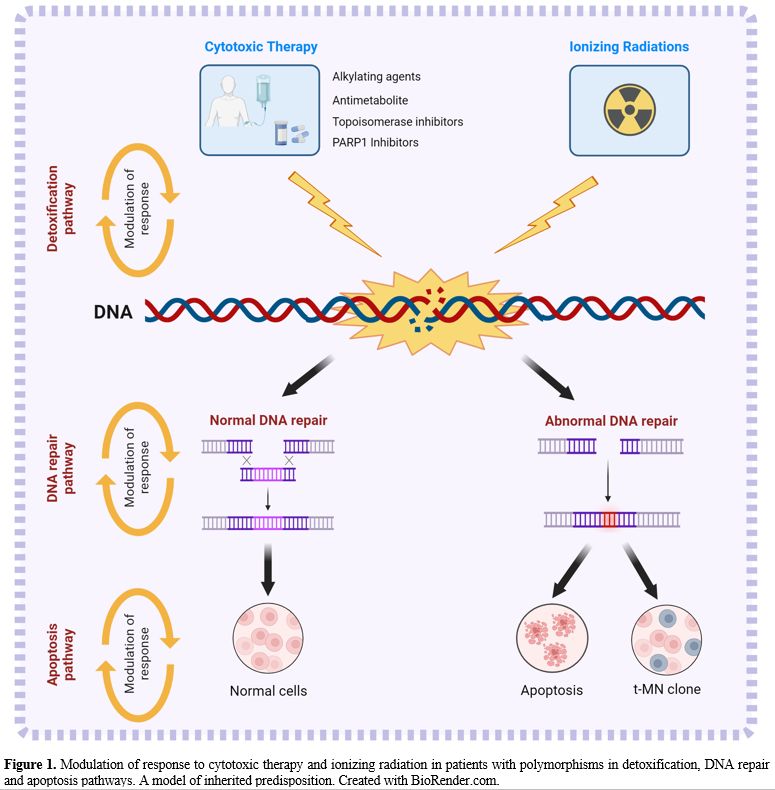 |
- Figure
1. Modulation of response to cytotoxic therapy and ionizing radiation
in patients with polymorphisms in detoxification, DNA repair and
apoptosis pathways.
- A model of inherited predisposition. Created with
BioRender.com.
|
Although
several Authors have contributed to delineate the role of these
polymorphisms, their association with t-MN development has not been
confirmed in large and independent study cohorts, probably because of
the lack of adequate controls not only matched for sex, age, and
primary disease but also for therapy and comparable follow-up.[11,44,45]
An
increased susceptibility to t-MN development has also been described in
individuals with inherited cancer predisposition syndromes such as
Fanconi anemia (FA) and Li-Fraumeni syndrome.
Fanconi anemia is associated with bi-allelic loss-of-function mutations in the FA pathway, including 21 FA or FA-like genes.
Voso
et al. reported a high frequency of FA gene variants in t-MN patients
(16%), with similar prevalence in t-MN secondary to lymphoproliferative
diseases and breast cancer, indicating that heterozygous carriers of FA
variants may have increased susceptibility to the DNA-damaging action
of cytotoxic therapy.[15] Similarly, Schwartz et al. identified TP53 germline variants in 15.5% of pediatric t-MN.[46]
Risk Factors in t-MN Pathogenesis: Acquired Predisposition
Clonal hematopoiesis (CH) can play a key role in the risk factors related to an acquired predisposition.
In
two independent works, Jaiswal et al. and Genovese et al. reported the
presence of somatic or acquired mutations in 1% of non-hematological
patients. These studies also highlighted that mutations were very rare
in patients under 40 (<1%) but progressively increased in older
individuals, achieving the percentage of about 20-30% in those aged 70
or older.[47,48] Since these mutations were present
in patients without detectable hematologic disorders, this phenomenon
has been defined as age-related clonal hematopoiesis (ARCH). In
contrast, clonal hematopoiesis of indeterminate potential (CHIP) was
defined by somatic mutations with Variant Allele Frequency (VAF)
greater than 2%.
Notably, genes mutated at higher frequency were identified in myeloid neoplasms.[47,48]
It
is becoming increasingly evident that t-MN with driver gene mutations
may arise in the background of CHIP under the positive selective
pressure of chemo and radiation therapies.
Hematopoietic stem
cells accumulate somatic mutations during their biological life, most
of which are nonpathogenic without functional consequences or potential
for clonal expansion.
Some mutated clones might gain
proliferation and survival advantages, triggering the clonal expansion
of a specific myeloid cell subset characterized by genetic alterations
such as cytogenetic abnormalities, somatic mutations, and/or copy
number variations. These genomic alterations represent a heterogeneous
condition that may promote the transition from a physiological state to
myeloid malignancy.
Although ARCH and CHIP are mostly considered
benign, they have been associated with an increased risk of t-MN. Their
presence may create a preexisting pool of altered cells more prone to
further genetic and malignant transformations and affect the
hematopoietic microenvironment, driving bone marrow niche alterations.[49]
Genovese
et al. showed that subjects with CHIP have a higher risk of progression
to hematological malignancies than subjects without mutated clones,
which appears proportional to the VAF of mutated genes. This risk is
about 11 to 13 times higher in individuals with clonal hematopoiesis,
and the overall transformation rate is about 1% per year.[48]
Gillis
and Colleagues, in a proof-of-concept case-control study, identified a
prevalence of CHIP in patients who developed therapy-related myeloid
neoplasms (62%) than that of control patients (27%), showing that
individuals carrying CHIP mutations were at increased risk of t-MN
compared to individuals without detectable CHIP mutations.[50]
Similarly,
our research group recently reported the high incidence of CHIP in
Chronic Lymphocytic Leukemia (CLL) patients who developed a t-MN after
treatment with chemo-(immuno)therapy, mostly Fludarabine,
Cyclophosphamide, Rituximab (FCR). We detected 30 pathogenic/likely
pathogenic variants in 10 of 13 patients with a t-MN (77%). In
contrast, CHIP variants were present in only 34 of 285 patients (12%)
from the CLL control cohort who received the same treatment. Of note,
backtracking the prevalence of CHIP in paired samples collected at the
time of CLL diagnosis, the same variants were identified in 62.5% of
patients.[24]
These data highlight the
potential role of CHIP as a risk factor for developing t-MN, suggesting
the screening for myeloid clonal states, especially in older patients,
before starting cytotoxic therapy.
Clonal Evolution in Therapy-Related Myeloid Neoplasm
Clonal
evolution is a dynamic process of expanding preexisting clones with or
without acquiring additional genetic alterations that may be crucial in
progressing from preleukemic states to t-MN. This process is directly
shaped by therapy that may promote clonal competition, favoring the
expansion of more aggressive and resistant clones, characterized by
proliferative and survival advantages.
The first evidence of clonal evolution in t-MN comes from the studies conducted by Wong et al., who described the role of TP53 mutations in the origin and evolution of t-MN in 2015.[51]
Sequencing the genomes of 22 cases of t-MN, the Authors identified 7 carriers of specific TP53
mutations. Backtracking these mutations in paired DNA samples collected
at the time of primary malignancy (Hodgkin and non-Hodgkin lymphoma),
they identified the same mutations at very low variant allele
frequencies (0.003–0.7%) in 4 of the 7 patients, concluding that rare
HSC carrying age-related TP53 mutations may be resistant to chemotherapy and expand preferentially after treatment.[51]
This paper was the first evidence that chemo and radiation therapy may
promote the clonal selection and expansion of preexisting mutant HSC,
favoring t‑MN development in a sort of Darwinian selection.
Two
years later, studying 14 t-MN patients with a primary hematologic
malignancy using ultra-deep NGS, we identified two distinct clonal
evolution models[17] (Figure 2).
Mutations
identified at the time of t-MN were tracked backward in bone marrow
samples preceding secondary leukemia occurrence in 8 paired DNA
samples. Somatic mutations were detectable before any cytotoxic
treatment in three patients, while the t-MN clone was acquired in the
remaining five patients.
In our study, we confirmed the key role of somatic mutations of the TP53 gene in the clonal evolution of t-MN and identified other genes, such as ASXL1, as pivotal players.
Of note, we also identified a t-MN patient with a particular pattern of clonal evolution characterized by IDH1 and SRSF2
somatic mutations. Both mutations were somatically acquired because
they were not detectable in the CD3+ T-lymphocyte population, and the
VAFs (38% and 35%, respectively) suggested their cohabitation in the
same clone.
The IDH1 mutation was originally present at similar VAF (35%) 9 years before t-MN onset. In contrast, the SRSF2 mutation was undetectable, suggesting a pre-leukemic role for IDH1 mutation and a pathogenic role for SRSF2 mutation in a susceptible individual (Figure 2A).
In
the second scenario, somatic mutations characterizing the t-MN clone, 5
out of 8 patients were not present at primary cancer diagnosis. They
appeared only after chemo and/or radiation therapy as a direct effect
of treatment, suggesting the dual role of cytotoxic therapy in t-MN
pathogenesis (Figure 2B).
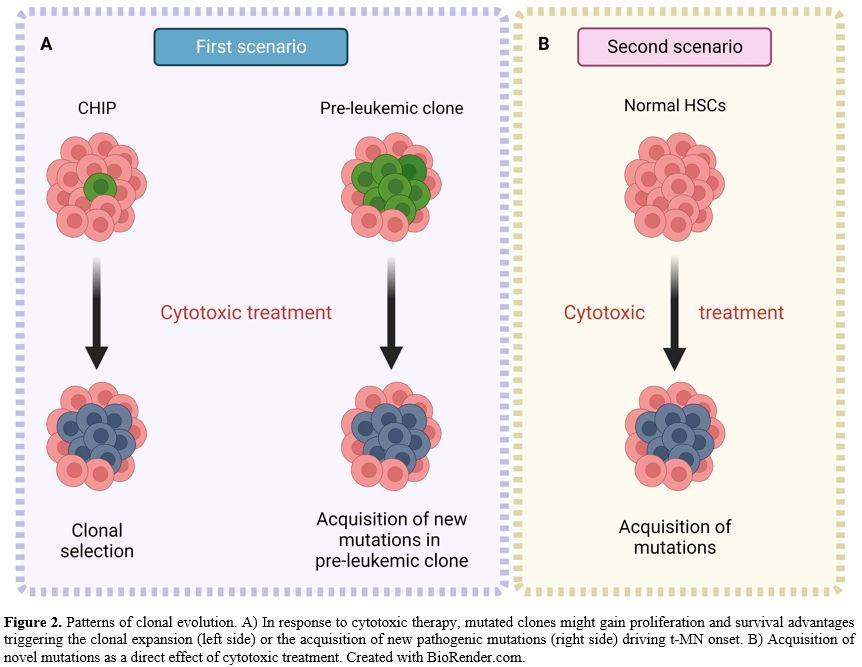 |
- Figure
2. Patterns of clonal evolution. A) In response to cytotoxic therapy,
mutated clones might gain proliferation and survival advantages
triggering the
- clonal expansion (left side) or the acquisition of new
pathogenic mutations (right side) driving t-MN onset. B) Acquisition of
novel mutations as a direct
- effect of cytotoxic treatment. Created with
BioRender.com.
|
To
date, NGS technology has become commonly used in research and
diagnostics too; thanks to it, the number of patients affected by t-MN
who have been mutationally screened has increased enormously.
Although many authors have demonstrated that the mutational burden of de novo myeloid neoplasm and t-MN are similar, all agree in identifying a higher incidence of TP53 mutations in t-MN patients.
TP53 mutations have been reported in 30-47 % of t-MN cases, more frequently associated with complex karyotype (80%).[52] In t-MN, these mutations may also occur associated with TP53 deletion, copy-neutral loss of heterozygosity, and in a multihit state.[53-56]
The TP53
gene encodes for the p53 tumor suppressor protein, which is activated
in response to cellular stress. Subsequently, it activates the
mechanisms of cell cycle arrest, senescence, and apoptosis, playing an
essential role in controlling cell proliferation and differentiation.
Many studies have shown the negative prognostic role of TP53
mutations in myeloid neoplasms, demonstrating poor response to standard
cytotoxic therapy and lower median overall survival and disease-free
survival compared to unmutated patients.[57-59]
Although revised diagnostic criteria for myeloid neoplasms (WHO and ICC) recommend major changes concerning TP53 mutations in relationship with their prognostic role, at least 2 mutations or 1 mutation with loss of TP53 wild-type or VAF≥ 50% as evidence of biallelic/multihit TP53 mutation, Shah et al. recently hypothesized a different prognostic role for TP53 mutations in the context of t-MN.[57] Analyzing 488 t-MN patients found that TP53mut t-MN with VAF≥ 10% had significantly shorter survival than wild-type patients, while TP53mut with VAF < 10% was comparable to wild types.
We
now know that not only the presence of clonal hematopoiesis may play a
role in the development of t-MNs but also that this role may be
directly related to the specific treatment patients undergo. In this
line, the direct link between clonal hematopoiesis, as a risk factor,
and the specific treatment, as a selective agent, in clonal evolution
and t-MN development is becoming evident.
For this reason,
several Authors are trying to focus on the study of a selected cohort
of patients affected by the same primary tumor and homogeneously
treated, comparing them with similar control cohorts.
Sperling et al. recently reported the landscape of 416 t-MN diagnosed and treated at MD Anderson Cancer Center[60]
to uncover the exposure relationships that provide selective advantage
to specific CH mutations. As expected, the Authors found a predominance
of TP53 and PPM1D mutations and mutations in DTA genes (DNMT3A, TET2, and ASXL1).
Complex karyotypes were enriched in patients treated with platinum
agents, while chromosome 5 and 7 abnormalities were more frequent in
patients treated with alkylating agents.
They also identified an enrichment of TP53
mutations in patients with a previous history of multiple myeloma (MM)
treated with thalidomide analogs and proteasome inhibitors.
Since TP53
mutations have been associated with resistance to lenalidomide therapy
in del(5q) MDS patients and secondary AML, the Authors tested, using
long-term in vitro competition assays, on HSPC from mice engineered by
CRISPR-Cas9 system, the effect of Thalidomide analogs on TP53 mutated cells.[61,62] They found that lenalidomide, but not pomalidomide, can induce the clonal selection of TP53 mutated HSPCs, while none of the other cells, PPM1D, TET2, and DNMT3A mutated, showed the same positive selection under treatment pressure. These data were also reproduced in “in vivo”
mouse models, highlighting the potential role of lenalidomide treatment
in t-MN development and suggesting the usefulness of CHIP screening in
the context of personalized therapies.
Summary and Future Prospective
Although
the main risk factors involved in therapy-related leukemogenesis seem
to be identified, their specific weight about the whole panoply of the
current cytotoxic therapies still needs to be well understood.
In
the near future, a comprehensive understanding of these heterogeneous
interactions can be achieved by studying homogeneous cohorts of
patients affected by the same primary disorder undergoing similar
treatment strategies.
In
these rare and precious study cohorts, it will be our task to use all
the available tools to identify patients at major risk of t-MN for whom
certain cytotoxic treatments should be avoided and replaced with less
leukemogenic approaches.
In
this line, high-throughput sequencing technologies, able to trace
clonal evolution in single cells, are the most promising tool to
achieve our goals.
In
the meantime, however, since CHIP has been recognized as a novel
predisposing factor in the pathogenesis of t-MN, somatic mutation
screening through NGS technologies should be carried out from the early
diagnostic stage of primary cancers to guide the choice of treatment
and minimize the risk of developing t-MN.
Acknowledgements
This
work was supported by AIRC 5×1000 call “Metastatic disease: the key
unmet need in oncology” to MYNERVA project (#21267) (MYeloid Neoplasms
Research Venture AIRC). A detailed description of the MYNERVA project
is available at http://www.progettoagimm.it and MURPNRR M4C2I1.3 PE6 project PE00000019 Heal Italia to MTV.
References
- Fianchi L, Criscuolo M, Fabiani E, Falconi G,
Maraglino AME, Voso MT et al. Therapy-related myeloid neoplasms:
clinical perspectives. Onco Targets Ther 2018; 11: 5909-5915. https://doi.org/10.2147/OTT.S101333 PMid:30271175 PMCid:PMC6149829
- Arber
DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The
2016 revision to the World Health Organization classification of
myeloid neoplasms and acute leukemia. Blood 2016; 127: 2391-2405. https://doi.org/10.1182/blood-2016-03-643544 PMid:27069254
- Khoury
JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF et al. The 5th
edition of the World Health Organization Classification of
Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms.
Leukemia 2022; 36: 1703-1719. https://doi.org/10.1038/s41375-022-01613-1 PMid:35732831 PMCid:PMC9252913
- Arber
DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka H-M et al.
International Consensus Classification of Myeloid Neoplasms and Acute
Leukemias: integrating morphologic, clinical, and genomic data. Blood
2022; 140: 1200-1228. https://doi.org/10.1182/blood.2022015850 PMid:35767897 PMCid:PMC9479031
- Leone
G, Fianchi L, Pagano L, Voso MT. Incidence and susceptibility to
therapy-related myeloid neoplasms. Chem Biol Interact 2010; 184: 39-45.
https://doi.org/10.1016/j.cbi.2009.12.013 PMid:20026017
- Takahashi
K, Wang F, Kantarjian H, Doss D, Khanna K, Thompson E et al.
Preleukaemic clonal haemopoiesis and risk of therapy-related myeloid
neoplasms: a case-control study. Lancet Oncol 2017; 18: 100-111. https://doi.org/10.1016/S1470-2045(16)30626-X PMid:27923552
- Nilsson
C, Linde F, Hulegårdh E, Garelius H, Lazarevic V, Antunovic P et al.
Characterization of therapy-related acute myeloid leukemia: increasing
incidence and prognostic implications. Haematologica 2023; 108:
1015-1025. https://doi.org/10.3324/haematol.2022.281233 PMid:36005563 PMCid:PMC10071134
- Larson
RA, Wang Y, Banerjee M, Wiemels J, Hartford C, Le Beau MM et al.
Prevalence of the inactivating 609C-->T polymorphism in the
NAD(P)H:quinone oxidoreductase (NQO1) gene in patients with primary and
therapy-related myeloid leukemia. Blood 1999; 94: 803-807. https://doi.org/10.1182/blood.V94.2.803 PMid:10397748
- Naoe
T, Takeyama K, Yokozawa T, Kiyoi H, Seto M, Uike N et al. Analysis of
genetic polymorphism in NQO1, GST-M1, GST-T1, and CYP3A4 in 469
Japanese patients with therapy-related leukemia/ myelodysplastic
syndrome and de novo acute myeloid leukemia. Clin cancer Res an Off J
Am Assoc Cancer Res 2000; 6: 4091-4095.
- Bolufer
P, Collado M, Barragan E, Calasanz M-J, Colomer D, Tormo M et al.
Profile of polymorphisms of drug-metabolising enzymes and the risk of
therapy-related leukaemia. Br J Haematol 2007; 136: 590-596. https://doi.org/10.1111/j.1365-2141.2006.06469.x PMid:17367411
- Fabiani
E, Fianchi L, Falconi G, Boncompagni R, Criscuolo M, Guidi F et al. The
BCL2L10 Leu21Arg variant and risk of therapy-related myeloid neoplasms
and de novo myelodysplastic syndromes. Leuk Lymphoma 2014; 55:
1538-1543. https://doi.org/10.3109/10428194.2013.845885 PMid:24047476
- Seedhouse
C, Bainton R, Lewis M, Harding A, Russell N, Das-Gupta E. The genotype
distribution of the XRCC1 gene indicates a role for base excision
repair in the development of therapy-related acute myeloblastic
leukemia. Blood 2002; 100: 3761-3766. https://doi.org/10.1182/blood-2002-04-1152 PMid:12393447
- Seedhouse
C, Faulkner R, Ashraf N, Das-Gupta E, Russell N. Polymorphisms in genes
involved in homologous recombination repair interact to increase the
risk of developing acute myeloid leukemia. Clin cancer Res an Off J Am
Assoc Cancer Res 2004; 10: 2675-2680. https://doi.org/10.1158/1078-0432.CCR-03-0372 PMid:15102670
- Churpek
JE, Marquez R, Neistadt B, Claussen K, Lee MK, Churpek MM et al.
Inherited mutations in cancer susceptibility genes are common among
survivors of breast cancer who develop therapy-related leukemia. Cancer
2016; 122: 304-311. https://doi.org/10.1002/cncr.29615 PMid:26641009 PMCid:PMC4707981
- Voso
MT, Fabiani E, Zang Z, Fianchi L, Falconi G, Padella A et al. Fanconi
anemia gene variants in therapy-related myeloid neoplasms. Blood Cancer
J. 2015; 5: e323. https://doi.org/10.1038/bcj.2015.44 PMid:26140431 PMCid:PMC4526773
- Swaminathan
M, Bannon SA, Routbort M, Naqvi K, Kadia TM, Takahashi K et al.
Hematologic malignancies and Li-Fraumeni syndrome. Cold Spring Harb Mol
case Stud 2019; 5. doi:10.1101/mcs.a003210. https://doi.org/10.1101/mcs.a003210 PMid:30709875 PMCid:PMC6371746
- Fabiani
E, Falconi G, Fianchi L, Criscuolo M, Ottone T, Cicconi L et al. Clonal
evolution in therapy-related neoplasms. Oncotarget 2017; 8:
12031-12040. https://doi.org/10.18632/oncotarget.14509 PMid:28076841 PMCid:PMC5355323
- Coombs
CC, Zehir A, Devlin SM, Kishtagari A, Syed A, Jonsson P et al.
Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic
Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell
Stem Cell 2017; 21: 374-382.e4. https://doi.org/10.1016/j.stem.2017.07.010 PMid:28803919 PMCid:PMC5591073
- Voso
MT, Falconi G, Fabiani E. What's new in the pathogenesis and treatment
of therapy-related myeloid neoplasms. Blood 2021; 138: 749-757. https://doi.org/10.1182/blood.2021010764 PMid:33876223
- Quintás-Cardama
A, Daver N, Kim H, Dinardo C, Jabbour E, Kadia T et al. A prognostic
model of therapy-related myelodysplastic syndrome for predicting
survival and transformation to acute myeloid leukemia. Clin Lymphoma
Myeloma Leuk 2014; 14: 401-410. https://doi.org/10.1016/j.clml.2014.03.001 PMid:24875590 PMCid:PMC4167474
- Ornstein
MC, Mukherjee S, Mohan S, Elson P, Tiu R V, Saunthararajah Y et al.
Predictive factors for latency period and a prognostic model for
survival in patients with therapy-related acute myeloid leukemia. Am J
Hematol 2014; 89: 168-173. https://doi.org/10.1002/ajh.23605 PMid:24123154
- Radivoyevitch
T, Sachs RK, Gale RP, Molenaar RJ, Brenner DJ, Hill BT et al. Defining
AML and MDS second cancer risk dynamics after diagnoses of first
cancers treated or not with radiation. Leukemia 2016; 30: 285-294. https://doi.org/10.1038/leu.2015.258 PMid:26460209
- Wolff
AC, Blackford AL, Visvanathan K, Rugo HS, Moy B, Goldstein LJ et al.
Risk of marrow neoplasms after adjuvant breast cancer therapy: the
national comprehensive cancer network experience. J Clin Oncol Off J Am
Soc Clin Oncol 2015; 33: 340-348. https://doi.org/10.1200/JCO.2013.54.6119 PMid:25534386 PMCid:PMC4302215
- Voso
M-T, Pandzic T, Falconi G, Denčić-Fekete M, De Bellis E, Scarfo L et
al. Clonal haematopoiesis as a risk factor for therapy-related myeloid
neoplasms in patients with chronic lymphocytic leukaemia treated with
chemo-(immuno)therapy. Br J Haematol 2022; 198: 103-113. https://doi.org/10.1111/bjh.18129 PMid:35277855
- Fianchi
L, Pagano L, Piciocchi A, Candoni A, Gaidano G, Breccia M et al.
Characteristics and outcome of therapy-related myeloid neoplasms:
Report from the Italian network on secondary leukemias. Am J Hematol
2015; 90: E80-5. https://doi.org/10.1002/ajh.23966 PMid:25653205
- Patel
AA, Rojek AE, Drazer MW, Weiner H, Godley LA, Le Beau MM et al.
Therapy-related myeloid neoplasms in 109 patients after radiation
monotherapy. Blood Adv 2021; 5: 4140-4148. https://doi.org/10.1182/bloodadvances.2021004964 PMid:34492705 PMCid:PMC8945635
- Boddu PC, Zeidan AM. Myeloid disorders after autoimmune disease. Best Pract Res Clin Haematol 2019; 32: 74-88. https://doi.org/10.1016/j.beha.2019.02.002 PMid:30927978 PMCid:PMC6541412
- Cristiano
A, Belardi R, Hajrullaj H, Fabiani E, Falconi G, Galossi E et al.
Correlation analysis between auto-immunological and mutational profiles
in myelodysplastic syndromes. Inflamm Res Off J Eur Histamine Res Soc .
[et al] 2023. doi:10.1007/s00011-023-01773-5. https://doi.org/10.1007/s00011-023-01773-5 PMid:37507570 PMCid:PMC10499973
- Burki TK. Azathioprine associated with myeloid neoplasm risk. Lancet. Oncol. 2017; 18: e140. https://doi.org/10.1016/S1470-2045(17)30096-7 PMid:28190763
- Le
Deley M-C, Suzan F, Cutuli B, Delaloge S, Shamsaldin A, Linassier C et
al. Anthracyclines, mitoxantrone, radiotherapy, and granulocyte
colony-stimulating factor: risk factors for leukemia and
myelodysplastic syndrome after breast cancer. J Clin Oncol Off J Am Soc
Clin Oncol 2007; 25: 292-300. https://doi.org/10.1200/JCO.2006.05.9048 PMid:17159192
- Kwong
Y-L. Azathioprine: association with therapy-related myelodysplastic
syndrome and acute myeloid leukemia. J Rheumatol 2010; 37: 485-490. https://doi.org/10.3899/jrheum.090834 PMid:20080917
- Li
K, Jing Y, Yang C, Liu S, Zhao Y, He X et al. Increased
leukemia-associated gene expression in benzene-exposed workers. Sci Rep
2014; 4: 5369. https://doi.org/10.1038/srep05369 PMid:24993241 PMCid:PMC4081871
- Bolton
KL, Ptashkin RN, Gao T, Braunstein L, Devlin SM, Kelly D et al. Cancer
therapy shapes the fitness landscape of clonal hematopoiesis. Nat Genet
2020; 52: 1219-1226. https://doi.org/10.1038/s41588-020-00710-0 PMid:33106634 PMCid:PMC7891089
- Preston
DL, Kusumi S, Tomonaga M, Izumi S, Ron E, Kuramoto A et al. Cancer
incidence in atomic bomb survivors. Part III. Leukemia, lymphoma and
multiple myeloma, 1950-1987. Radiat Res 1994; 137: S68-97. https://doi.org/10.2307/3578893
- Hsu
W-L, Preston DL, Soda M, Sugiyama H, Funamoto S, Kodama K et al. The
incidence of leukemia, lymphoma and multiple myeloma among atomic bomb
survivors: 1950-2001. Radiat Res 2013; 179: 361-382. https://doi.org/10.1667/RR2892.1 PMid:23398354 PMCid:PMC3875218
- Rassool
F V, Gaymes TJ, Omidvar N, Brady N, Beurlet S, Pla M et al. Reactive
oxygen species, DNA damage, and error-prone repair: a model for genomic
instability with progression in myeloid leukemia? Cancer Res 2007; 67:
8762-8771. https://doi.org/10.1158/0008-5472.CAN-06-4807 PMid:17875717
- Sallmyr
A, Fan J, Rassool FV. Genomic instability in myeloid malignancies:
increased reactive oxygen species (ROS), DNA double strand breaks
(DSBs) and error-prone repair. Cancer Lett 2008; 270: 1-9. https://doi.org/10.1016/j.canlet.2008.03.036 PMid:18467025
- Wang L, He X, Bi Y, Ma Q. Stem cell and benzene-induced malignancy and hematotoxicity. Chem Res Toxicol 2012; 25: 1303-1315. https://doi.org/10.1021/tx3001169 PMid:22540379
- Spatari
G, Allegra A, Carrieri M, Pioggia G, Gangemi S. Epigenetic Effects of
Benzene in Hematologic Neoplasms: The Altered Gene Expression. Cancers
(Basel) 2021; 13. doi:10.3390/cancers13102392. https://doi.org/10.3390/cancers13102392 PMid:34069279 PMCid:PMC8156840
- Singhal
D, Hahn CN, Feurstein S, Wee LYA, Moma L, Kutyna MM et al. Targeted
gene panels identify a high frequency of pathogenic germline variants
in patients diagnosed with a hematological malignancy and at least one
other independent cancer. Leukemia 2021; 35: 3245-3256. https://doi.org/10.1038/s41375-021-01246-w PMid:33850299
- Baranwal
A, Hahn CN, Shah MV, Hiwase DK. Role of Germline Predisposition to
Therapy-Related Myeloid Neoplasms. Curr Hematol Malig Rep 2022; 17:
254-265. https://doi.org/10.1007/s11899-022-00676-2 PMid:35986863
- Shih
AJ, Jun T, Skol AD, Bao R, Huang L, Vora S et al. Inherited cancer
predisposing mutations in patients with therapy-related myeloid
neoplasms. Br J Haematol 2023; 200: 489-493. https://doi.org/10.1111/bjh.18543 PMid:36349721
- McNerney
ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when
genetics and environment collide. Nat Rev Cancer 2017; 17: 513-527. https://doi.org/10.1038/nrc.2017.60 PMid:28835720 PMCid:PMC5946699
- Allan
JM, Wild CP, Rollinson S, Willett E V, Moorman A V, Dovey GJ et al.
Polymorphism in glutathione S-transferase P1 is associated with
susceptibility to chemotherapy-induced leukemia. Proc Natl Acad Sci U S
A 2001; 98: 11592-11597. https://doi.org/10.1073/pnas.191211198 PMid:11553769 PMCid:PMC58774
- Seedhouse
C, Bainton R, Lewis M, Harding A, Russell N, Das-Gupta E. The genotype
distribution of the XRCC1gene indicates a role for base excision repair
in the development of therapy-related acute myeloblastic leukemia.
Blood 2002; 100: 3761-3766. https://doi.org/10.1182/blood-2002-04-1152 PMid:12393447
- Schwartz
JR, Ma J, Kamens J, Westover T, Walsh MP, Brady SW et al. The
acquisition of molecular drivers in pediatric therapy-related myeloid
neoplasms. Nat Commun 2021; 12: 985. https://doi.org/10.1038/s41467-021-21255-8 PMid:33579957 PMCid:PMC7880998
- Jaiswal
S, Fontanillas P, Flannick J, Manning A, Grauman P V, Mar BG et al.
Age-related clonal hematopoiesis associated with adverse outcomes. N
Engl J Med 2014; 371: 2488-2498. https://doi.org/10.1056/NEJMoa1408617 PMid:25426837 PMCid:PMC4306669
- Genovese
G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF et al.
Clonal hematopoiesis and blood-cancer risk inferred from blood DNA
sequence. N Engl J Med 2014; 371: 2477-2487. https://doi.org/10.1056/NEJMoa1409405 PMid:25426838 PMCid:PMC4290021
- Bowman
RL, Busque L, Levine RL. Clonal Hematopoiesis and Evolution to
Hematopoietic Malignancies. Cell Stem Cell 2018; 22: 157-170. https://doi.org/10.1016/j.stem.2018.01.011 PMid:29395053 PMCid:PMC5804896
- Gillis
NK, Ball M, Zhang Q, Ma Z, Zhao Y, Yoder SJ et al. Clonal haemopoiesis
and therapy-related myeloid malignancies in elderly patients: a
proof-of-concept, case-control study. Lancet Oncol 2017; 18: 112-121. https://doi.org/10.1016/S1470-2045(16)30627-1 PMid:27927582
- Wong
TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS et al. Role of
TP53 mutations in the origin and evolution of therapy-related acute
myeloid leukaemia. Nature 2015; 518: 552-555. https://doi.org/10.1038/nature13968 PMid:25487151 PMCid:PMC4403236
- Leone
G, Fabiani E, Voso MT. De Novo and Therapy-Related Myelodysplastic
Syndromes: Analogies and Differences. Mediterr J Hematol Infect Dis
2022; 14: e2022030. https://doi.org/10.4084/MJHID.2022.030 PMid:35615324 PMCid:PMC9083943
- Ok
CY, Patel KP, Garcia-Manero G, Routbort MJ, Fu B, Tang G et al.
Mutational profiling of therapy-related myelodysplastic syndromes and
acute myeloid leukemia by next generation sequencing, a comparison with
de novo diseases. Leuk Res 2015; 39: 348-354. https://doi.org/10.1016/j.leukres.2014.12.006 PMid:25573287 PMCid:PMC5548131
- Lindsley
RC, Mar BG, Mazzola E, Grauman P V, Shareef S, Allen SL et al. Acute
myeloid leukemia ontogeny is defined by distinct somatic mutations.
Blood 2015; 125: 1367-1376. https://doi.org/10.1182/blood-2014-11-610543 PMid:25550361 PMCid:PMC4342352
- Singhal
D, Wee LYA, Kutyna MM, Chhetri R, Geoghegan J, Schreiber AW et al. The
mutational burden of therapy-related myeloid neoplasms is similar to
primary myelodysplastic syndrome but has a distinctive distribution.
Leukemia 2019; 33: 2842-2853. https://doi.org/10.1038/s41375-019-0479-8 PMid:31089247
- Bernard
E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS
et al. Implications of TP53 allelic state for genome stability,
clinical presentation and outcomes in myelodysplastic syndromes. Nat
Med 2020; 26: 1549-1556. https://doi.org/10.1038/s41591-020-1008-z PMid:32747829 PMCid:PMC8381722
- Shah
MV, Tran ENH, Shah S, Chhetri R, Baranwal A, Ladon D et al. TP53
mutation variant allele frequency of ≥10% is associated with poor
prognosis in therapy-related myeloid neoplasms. Blood Cancer J 2023;
13: 51. https://doi.org/10.1038/s41408-023-00821-x PMid:37041128 PMCid:PMC10090194
- Shah
M V, Hahn CN, Tran ENH, Sharplin KM, Chhetri R, Baranwal A et al. TP53
Mutation Status Defines a Distinct Clinicopathological Entity of
Therapy-Related Myeloid Neoplasm, Characterized By Genomic Instability
and Extremely Poor Outcome. Blood 2022; 140: 9798-9799. https://doi.org/10.1182/blood-2022-165859
- Weinberg
OK, Siddon A, Madanat YF, Gagan J, Arber DA, Dal Cin P et al. TP53
mutation defines a unique subgroup within complex karyotype de novo and
therapy-related MDS/AML. Blood Adv 2022; 6: 2847-2853. https://doi.org/10.1182/bloodadvances.2021006239 PMid:35073573 PMCid:PMC9092405
- Sperling
AS, Guerra VA, Kennedy JA, Yan Y, Hsu JI, Wang F et al. Lenalidomide
promotes the development of TP53-mutated therapy-related myeloid
neoplasms. Blood 2022; 140: 1753-1763. https://doi.org/10.1182/blood.2021014956 PMid:35512188 PMCid:PMC9837415
- Jädersten
M, Saft L, Smith A, Kulasekararaj A, Pomplun S, Göhring G et al. TP53
mutations in low-risk myelodysplastic syndromes with del(5q) predict
disease progression. J Clin Oncol Off J Am Soc Clin Oncol 2011; 29:
1971-1979. https://doi.org/10.1200/JCO.2010.31.8576 PMid:21519010
- Martinez-Høyer
S, Deng Y, Parker J, Jiang J, Mo A, Docking TR et al. Loss of
lenalidomide-induced megakaryocytic differentiation leads to therapy
resistance in del(5q) myelodysplastic syndrome. Nat Cell Biol 2020; 22:
526-533.
https://doi.org/10.1038/s41556-020-0497-9 PMid:32251398