Ugo Testa1, Giuseppe Leone3, Elvira Pelosi1, Germana Castelli1 and Stefan Hohaus2,3.
1 Istituto Superiore di Sanità, Roma.
2
Dipartimento Di Diagnostica per Immagini, Radioterapia Oncologica Ed
Ematologia, Fondazione Policlinico Universitario Agostino Gemelli
IRCCS, Roma, Italy. Sezione Di Ematologia.
3 Dipartimento Di Scienze Radiologiche Ed Ematologiche, Università Cattolica Del Sacro Cuore, Roma, Italy.
Published: November 01, 2023
Received: October 09, 2023
Accepted: October 16, 2023
Mediterr J Hematol Infect Dis 2023, 15(1): e2023066 DOI
10.4084/MJHID.2023.066
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Large B-cell lymphomas (LBCLs) are
among the most frequent (about 30%) non-Hodgkin’s lymphoma. Despite the
aggressive behavior of these lymphomas, more than 60% of patients can
be cured with first-line chemoimmunotherapy using the R-CHOP regimen.
Patients with refractory or relapsing disease show a poor outcome even
when treated with second-line therapies.
CD19-targeted chimeric
antigen receptor (CAR) T-cells are emerging as an efficacious
second-line treatment strategy for patients with LBCL. Three
CD19-CAR-T-cell products received FDA and EMA approval. CAR-T cell
therapy has also been explored for treating high-risk LBCL patients in
the first-line setting and for patients with central nervous system
involvement.
Although CD19-CAR-T therapy has transformed the care
of refractory/relapsed LBCL, about 60% of these patients will
ultimately progress or relapse following CD19-CAR-T; therefore, it is
fundamental to identify predictive criteria of response to CAR-T
therapy and to develop salvage therapies for patients relapsing after
CD19-CAR-T therapies. Moreover, ongoing clinical trials evaluate
bispecific CAR-T cells targeting both CD19 and CD20 or CD19 and CD22 as
a tool to improve the therapeutic efficacy and reduce the number of
refractory/relapsing patients.
|
Introduction
Chimeric
antigen receptors (CARs) are engineered receptors that enable T
lymphocytes with the capacity to specifically recognize an antigen and
induce a cytotoxic reaction against tumor cells based on their
expression of this antigen.
Antitumor adaptive therapies represent
a key strategy in the treatment of tumors. Some of these
immunotherapies were based on the use of genetically engineered cells.
Particularly, two different types of immunotherapies have been
developed using genetically modified T lymphocytes: (i) T-cell
receptor-engineered cells enabled to recognize specific membrane
antigens in a HLA-restricted manner; (ii) CAR-T transduced T
lymphocytes that interact with specific membrane antigens in a
HLA-unrestricted manner and antibody-specific manner.
The
molecular architecture of a CAR molecule implies four components: i) an
extracellular target antigen-binding domain (ABD); ii) a hinge region;
iii) a transmembrane region; iv) one or more intracellular signaling
domains. ABD is the part of the CAR molecule that confers specificity
in antigen recognition and is usually derived from the variable heavy (VH) and light (VL)
chains of monoclonal antibodies, connected through a flexible linker to
generate a single-chain variable fragment (scFv). The hinge component
is a spacer region that extends the ABD from the ABD to the
transmembrane region and confers sufficient flexibility to avoid steric
hindrance. The transmembrane region is required to anchor the CAR
molecule to the cell membrane of T lymphocytes. The intracellular
signaling domains play an important role in the modulation of CAR-T
cell activity; a large part of CARs is based on the activation of T
lymphocytes using CD3-derived immunoreceptor tyrosine-based activation
motifs.[1]
The procedure for generating CAR-T
cells evolved over time with 5 different CAR-T generations from the
first procedures in late 1990 to the most recent developments.[1] The first generation of CAR-T was based on the CD3-ζ
intracellular domain, in the absence of costimulatory domains; the
second generation of CAR-T cells contained a costimulatory domain, such
as CD28, in the intracellular domain; the third generation was based on
the presence of multiple costimulatory domains; the fourth generation
involved the production of T cell redirected for general universal
cytokine-mediated killing (TROCKs), a property obtained through IL-12
production, either constitutive or after CAR-T activation; the fifth
generation also included a STAT3 binding site required for generation
of three activation signals acting on the cell signaling, costimulatory
and cytokine signaling domains.[1] Compared to the
first generation, the consistent advantages of second- and
third-generation CAR-T cells consisted of enhanced proliferation,
cytotoxicity, and lifespan in vivo.[1] The last generations of CAR-T cells showed superior in vivo
persistence and enhanced antitumor effects in leukemia and solid tumor
models compared to initial CAR-T cell generations and are expected to
demonstrate superior antitumor effects with reduced toxicity in the
clinic.[2]
The key role of CD19 targeting by CAR-T for the therapy of B-cell malignancies.
The human CD19 antigen is a B-lymphocyte antigen belonging to the
immunoglobulin superfamily, whose expression is restricted to the
B-cell lineage starting from the early stages of B cell development
corresponding to heavy chain immunoglobulin rearrangement to the late
stages of B cell differentiation; CD19 expression increases during B
cell differentiation.[3-4] On the cell membrane of B-lymphoid cells, CD19 forms a transduction complex with CD21, CD81, and Leuk-13.[5] Furthermore, CD81 regulates the expression of CD19 during B cell development.[6]
FDA and EMA approved CAR-T cell therapies for B lymphoid malignancies.
Four CAR products are commercially available for patients with B cell
lymphomas: Axicabtagene ciloleucel (Axi-Cel), Brexacubtagene autoleucel
(Brexa-Cel), Lisocabtagene maroleucel (Liso-Cel) and Tisagenlecleucel
(Tisa-Cel); two for B-ALL: Brexa-Cell and Tisa-Cel. All these products
are based on a second-generation CAR construct (see Figure 1)
and involve the presence of an intracellular component containing a
T-cell activation domain (CD3ζ) and a costimulatory domain (CD28 in
Axi-Cell and Brexa-Cel, 4-1BB in Tisa-Cel and Liso-Cel).
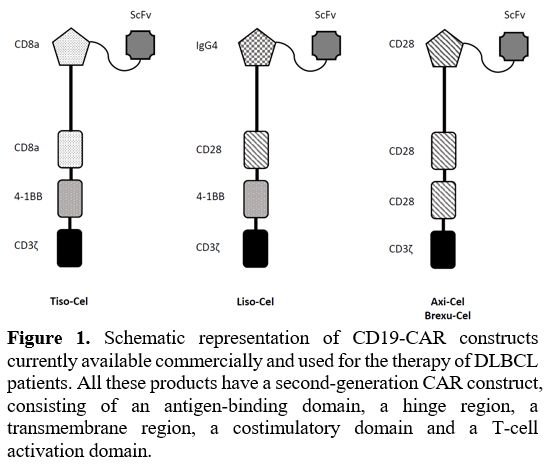 |
- Figure
1. Schematic representation of CD19-CAR constructs currently available
commercially and used for the therapy of DLBCL patients.
- All these
products have a second-generation CAR construct, consisting of an
antigen-binding domain, a hinge region, a transmembrane
- region, a
costimulatory domain and a T-cell activation domain.
|
The
structure of Axi-Cel and Brexa-Cel is identical, but their
manufacturing process is different in that the procedure of production
of Brexa-Cel also implies a step of removal of malignant cells from the
apheresis sample; both these CAR genes are delivered to T lymphocytes
using a gammaretrovirus. The CAR gene used for Tisa-Cel and Liso-Cel is
delivered using lentiviruses; particularly, Liso-Cel is delivered to a
defined CD4/CD8 T cell composition.
Clinical studies on CAR-T cells in DLBCL.
Large B cell lymphomas are among the most frequent (about 30%)
non-Hodgkin's lymphoma. Gene expression studies, based on
cell-of-origin, have identified two main subtypes of DLBCLs with
distinct clinical and molecular features: activated B-cell (ABC) and
germinal center B cell (GCB).[7] Analysis of genomic
alterations has shown an additional heterogeneity of ABC and GCB
subgroups. Within GCB-DLBCLs, lymphomas with EZH2 mutations and BCL2 translocations have a poor outcome. Lymphomas harboring both MYC and BCL2 and/or BCL6
rearrangements are identified as high-grade B-cell lymphomas double-hit
or triple-hit (HGBL-DH and HGBL-TH) and show an aggressive
clinical-biological phenotype. Within the ABC-DLBCL group, lymphomas
with NOTCH1 mutations or co-occurring MYD88 and CD79B mutations have a poor prognosis.[7]
Clinical studies using CAR-T in DLBCL
Several
studies have explored the safety and efficacy of CAR-T in diffuse large
B-cell lymphomas (DLBCL). These patients may be cured with first-line
therapy; however, up to 30% to 40% of them may become refractory or
relapse.
Clinical studies with Axi-Cel.
The ZUMA-1 and ZUMA-7 trials evaluated the safety and efficacy of
Axi-Cel in different clinical settings of patients with LBCL. The
ZUMA-7 study is a phase III trial in which patients with early relapsed
or refractory DLBCL were randomized to receive either Axi-Cel (180
patients) or standard care (179 patients) that consisted of salvage
chemotherapy followed by high-dose chemotherapy with autologous stem
cell transplantation (ASCT) in second-line therapy.[8]
At the latest median follow-up explored (47.2 months), the following
results were observed: 82 deaths in the Axi-Cel group and 95 in the
standard-care group; median overall survival (mOS) not reached in the
Axi-Cel group and 31.1 months in the standard-care group; median
progression-free survival (mPPFS) was 14.7 months in the Axi-Cel group
and 3.7 months in the standard-care group.[8-9] A
subgroup analysis of the ZUMA-7 study limited to patients 65 years of
age or older showed a higher rate of ORR and CRR in the Axi-Cel group
compared to the standard care group (ORR 88% vs 52%, respectively; CRR
75% vs 33%, respectively).[10] In particular, no
grade 5 cytokine release syndrome or neurologic events occurred in this
group of patients who are more fragile and at risk for complications.[10]
The
ZUMA-7 trial was preceded by the ZUMA-1 trial exploring the efficacy of
Axi-Cel in third-line therapy. ZUMA-1 was a single-arm phase I/II study
enrolling LBCL patients with refractory or relapsed disease after
autologous stem cell transplantation; the patients received a target
dose of 2x106 CAR-T cells per Kg of body weight after conditioning chemotherapy with fludarabine and cyclophosphamide.[11]
One hundred one patients were enrolled in this study, and after a
median follow-up of 63.1 months, the following results were observed:
83% of ORR, with 58% of CRR; mOS was 25.8 months, with a
disease-specific survival at 5 years of 51%.[11-12] These results suggested a curative potential of Axi-Cel in a subset of LBCL patients.[11-12]
A comparison of 2-year outcomes with CAR-T cells of patients enrolled
in the ZUMA-1 trial showed better results observed with CAR-T cell
therapy compared to salvage chemotherapy in a comparable group of
patients (ORR 83% vs. 34% and CRR 54% vs. 20%, respectively).[13] The 2-year survival rate was 54% with Axi-Cel and 20% with salvage therapy.[13]
The
analysis of the clinical results observed in a real-world setting of
275 relapsed-refractory DLBCL patients receiving Axi-Cel confirmed the
results observed in the ZUMA-1 study, with ORR 82% and CRR 64%. At a
median follow-up of 12.9 months, the PFS was 8.3 months.[14]
The
58% achieved a complete response rate following treatment with Axi-Cel
in LBCL patients participating to the ZUMA-1 study offered the
opportunity to explore the existence of tumor-related and
tumor-associated parameters differentially expressed in responding and
non-responding patients. Thus, many studies have shown the existence of
several clinical, biochemical, and biological parameters that either
negatively or positively affect the response to Axi-Cel.
A high tumor burden, measured through evaluation of baseline metabolic tumor volume (MTV) on 18F fluorodeoxyglucose positron emission tomography, was associated with decreased efficacy of Axi-Cel in LBCL patients.[15]
A second study based on the analysis of patients enrolled in the ZUMA-1
study subdivided patients into three subgroups (responders,
non-responders, and relapsed). Low baseline tumor burden, high CAR-T
cells/tumor burden ratio, low systemic inflammation, and high product
CD8 and CCR7+, CD45RA+ T cells were associated with better tumor response.[16]
A third study showed that resistance to Axi-Cel is related to immune
dysregulation that is frequently observed in LBCL and leads to
insufficient in vivo Axi-Cel
expansion consisting of high blood levels of monocytic myeloid-derived
suppressor cells (M-MDSCs) and tumor interferon signaling, giving rise
also to expression of immune checkpoint ligands.[17]
Finally, a fourth study provided evidence that tumor immune contexture
is a major determinant of Axi-Cel efficacy. In particular, clinical
response and overall survival were associated with immunological
parameters that can be identified using Immunoscore (tumor-infiltrating
T cell density) and Immunosign 21 (immune-related gene expression
profile).[18] Furthermore, circulating CAR-T cell
levels were associated with post-treatment T cell exhaustion in the
tumor microenvironment.[18]
Clinical studies with Liso-Cel.
Several studies have evaluated Liso-Cel in the treatment of relapsed or
refractory LBCLs. The phase I TRASCEND study evaluated 269 LBCL
patients with relapsed/refractory disease who received at least two
previous lines of therapy and were treated with Liso-Cel using three
different dose levels (50x106 or 100x106 or 150x106 CAR-T cells).[19]
The first results of this study showed high response rates (ORR 73%,
CRR 53%), with a low incidence of grade 3 or worse cytokine release
syndrome and neurological events.[19]
The
TRANSFORM phase III trial randomized 184 LBCL relapsed/refractory
patients, candidates for autologous SCT, to treatment with either
standard-of-care therapy or Liso-Cel (100x106 CAR-T cells).[20]
With a median follow-up of 6.2 months, the EFS was 10.1 months in the
Liso-Cel group compared to 2.3 months in the standard-of-care group.[20]
An analysis of the results observed in this trial after a follow-up of
17.5 months showed: a CRR of 74% in the Liso-Cel arm compared to 43% in
the SOC arm; a PFS not reached in the Liso-Cel group compared to 6.2
months in the SOC group; a mOS not reached in the Liso-Cel arm compared
to 29.9 months in the SOC arm.[21] The safety profile
of treatment with Liso-Cel was favorable, with grade 3 cytokine release
syndrome and neurological events occurring in 1 and 4% of patients,
respectively.[21]
An analysis of the parameters
related to the quality of life (QoL) showed that the Liso-Cel arm
showed a higher improvement in QoL parameters and a lower deterioration
than the SOC arm.[22]
Olson and coworkers
explored tumor biology and microenvironment from lymph node biopsies of
DLBCL patients undergoing treatment with Liso-Cel. The authors compared
gene expression profiles between responding and non-responding
patients.[23] Tumor microenvironment and
tumor-associated macrophage stromal gene signatures had been previously
associated with adverse outcomes to standard chemoimmunotherapy
treatment in DLBCL.[24] Their study was carried out
on 78 patients with DLBCL included in the TRASCEND NHL 001 trial and
showed that pre-treatment biopsies from patients achieving a complete
response showed higher expression levels of T-cell and
stroma-associated genes and lower expression of cell-cycle-related
genes in the responding patients, post-treatment biopsies had higher
levels of CAR-T-cell densities and CAR gene expression, general immune
infiltration, and immune activation.[23]
Clinical studies with Tisa-Cel.
Other studies have explored the safety and efficacy of Tisa-Cel in
adult relapsed/refractory DLBCL patients. The phase II multicentre
JULIET study initially explored the safety and the efficacy of Tisa-Cel
in a group of 93 patients with relapsed/refractory DLBCL, showing 52%
of ORR, with 40% of CR and 12% of PR; 22% of patients displayed a
cytokine release syndrome and 12% neurologic events.[25]
Analysis of long-term outcomes in the JULIET trial extended to 115
patients treated with Tisa-Cel showed the following results: an ORR and
a CRR in 53% and 39% of cases, respectively; mPFS and mOS were 2.9
months and 11.1 months, respectively; among 34 patients with CR at 6
months, only 3 relapsed within 12 months. Post-hoc analysis showed that
PFS and OS were not reached among patients reaching a CR after 6
months.[26]
A comparison of the results of the
JULIET trial with historical results (CORAL study) observed in a
similar patient group treated with standard chemotherapy supported the
superiority of CAR-T-based treatment compared to chemotherapy (mOS of
12.48 months compared to 4.40 months, respectively).[27]
The
BELINDA phase III clinical trial randomized 322 patients with
aggressive B-cell lymphomas (about 70% of the patients had DLBCLs) to
treatment with Tisa-Cel or with standard of care (salvage chemotherapy
and autologous HSCT). The ORR and EFS were similar in the two groups of
patients.[28] Tisa-Cel was not superior to standard salvage therapy in this trial.
In
a retrospective study, 418 relapsed/refractory DLBCL patients, included
in the French DESCART-T registry, were treated with CAR-T cell therapy
either using Axi-Cel or Tisa-Cel. Treatment results were compared after
1:1 propensity score matching. With a median follow-up of 11.7 months,
the 1-year PFS was 46.6% for Axi-Cel and 33.2% for Tisa-Cel, 1-year OS
was 63.5% for Axi-Cel and 48.8% for Tisa-Cel, the ORR was 80% for
Axi-Cel compared to 66% for Tisa-Cel and the CRR was 60% for Axi-Cel
compared to 42% for Tisa-Cel.[29] However, immune
effector cell-associated neurotoxicity syndrome (ICANS) and cytokine
release syndrome (CRS) were more frequent in the Axi-Cel than in the
Tisa-Cel group.[29] In conclusion, this retrospective study supports a higher efficacy and higher Axi-Cel toxicity than Tisa-Cel.[29]
The
analysis of the safety profile of DLBCL patients treated with
Tisa-Cel-based CAR-T in the context of the JULIET trial showed the
occurrence of manageable long-term (LT) adverse events: 14% of
responding patients displayed LT cytopenias lasting 90 days; patients
treated with rituximab displayed hypogammaglobulinemia that in some
patients was exacerbated by CAR-T treatment; few responding patients
had LT infections (severe or opportunistic infections).[30]
The
phase Ib PORTIA study explored the safety and the efficacy of Tisa-Cel
in association with the anti-PD-1 inhibitor pembrolizumab in
relapsed/refractory DLBCL patients; the patients enrolled in this study
were subdivided into three cohorts: 4 patients were treated with
pembrolizumab on day 15, 4 on day 8 and 4 patients on day -1, for CAR-T
cell infusion.[31] The best response rates were
observed in patients treated with pembrolizumab before Tisa-Cel, but
definitive conclusions cannot be drawn given the limited number of
patients studied; the drug association displayed a manageable safety
profile; pembrolizumab did not stimulate the cellular expansion of
Tisa-Cel but delayed peak expansion in the day -1 cohort.[31]
Comparative analysis of the results obtained in phase III studies on CAR-T cell therapy in the second-line.
Three prospective randomized phase III clinical trials, ZUMA-7
(Axi-Cel), TRANSFORM (Liso-Cel), and BELINDA (Tiso-Cel), have compared
CAR-T cell therapy to standard of care (high-dose chemotherapy with
auto-HSCT) in DLBCL patients with early relapsed/refractory disease
(<12 months after chemoimmunotherapy). The ZUMA-7 study reported an
improved event-free survival in the Axi-Cel arm compared to the SOC arm
(8.3 months vs 2.0 months).[8-9] In the TRANSFORM study, median EFS was longer in the Liso-Cel arm than in the SOC arm (not reached vs 2.4 months).[19-20] In contrast, in the BELINDA study, no difference in median EFS was observed between Lisa-Cel and SOC arm (3.0 vs 3.0 months).[28]
Based on these data in 2022, the FDA and EMA approved Axi-Cel and
Lisa-Cel for DLBCL patients with refractory disease or relapsed within
12 months of first-line treatment. In the BELINDA study, the Tisa-Cel
arm included a higher proportion of patients with intermediate or
higher IPI scores and double-hit lymphoma compared to the SOC arm
(65.4% vs 57.5%, respectively).[28]
CAR-T cell therapy in first-line treatment of DLBCL.
In addition to the studies carried out in second and third-line
refractory/relapsed DLBCL patients, a recent study explored the safety
and the efficacy of Axi-Cel in first-line therapy in a population of
high-risk DLBCL patients.[32] This study enrolled 40
patients: 22 with DLBCL, 16 with double- or triple-hit lymphomas, and 2
with high-grade B-cell lymphoma (HGBL) not otherwise specified.[32] Double-hit or triple-hit lymphomas correspond to a subtype of DLBCLs with MYC rearrangement concurrent with a rearrangement in BCL2, BCL6, or both. They are associated with poor outcome after standard chemotherapy treatment.[7]
All treated patients received two cycles of one previous systemic
therapy, most commonly R-CHOP or DA-EPOCH-R; 40% had PD, suggesting a
primary chemorefractory disease.[32] The following
results were observed: a CRR of 78%, with a median time to CR of 30
days; an ORR of 89%, with a median time to objective response of 29
days; the estimated rates for DoR, PFS, EFS, and OS at 12 months were
81%, 75%, 73%, and 91%, respectively.[32] Grade 3 cytokine release syndrome and grade 3 neurologic events were observed in 8% and 18% of patients, respectively.[32]
Given
the results observed in the ZUMA-12 study, the ZUMA-23 trial was
proposed as a phase III randomized controlled trial involving the
evaluation of Axi-Cel as a first-line regimen in comparison with
standard of care in about 300 LBCL adult patients with a high-risk
disease, defined as International Prognostic Index 4-5.[33]
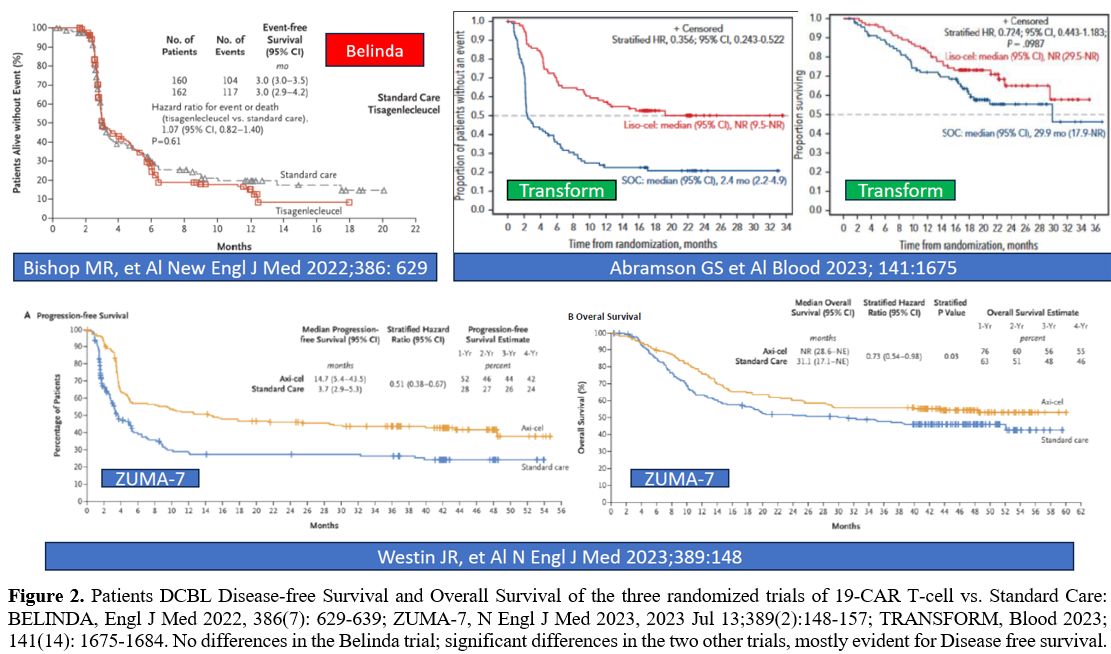 |
- Figure 2. Patients
DCBL Disease-free Survival and Overall Survival of the three randomized
trials of 19-CAR T-cell vs. Standard Care: BELINDA, Engl J Med
- 2022,
386(7): 629-639; ZUMA-7, N Engl J Med 2023, 2023 Jul 13;389(2):148-157;
TRANSFORM, Blood 2023; 141(14): 1675-1684. No differences in the
- Belinda trial; significant differences in the two other trials, mostly
evident for Disease free survival.
|
CAR-T with double targeting.
Failure to achieve sustained responses is observed in about 60-70% of
relapsed/refractory LBCL patients treated with CD19-directed CAR-T
cells. This result is due to different mechanisms: (i) CD19-negative
relapse due to antigen downregulation or loss of CAR-T selection
pressure; (ii) impaired CAR-T cell expansion and T-cell exhaustion;
(iii) overexpression of programmed cell death protein 1 (PD-1) and/or
high expression of PD-L1. One of the possible strategies to bypass
these resistance mechanisms consists of dual antigen targeting, such as
dual targeting of CD19 and CD22 with bicistronic CAR-T cells. Thus,
AUTO3, a dual-targeting, humanized, second-generation autologous
CD19/CD22 CAR-T product, was developed using a bicistronic vector
encoding CD19 CAR and CD22 CAR within a single construct.[34]
In the phase I ALEXANDER trial AUTO3 plus PD-1 blockade with
pembrolizumab was evaluated as third-line therapy in 52 adult patients
with relapsed/refractory LBCL.[34] AUTO3 administration was well tolerated with only rare events of drug-related toxicity.[34]
The ORR was 66%, with 54% of CR; the mDoR was 8.3 months; for patients
with CR, the mDoR was not reached; mPFS was 3.3 months and the mOS was
13.8 months.[34]
The results of this study
indicated that dual-targeting CAR-T and pembrolizumab as third-line
therapy was able to induce a significant therapeutic response in 55% of
relapsed/refractory LBCL patients and future studies will attempt to
develop a new generation of AUTO3, endowed with a higher capacity of in-vivo expansion.[34]
Other
studies have explored another strategy based on the combined targeting
of CD19 and CD20 on the surface of B-lymphoid cells. In this context,
Tong et al. reported the development of optimized tandem CD19/CD20
CAR-engineered T cells (TanCAR7 T cells) that target these two antigens
simultaneously or separately.[35] TanCAR7 T cells were shown to possess a marked antitumor activity in vitro;
furthermore, early clinical evidence demonstrated an acceptable safety
profile and clinical efficacy: 28 patients with relapsed/refractory
NHLs, including 16 patients with DLBCL, were treated with TanCAR7 T
cells.[35] 75% of patients with DLBCL had a response,
with an mPFS not reached, and 75% of patients showed no disease
progression at 12 months after infusion.[35]
Concerning the safety profile, 14% of patients displayed a grade 3 CRS
and no cases of grade 3 T-cell-related encephalopathy syndrome were
observed.[35]A more recent study reported the results obtained with this strategy in 87 NHL patients (66% with DLBCL).[36]
Among DLBCL patients, 78% of objective responses were observed, with a
CRR of 71%, a median DoR not reached 74% of patients remaining in
remission 12 months after having a response, a median PFS of 23.5
months, and 59% of patients showing no disease progression at 12 months
after infusion.[36] The mOS was not reached, and the probability of survival was 88% at 6 months and 75% at 12 months.[36]
The
DALY II multicenter trial evaluated the safety and efficacy of
bispecific targeted CD20/CD19 therapy with ZAMTO-CEL in 22 patients
with relapsed/refractory DLBCL (68% with high-risk disease with IPI ≥3
and 13% exposed to previous CAR-T cell therapy).[37] At 3 months post-treatment, the ORR was 52% with 38% CR and 14% PR.[37]
Role of bridging therapy during CAR-T cell therapy.
Bridging therapy (BT) is the anticancer therapy administered in
patients during CAR-T cell manufacturing. It is a tool to stabilize or
debulk disease between leukapheresis and CAR-T cell administration.[38]
However, there is no clear indication for administering or not BT, and
the use of BT is guided by physician and patient preferences. A recent
study explored BT modality and response in 375 patients undergoing
treatment with either Axi-Cel or Lisa-Cel; most patients received BT as
chemotherapy or radiotherapy.[39] This analysis
showed that complete or partial response to BT conferred a 42%
reduction in disease progression and death after CD19 CAR-T therapy.[39] The best responses to BT were observed in patients treated with polatuzumab-containing chemotherapy regimens.[39]
The
decision to perform bridging therapy should be individualized at the
level of the single patient, taking into account several factors, such
as tumor burden, number and types of previous lines of therapy, and the
expected timing for CAR-T cell infusion compared to apheresis.[38]
Hubblings
and coworkers have evaluated the role of bridging therapy based on
radiotherapy in a group of 33 DLBCL patients undergoing CD19 CAR-T cell
therapy.[40] Bridging radiotherapy induced a
significant reduction of the diameter of lymphoma lesions, MTV, SUV
(standard uptake value), and LDH levels, all predictors of poor
outcomes post-CAR-T therapy outcomes.[40] Therefore, bridging radiotherapy may help to convert poor-risk LBCL patients into patients with better risk.[40]
Clonal Hematopoiesis and CAR-T Cell Therapy
A
recent study explored the potential risk caused by clonal hematopoiesis
(CH) in DLBCL patients undergoing treatment with anti-CD19 CAR-T cells.
CH is a condition of clonal expansion of hematopoietic stem/progenitor
cells bearing somatic gene mutations.[41] CH is
associated with an increased risk of hematological malignancies,
cytopenias, and nonhematological conditions such as atherosclerosis and
cardiovascular and cerebrovascular disease.[41]
Sinai
et al. explored 114 LBCL patients undergoing treatment with CAR-T cells
(105 with Axi-Cel and 9 with Tisa-Cel) for CH detected in 36.8% of
cases. The two most frequently mutated genes were PPMN1D (19/114) and TP53 (13/114).[42]
The incidence of therapy-related neurotoxicity was higher in
CH-positive than in CH-negative patients (45.2% vs 25.0%,
respectively). Higher neurological toxicities were preferentially
associated with DNMT3A, TET2, and ASXL1 genes.[41]
A higher incidence of grade ≥3 cytokine release syndrome was observed
in the CH-positive than in CH-negative patients (17.7% vs 4.2%,
respectively).[42] Finally, the 24-month cumulative
incidence of therapy-related myeloid neoplasms after CAR-T cell therapy
was higher in CH-positive than in CH-negative patients (19% vs 4.2%,
respectively).[42]
Other studies have reported myeloid malignancies development in LBCL patients undergoing anti-CD19 CAR-T cell treatments.[43-44]
The precise mechanism behind the increased risk of tMN has to be
elucidated and remains subject to speculation. It remains unclear
whether the particular immune dysregulation in patients after CAR-T
plays an important role or whether the occurrence of tMN is simply the
consequence of genetic damage induced by the precedent lines of therapy
in these mostly heavily pretreated patients.
Autologous Stem Cell Transplantation vs CAR-T Cell Treatment for DLBCL Patients in Partial Remission
Randomized
clinical trials have evaluated CAR-T treatment's safety and efficacy in
a subset of DLBCL patients with early treatment failure. These patients
were randomized to salvage therapy followed by auto-HSCT consolidation
in responding patients or directly to CAR-T treatment without
attempting salvage therapy. However, the efficacy of auto-HSCT and
CAR-T treatment was not comparatively evaluated in a population of
DLBCL patients achieving a partial response to initial standard
therapy. DLBCL patients achieving only partial response after
chemoimmunotherapy show durable remissions after autologous HSCT with a
5-year PFS of 41% and an OS of 51%-63%.[45]
In a
retrospective analysis carried out in a group of patients with DLBCL
patients in partial response post-salvage therapy, auto HSCT (266
patients) and CAR-T cell therapy with Axi-Cel (145 patients) gave
2-year PFS of 52% vs 42% and OS of 69% vs 47%, respectively.[46]
Therefore, this study showed a slightly longer PFS and OS in DLBCL
patients in partial response after salvage therapy treated with
auto-HSCT compared to CAR-T cell therapy.[46]
At variance with the findings of this study, Akhtar et al. performed a retrospective analysis on 125 older (≥65
years) DLBCL patients in partial response after salvage therapy
undergoing treatment with either auto-HSCT or CAR-T infusion: no
statistically significant differences between auto-HSCT and CAR-T
groups in 1-year OS (68% vs 72%, respectively) and 1-year PFS (56% vs
59%, respectively) were observed.[47] Furthermore,
patients in the CAR-T group showed a trend to lower non-relapse
mortality compared to those in the auto-HSCT group.[47]
According to these observations, the authors suggest that in older
patients with refractory/relapsed DLBCL patients achieving a partial
response to salvage chemotherapy, CAR-T treatment resulted in outcomes
comparable to auto-HSCT.[47]
Allogeneic HSCT and CAR-T Therapy after Auto-HSCT Failure in DLBCL
A
retrospective noncomparative registry study analyzed outcomes in 584
patients with DLBCL undergoing a reduced intensity allo-HSCT or CAR-T
therapy with Axi-Cel after a prior auto-HSCT failure. The 1-year
relapse, non-relapse mortality, overall survival, and progression-free
survival after auto-HSCT failure were: for CAR-T treatment, 39.5%,
4.8%, 73.4%, and 55.7%, respectively; for the allo-HSCT cohort, 26.2%,
20.0%, 65.6%, and 53.8%, respectively.[48] Therefore,
both CAR-T cell treatment and allo-HSCT can provide durable remissions
in a subset of DLBCL patients relapsing after auto-HSCT.[48]
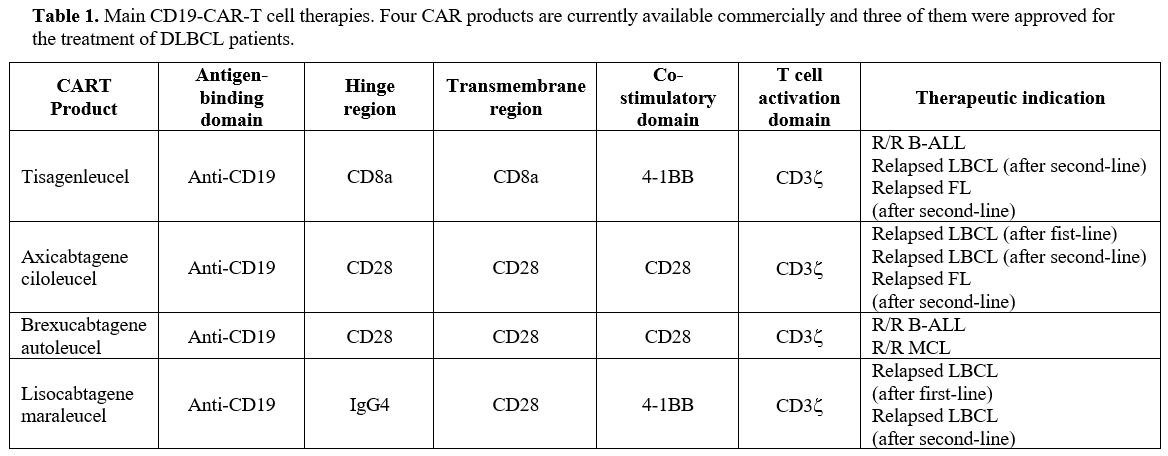 |
- Table 1. Main
CD19-CAR-T cell therapies. Four CAR products are currently available
commercially and three of them were approved for the treatment of
- DLBCL
patients.
|
Therapy of DLBCL after CAR-T Failure
Over
60% of LBCL patients ultimately progress or relapse following
CD19-CAR-T cell therapy. The treatment of patients relapsing after
CAR-T cell therapy failure is extremely challenging and largely
undefined.Tomas
et al. explored 182 LBCL patients experiencing disease recurrence or
progression after CAR-T therapy; 74% received anticancer treatment
post-CAR-T failure, with a mOS of 8 months.[49] Most
of these patients were treated with standard chemotherapy,
polatuzumab-based therapies, or lenalidomide-based therapies: no CRs
were observed in patients treated with conventional chemotherapy,
while ≥30% CRs were observed among patients treated with polatuzumab- or lenalidomide-based therapies.[49]
Factors associated with poor overall survival among patients treated
post-CAR-T failure were represented by pre-CAR-T bulky disease, lack of
response to CAR-T therapy, age >65 years, and elevated LDH at
post-CAR-T treatment: the presence of ≥2 of these factors was associated with lower OS compared to ≤1.[49]Another study retrospectively evaluated 83 patients with LBCL receiving an allo-HSC after anti-CD19 CAR-T cell therapy failure.[49]
The median number of lines of therapy between CAR-T infusion and
allo-HSCT was 1; low-intensity conditioning was used in 77% of cases,
and peripheral blood was the most common graft source; the most common
donor types were matched unrelated donor (39%), followed by
haploidentical (30%) and matched-related donor (26%).[50] One year OS, PFS, and GVHD were 59%, 45%, and 39%, respectively.[50] These findings concluded that allo-HSCT after CAR-T failure can provide durable remissions in a subset of patients.[50]
CAR-T Therapy in DLBCL: Prognostic Factors and Mechanisms of Relapse
Identifying
a subgroup of DLBCL patients who benefit from anti-CD19 CAR-T cell
therapy remains a key challenge. The clinical trials with Axi-Cel and
Tisa-Cel failed to identify clinical covariates predictive of efficacy.
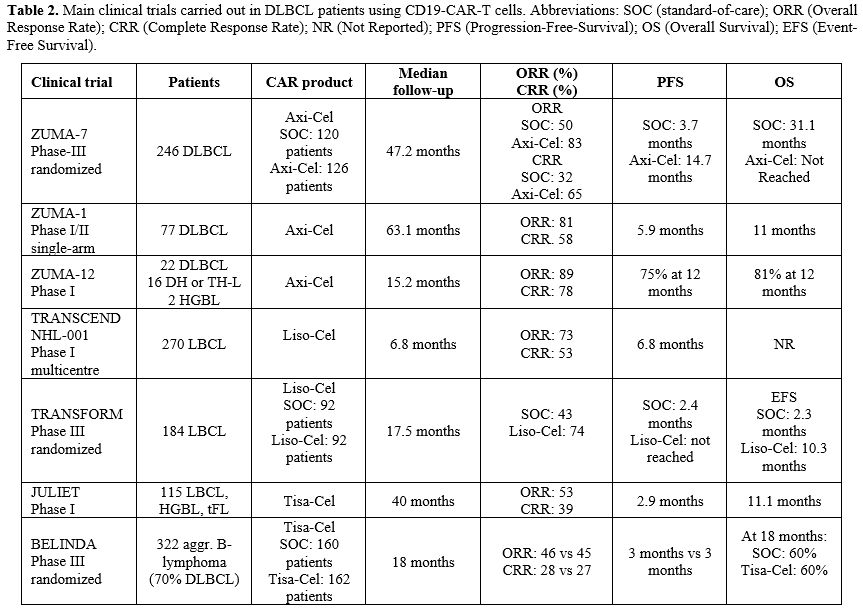 |
- Table 2. Main clinical
trials carried out in DLBCL patients using CD19-CAR-T cells.
Abbreviations: SOC (standard-of-care); ORR (Overall Response Rate);
- CRR
(Complete Response Rate); NR (Not Reported); PFS
(Progression-Free-Survival); OS (Overall Survival); EFS (Event-Free
Survival).
|
Clinical factors.
Vercellino and coworkers have investigated the predictive factors for
early progression after CAR-T cell therapy in 116 refractory/relapsed
LBCL patients; 55 of these patients failed treatment, and 49% of these
patients relapsed within the early months after CAR-T cell therapy and
therefore are early progressors.[51] Risk factors
identified for early progression at the time of diagnosis and at the
time of treatment are represented by extranodal site involvement (≥2 sites) and lymphoma tumor burden as measured by total metabolic tumor volume (TMTV) assessment and LDH levels.[51]
As
discussed above, the tumor burden is a major determinant of outcomes of
DLBCL patients at the moment of CAR-T cell therapy with Axi-Cel.[14-15]
In line with these observations, Nastoupil et al., in a retrospective
analysis, showed an association between achieving CR at 12 months after
Axi-Cel treatment and no need for bridging therapy.[13]
Since the need for bridging therapy reflects either a higher tumor
burden or a more rapidly progressive disease, it is evident why it
emerges as a negative prognostic factor.
Hirayama and coworkers
have explored some prognostic factors associated with durable responses
in patients with aggressive NHL (mostly
DLBCL and HGBL-DH or TH). These patients received
lymphodepletion with cyclophosphamide and fludarabine, followed by
CAR-T cell infusion.[52] This analysis identified
lower serum lactate dehydrogenase and a favorable cytokine profile
(defined as serum day 0 monocyte chemoattractant protein-1 (MCP-1) and
interleukin-7 (IL-7) above the median level) as serum biomarkers
associated with a better PFS.[52]
CAR-T cell
therapy is associated with two main early toxicities represented by
cytokine release syndrome and neurotoxicity; the frequency and severity
of these toxicities are partly associated with baseline disease and
patient characteristics. Both the Cumulative Illness Rating Scale
(CIRS) and the International Prognostic Index (IPI) are associated with
outcomes in DLBCL patients after CAR-T cell therapy.[53-54] A recent study used the CIRS to define a prognostic score predictive of outcomes of CAR-T cell therapy in DLBCL patients.[55] Particularly, a CIRS ≥3
in the respiratory, upper gastrointestinal, hepatic, or renal system,
defined as "severe 4", predicted shorter PFS and OS and a CRS of
grade ≥3.[55]
Therefore, a simplified CIRS-derived comorbidity index may predict
adverse outcomes in DLBCL patients undergoing CAR-T cell therapy.
CD19
antigen escape is one of the mechanisms of relapse observed in some
DLBCL patients relapsing after CAR-T cell therapy. In this context,
Plaks and coworkers explored [20] DLBCL patients
treated in the ZUMA-1 trial with Axi-Cel for CD19 expression at RNA and
protein level and for CD19 gene mutational status.[56]
30% of these patients showed a relapse characterized by negative/low
CD19 expression; the mechanism responsible for the generation of a
CD19-negative relapse seems to be related to indirect treatment-related
selection of tumor cells with low-very low CD19 protein expression in
the context of removal of antigen-positive tumor cells rather than
alternative splicing or CD19 mutation.[56]
Tumor-related genomics.
Typical tumor-related features, such as double- or triple-hit
translocations, activated B-cell-like, and cells of origin phenotype,
are not informative of outcomes in LBCL patients undergoing CAR-T cell
treatment. To identify tumor-related factors that could be associated
with response to CAR-T cell therapy, Shouval et al. have characterized
the mutational profile of 153 LBCL patients undergoing CD19-CAR-T cell
therapy; 37% of these patients displayed TP53 alterations (either mutations and/or copy number alterations): the 1-year OS of TP53-altered LBCL was 44% compared to 1-year OS of 76% among TP53WT patients.[57] Transcriptomic studies showed that TP53
alterations are associated with dysregulation of pathways associated
with CAR-T-cell cytotoxicity and reduced CD8 T-cell tumor infiltration.[57]
Jain
and coworkers have analyzed the genomic profile of 49 LBCL patients
undergoing CAR-T cell therapy by whole-genome sequencing.[58]
The analysis showed that the pre-treatment presence of complex
structural variants, APOBEC mutational signatures, and genomic damage
deriving from reactive oxygen species predict CAR-T resistance;
furthermore, the recurrent 3p21.31 chromosomal deletion englobing the RHOA tumor suppressor gene was markedly enriched in patients with failure to CAR-T cell therapy.[58]
Zhou
et al. have used low-pass whole genome sequencing of ct-DNA to explore
copy number alterations (CNAs) in pre-treatment plasma samples of 122
LBCL patients before CAR-T cell therapy.[59] A high
focal CNA score, denoting genomic instability, was the most significant
pre-treatment CNA associated with inferior 3-month CRR, PFS, and OS.[59]
Among the 34 unique focal CNAs observed in these patients, deletion at
10q23.2, determining the loss of the FAS death receptor, was most
significantly associated with poor outcomes.[59]
Other
studies have evaluated the residual tumor disease in DLBCL patients
undergoing CAR-T cell therapy using noninvasive monitoring for
treatment response and predicting disease relapse after therapy.
Routine surveillance by tumor imaging for DLBCL patients achieving
remission is of limited utility. In contrast, molecular disease
evaluation by immunoglobulin high-throughput sequencing from peripheral
blood provides a more sensitive strategy for surveillance. Molecular
disease can be detected in peripheral blood cells and plasma; molecular
disease detection often precedes PET/CT detection of relapse in
patients initially achieving remission.[60]
Frank
et al. have evaluated the role of monitoring circulating tumor DNA in
detecting relapse following CAR-T cell therapy with Axi-Cel. 69 LCBL
patients with a tumor clonotype were explored by analysis of ctDNA:
high pre-treatment ctDNA concentrations were associated with
progression after Axi-Cel infusion and development of CRS and immune
effector cell-associated neurotoxicity syndrome; 70% of patients with
durable response compared to 13% of progressing patients showed
non-detectable ctDNA one week after Axi-Cel infusions. At day 28,
patients with detectable ctDNA compared to those with undetectable
ctDNA had a PFS of 3 months vs not reached and an OS of 19 months vs
not reached; ctDNA was detected at or before radiographic relapse in
94% of patients, while 100% of durably responding patients had
undetectable ctDNA; in patients with radiographic PR or stable disease,
10% of those with concurrently undetectable ctDNA relapsed and 92% of
those with concurrently detectable ctDNA relapsed.[61]
A
recent study by Sworder et al. provided fundamental information on the
genomic mechanisms of resistance to CAR-T cell therapy observed in 138
relapsed/refractory LBCL patients undergoing treatment with Axi-Cel.[62]
In this study, a peculiar methodology was developed for the
simultaneous assessment of ctDNA, cell-free CAR19 (cfCAR19) retroviral
fragments (for evaluation of CAR-T cell expansion and functional
persistence in vivo after
their infusion), and cell-free T cell receptor rearrangements (cfTCR)
that enabled noninvasive profiling and integration of tumor dynamics
and of T cell expansion and TCR diversification in CAR19 patients.[62]
Baseline and dynamic ctDNA levels were prognostic for outcome: patients
experiencing disease progression had significantly higher pre-treatment
ctDNA levels; at 4 weeks post-infusion, patients that achieved a ctDNA
major molecular response showed significantly better outcomes.[62]
The analysis of cfCAR19 showed similar levels between patients
responding or not to CAR-T cell therapy, without any significant
difference between these two groups.[62] However,
cfTCR levels at 4 weeks after CAR19 infusion were higher in patients
with durable response than in patients with disease progression.[62]
The analysis of the mutational profile showed that mutations in several
genes are significantly associated with inferior event-free survival,
such as alterations of TMEM30A, IRF8, PAX5, TP53, and DXT1 genes; other mutations appeared in patients relapsing after CAR-T therapy, such as multiple CD19 alterations and PPM1D mutations.[62] Relapsing patients also displayed gene amplifications of PD-L1 or PD-L2.
These somatic mutations affect CAR-T cell therapy at various levels,
including CAR-T cell expansion, persistence, and tumor
microenvironment. Resistant DLBCL tumors may display either abundant
infiltrating CAR-T cells or low/absent CAR-T cells: tumors with high
infiltration demonstrate different microenvironmental and inflammatory
signatures compared to tumors with low CAR-T infiltration, thus
suggesting different mechanisms of resistance.[62]
Gene expression studies.
Several biological mechanisms contribute to the heterogeneity of DLBCL,
such as cell-of-origin subtypes, genomic alterations, and differences
in composition and activation of cellular elements present in the tumor
microenvironment.
Gene expression profiling (GEP) studies have
refined the molecular classification of DLBCL. GEP studies have
characterized the consistent heterogeneity in the lymphoma
microenvironment. In this context, Kotlov and coworkers have analyzed
the publicly available gene expression profiles of 4655 DLBCL patients;
using this approach, they have identified 25 functional gene expression
signatures (FGES) corresponding to
subtypes of the microenvironment, non-cellular components of the tumor
microenvironment, biological processes, and signaling pathways.[63]
According to these FGES, four types of lymphoma microenvironment were
identified: a germinal center-like LME1 (15%), with the presence
of FGES
of cell types present in germinal centers; a mesenchymal LME2 (33%),
due to the presence of stromal and extracellular matrix pathways; an
inflammatory LME3 (25%), due to the presence of FGES
associated with inflammatory cells and pathways; and a depleted LME4
(27%), due to the low presence of microenvironment-related FGES and to the presence of proliferation-related FGES.[63]
The four LME categories of DLBCLs are associated with specific genomic
alterations and distinct clinical outcomes: a better PFS and OS for
LME1 and LME2 than for LME3 and LME4.[63]
Steen
and coworkers have implemented a machine learning algorithm, termed Eco
Typer, to integrate transcriptomic deconvolution and single-cell RNA
sequencing to define states and ecosystems present in DLBCLs.[64]
B-cell states were defined by COO subtypes GBC and ABC and subdivided
into centrocytes, centroblasts, memory B cells, and plasmablasts. This
approach identified five different cell states of malignant B cell
differentiation associated with differences in prognosis.[64]
Several
studies have shown that the heterogeneous characteristics of the TME in
LBCL are associated with clinical responses to anti-CD19 CAR-T cell
therapy. Scholler et al. have explored the dynamic changes in TME
occurring in LBCL patients undergoing treatment with Axi-Cel in the
context of the ZUMA-1 trial.[18] In this analysis the
patients were subdivided into two groups, responders and
non-responders; responders showed an early and rapid increase of
cytotoxic T cell-related genes, such as CD8α, T cell growth factor
genes such as IL-15, interferon-γ-regulated immune checkpoint encoding
genes (CD274, CD276 and CTLA-4), myeloid-related genes and chemokines;
in non-responders, no increase in immune-related genes was observed,
except for pro-inflammatory chemokines such as CXCL10 and CXCL11.[18]
Immunohistochemical studies in a few patients have confirmed these
observations by gene expression analysis, showing higher T cell
densities among responders reflecting pre-treatment T cell density.[18]
The infiltration of TME with exhausted T cytotoxic lymphocytes observed
in non-responding patients correlated with poor CAR-T cell expansion in
blood. The pre-treatment quantification of tumor-infiltrating T cell
density by Immunoscore and of a panel of immune genes by Immunosign 21
positively correlated with overall survival after CAR-T cell therapy.[18]
Batlevi
and coworkers extensively characterized 49 DLBCL patients treated with
CD19-CAR-T cell therapy using whole exome sequencing performed on tumor
samples, defining the cell of origin, assessing double hit gene
signatures and the lymphoma microenvironment, analyzing gene expression
according to Kotlov et al.[63] In these patients, the
overall response at 3 months was 77.6%, with 59.2% CR and 18.4% PR, PFS
at 6 months was 49%; prognostic biomarkers to CAR-T therapy, such as
LDH levels, MTV, and SUV were confirmed.[65] The major findings of this study were that: PIK3CA amplification was associated with improved PFS; increased MHCII expression, associated with centrocyte-like phenotype,[66]
was higher in DLBCL patients with GCB phenotype responsive to
CD19-CAR-T therapy; DLBCL patients with GCB phenotype and with higher SMAD1
expression are usually responsive to CD19-CAR-T cell therapy;
germinal-center-like and mesenchymal LME subtypes exhibited increased
OS compared to those with inflamed and depleted LME subtypes.[65]
Haradhval
et al. used single-cell RNA sequencing to explore cellular dynamics
associated with response to CAR-T therapy for DLBCL using Axi-Cel or
Tisa-Cel.[66] Axi-Cel and Tisa-Cel, as discussed
above, differ for many characteristics related to differences in CAR
design (4-1BB vs. CD28 costimulatory domain, CD8 vs. CD28 transmembrane
domain for Tisa-Cel vs Axi-Cel, respectively), in vectors used for
their delivery, and in manufacturing processes (fresh vs frozen
apheresis products, activation by antibody-coated beads vs soluble
antibody and cytokines). This study showed that Tisa-Cel responses were
associated with the expansion of central-memory CD8 cell populations,
while Axi-Cel responders displayed more heterogeneous cell populations.[67]
Despite these differences in cell types associated with response, both
Axi-Cel and Tisa-Cel CAR-T cells displayed at day seven after infusion
a remarkable increase in the expression of genes related to cellular
proliferation and activation.[67] In Axi-Cel non-responders, a population of regulatory T-cells with CAR transcripts in the infusion product was expanded in vivo and could exert an immunosuppressive activity.[67]
Gene
expression studies have also contributed to understanding the
consistent heterogeneity of CAR-T products obtained from different
DLBCL patients. Deng and coworkers analyzed the cellular and molecular
features of CAR-T infusion cell products prepared using Axi-Cel to
identify transcriptomic (by single-cell RNA sequencing) features
associated with efficacy and toxicity in 24 LBCL patients. 50% of these
patients had progressive disease, 4% a partial response and 38% a
complete response.[68] Patients achieving a complete
response at 3 months had 3-fold higher frequencies of CD8 T cells
expressing memory signatures compared to patients with partial
responses or progressive disease.68 Molecular responses at day 8
post-infusion were significantly associated with the clinical response
signature of CD8 T-cell exhaustion associated with a poor molecular
response.[67] Finally, a rare cell population with monocytic features was associated with ICANS occurrence.[68]
Hematologic toxicity.
Hematologic toxicity is frequently observed in DLBCL patients
undergoing CAR-T cell therapy. In 258 patients receiving CD19-CAR-T
cell therapy, profound neutropenia was observed in 72% of cases and
prolonged neutropenia in 64% of patients; in these patients, predictive
biomarkers of hematologic toxicity were baseline cytopenia
(thrombocytopenia) and inflammatory state (hyperferritinemia). [69]
According to these observations, a predictive model for hematologic
toxicity (CAR-HEMATOTOX) was generated based on markers associated with
hematopoietic reserve, such as platelet count, absolute neutrophil
count, and hemoglobin level, and baseline inflammation markers, such as
C-reactive protein and ferritin.[68] A high CAR-HEMATOTOX score predicted a longer neutropenia duration and a higher incidence of thrombocytopenia and anemia.[69]
Infectious
complications represent the key determinant of non-relapse mortality
after CAR-T cells. They are favored not only by neutropenia but also by
the immune disturbance caused by the T-CAR cells. The temporal
distribution of these risk factors shapes different infection patterns
early versus late post-CAR-T-cell infusion. Furthermore, due to the
expression of their targets on B lineage cells at different stages of
differentiation, CD19 and B-cell maturation antigen (BCMA), CAR-T cells
induce distinct immune deficits that could require different prevention
strategies. Infection incidence is the highest during the first-month
post-infusion and decreases afterward. However, infections remain
relatively common even a year after infusion. Bacterial infections
predominate early after CD19, while an equal distribution between
bacterial and viral causes is seen after BCMA CAR-T-cell therapy, and
fungal infections are universally rare. Cytomegalovirus (CMV) and other
herpesviruses are increasingly reported.[70]
Toxicity associated with the immune effector response.
CAR T cells can result in significant toxicities directly associated
with the induction of powerful immune effector responses. Cytokine
release syndrome (CRS), neurotoxicity,[71-73] or more rarely cardiotoxicity[74]
represent the most frequent manifestations of this toxicity, which is
in relationship with the immunological effects of CAR T cells.[71-73]
Toxicities may be related both to the activation of T cells with the
release of high levels of cytokines and the interaction between CAR and
CAR-target antigens expressed on non-malignant cells. Cytokine release
syndrome (CRS) and immune effector cell-associated neurotoxicity
syndrome (ICANS) are well-known inflammatory side effects of CAR T-cell
therapy.[71-73] CRS typically presents with
constitutional symptoms such as fever, myalgia, and arthralgia or
constitutional symptoms such as rigors, fatigue, malaise, and anorexia.
However, rapid progression to hemodynamic instability, respiratory
failure, organ dysfunction, shock, and hemophagocytic
lymphohistiocytosis has also been reported.[71,72]
Depending on the product, CRS typically occurs within 1–2 weeks of
CART-cell infusion but can occur as early as a few hours post-infusion.[71,72]
CRS has a reported incidence between 37% and 93% across different
studies. Factors associated with CRS include the product type, tumor
burden, disease indication, elevation in baseline inflammatory markers
(i.e., ferritin, C-reactive protein), and concomitant infection. In
cases where CRS develops early and is higher grade, severe ICANS is
more likely. This occurrence may be partly associated with a high dose
of CART cells or usually robust and rapid CART-cell expansion. Notably,
ICANS can also infrequently develop in the absence of CRS. ICANS
presents as dysgraphia, word-finding difficulties, headache, tremor,
confusion, somnolence, expressive aphasia, seizure, and coma. Rarely,
death from cerebral edema has been reported (1%–2% estimated
incidence).[71-73] Rubin et al.[73]
reported that among 100 treated cases, the most commonly occurring
neurological symptoms were encephalopathy (57%), headache (42%), tremor
(38%), aphasia (35%) and focal weakness (11%). Focal neurological
deficits were frequently observed after chimeric antigen receptor
T-cell therapy and are associated with regional EEG abnormalities,
FDG-PET hypometabolism, and elevated velocities on transcranial Doppler
ultrasound. In contrast, structural imaging was typically normal, ICANS
may co-occur with CRS or immediately following CRS. Neurologic signs
and symptoms typically begin 3–6 days after CART-cell infusion and peak
around day 7 or 8, with complete symptom resolution by days 14–21. The
rate and grade of ICANS toxicity varies greatly among CAR T-cell
products. ICANS occurs in 20%–70% of patients treated with CD19 CART
cells.[71-73] Inflammatory cytokines released by
macrophages, specifically IL-6 and IL-1, have been widely identified as
critical components in the pathogenesis of CRS and ICANS, respectively.[71-72]
Elevated serum levels of IL-6 are one of the most correlated findings
with CRS Activated CART cells release IFN-γ, TNF-α, and
granulocyte-macrophage colony-stimulating factor (GM-CSF) to induce
tumor cell cytolysis. However, these cytokines also activate
macrophages, which release IL-6 and TNF-α.[71,72]
Cardiotoxicity
hits about 10% of patients and manifests as cardiomyopathy, heart
failure, arrhythmias, and myocardial infarction. Patients undergoing
T-cell therapies should be screened for cardiovascular conditions that
may not be able to withstand the hemodynamic perturbations imposed by
CRS.[74]
Brammer JE et al.[75]
report 102 CAR-T-treated patients; of them, 90 were identified as
treated with single-agent therapy, of which 88.9% developed toxicity
(80 CRS, 41 neurotoxicity, and 17 cardiotoxicity), including 28.9% with
high-grade (≥3) events. The most common manifestations were hypotension
at 96.6% and fever at 94.8%. Among patients with cardiac events, there
was a non-significant trend toward a higher prevalence of concurrent or
preceding high-grade (≥3) CRS. 50.0% required tocilizumab or
corticosteroids. The median time to toxicity was 3 days; high-grade CRS
development was associated with cardiac and neurotoxicity. In
multivariable regression, accounting for disease severity and
traditional predictors of disease response, moderate (maximum grade 2)
CRS development was associated with higher complete response at 1 year
(HR: 2.34; p=0.07), and longer PFS (HR: 0.41; p=0.02, in landmark
analysis), and OS (HR: 0.43; p=0.03). Among those with CRS, relative
blood pressure (HR: 2.25; p=0.004), respectively, was also associated
with improved PFS. No difference in disease outcomes or maximum
toxicity grade (CRS, neurotoxicity, or cardiotoxicity) was observed
based on the presence or absence of early CRS-directed therapies.
Therefore, moderate toxicity predicts a good outcome. Nonhematological
toxicity also depends on tumor burden; patients with DLBCL without
residual lymphoma at the time of CD19 CAR T-cell therapy show low
toxicity and excellent outcomes.[76]
Anti-inflammatory therapy, specifically targeting IL6, has become the cornerstone of CRS management.[77,78]
Tocilizumab, a humanized IgG1k anti-IL-6R antibody, binds to both
soluble and membrane-bound IL-6R, blocking the downstream signal
transduction pathways implicated in CRS. It is currently the only
anti-IL6 therapy approved by the FDA for treating severe or
life-threatening CAR T cell–induced CRS.[77,78] While
it is approved for severe or life-threatening CRS, current guidelines
and product information recommend initiating tocilizumab for treating
grade ≥ 2 or grade 1 CRS in patients at high risk of early and severe
CRS or those whose symptoms persist greater than 24 h. For severe
(grade ≥ 3) or refractory CRS, the addition of steroids is recommended.[78] A recent analysis[77]
of the ZUMA-1 study of axicabtagene-ciloleucel (axi-cel) shows
prophylactic corticosteroids and earlier corticosteroid and/or
tocilizumab intervention resulted in no grade 3 or higher CRS, a low
rate of grade 3 or higher NEs and high response rates in this study
population. 95% and 80% objective and complete response rates,
respectively, for patients who received prophylactic steroids
(dexamethasone 10 mg on day 0 (pre-infusion), day 1 and 2) or early
addition of steroids to tocilizumab for CRS[77]
Although tocilizumab and steroids are first-line interventions for
prevention and treatment of CRS and ICANS, with high response, data for
outlining the treatment of refractory CRS and/or ICANS, are lacking.
However, there is an emerging use of anakinra and an improvement of
mitigation strategies and supportive care measures to ameliorate
outcomes of patients who develop these refractory toxicities.[79]
Conclusions
The
studies carried out in the last ten years have clearly supported and
defined a role for CD19-CAR-T cells in the therapy of DLBCL patients
with refractory/relapsed disease. This therapeutic role was established
for patients with refractory disease and early relapse. For DLBCL
patients with partial response after salvage therapy, CD19-CAR-T cells
also have shown consistent therapeutic activity, but additional studies
are required to compare their efficacy to auto-HSCT carefully.
Similarly, CD19-CAR-T cells have shown efficacy in treating high-risk
DLBCL patients in first-line, but additional studies are required to
assess their efficacy compared to standard treatments. At present,
there are no data suggesting which of the four CAR products
commercially available for patients with B cell lymphomas: Axicabtagene
ciloleucel (Axi-Cel), Brexacubtagene autoleucel (Brexa-Cel),
Lisocabtagene macrolevel (Liso-Cel) and Tisagenlecleucel (Tisa-Cel),
could be the best in term of efficacy and side effects. The results of
the contemporary publication in the NEJM of two randomized trials
employing one the Tisa-Cel,[28] the other the Axi-Cel,[8]
could induce to think a superior efficacy of Axi-Cel; however, the
criteria for enrollment of patients are different, so no comparison is
possible.[80,81] Another problem is the cost of this
therapy. Utilization of CAR T cell therapy is very expensive, but
papers comparing the Cost-Effectiveness ratio of the different products
are rare.[82] However, the best standard salvage care requires fewer resources in comparison with CAR-T.[83]Although
the efficacy of CD19-CAR-T cell therapy in refractory/relapsed DLBCL
patients was well documented, only about 40% of relapsed/refractory
DLBCL patients are responding to this treatment, and the remaining are
refractory or rapidly relapse. Several strategies seem to be required
to improve the outcomes of these patients: (i) decrease tumor burden
using novel bridging therapies that include chemoimmunotherapy or
radiation therapy prior to CAR-T cell therapy; (ii) use CAR-T cells
engineered with double targeting activity, such as CD19/CD20 or
CD19/CD22; (iii) optimize CAR-T cell expansion and persistence by
increasing the number of infusions or the dosing of infused cells; (iv)
modify the therapy in patients who do not show an adequate clearing of
lymphoma cells following CAR-T cell infusion; (v) define alternative
treatments in DLBCL patients displaying genomic alterations predicting
resistance to CAR-T cell therapy. References
- Wei W, Wang D, Chen X, Liang D, Zou L, Zhao X.
Chimeric antigen receptor T-cell therapy for T-ALL and AML. Front Oncol
2022; 12: 967754. https://doi.org/10.3389/fonc.2022.967754 PMid:36523990 PMCid:PMC9745195
- Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J 2021; 11: 69. https://doi.org/10.1038/s41408-021-00459-7 PMid:33824268 PMCid:PMC8024391
- Sato
S, Ono N, Steeber DA, Pisetsky DS, Tedder TF. CD19 regulates B
lymphocyte signaling thresholds critical for the development of B-1
lineage cells and autoimmunity. J Immunol 1996; 157(10): 4371-4378. https://doi.org/10.4049/jimmunol.157.10.4371 PMid:8906812
- Sato
S, Steeber DA, Jansen PJ, et al. CD19 expression levels regulate B
lymphocyte development: human CD19 restores normal function in mice
lacking endogenous CD19. J Immunol 1997; 158(10): 4662-4669. https://doi.org/10.4049/jimmunol.158.10.4662 PMid:9144478
- Tedder
TF, Inaoki M, Sato S. The CD19-CD21 complex regulates signal
transduction thresholds governing humoral immunity and autoimmunity.
Immunity 1997; 6(2): 107-118. https://doi.org/10.1016/S1074-7613(00)80418-5 PMid:9047233
- Shoham
T, Rajapaska R, Boucheix C, Rubinstein E, Poe JC, Tedder TF, Levy S.
The tetraspanin CD81 regulates the expression of CD19 during B cell
development in a postendoplasmic reticulum compartment. J Immunol 2003;
171(8): 4062-4072. https://doi.org/10.4049/jimmunol.171.8.4062 PMid:14530327
- Sehn LH, Salles G. Diffuse large B-cell lymphoma. N Engl J Med 2012; 384: 842- 858. https://doi.org/10.1056/NEJMra2027612 PMid:33657296 PMCid:PMC8377611
- Locke
FL, Miklos DB, Jacobson CA, Perales MA, Kersten MJ, Oluwole OO, Ghobadi
A, Papoport AP, et al. Axicabtagene ciloleucel as second-line therapy
for large B-cell lymphoma. N Engl J Med 2022; 386(7): 640-654. https://doi.org/10.1056/NEJMoa2116133 PMid:34891224
- Westin
JR, Oluwole OO, Kersten MJ, Miklos DB, Perales MA, Ghobadi A, Rapoport
AP, Sureda A, Jacobson CA, et al. Survival with axicabtagene ciloleucel
in large B-cell lymphoma. N Engl J Med 2023, 2023 Jul 13;389(2):148-157
https://doi.org/10.1056/NEJMoa2301665 PMid:37272527
- Westin
JR, Locke FL, Dickinson M, Ghobadi A, Elsawy M, van Meerten T, Miklos
DB, Ulrickson ML, Perales MA, et al. Safety and efficacy of
axicabtagene ciloleucel versus standard of care patients 65 years of
age or older with relapsed/refractory large B-cell lymphoma. Clin
Cancer Res 2023; 29(10): 1894-1905. https://doi.org/10.1158/1078-0432.CCR-22-3136 PMid:36999993 PMCid:PMC10183830
- Locke
FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, Lin Y,
Braunscweig I, Hill BT, Timmerman JM, et al. Long-term safety and
activity of axicabtagene ciloleucel in refractory large B-cell lymphoma
(ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol
2019; 20(1): 31-42. https://doi.org/10.1016/S1470-2045(18)30864-7 PMid:30518502
- Neelapu
SS, Jacobson, C.A.; Ghobadi A.; Miklos, D.B.; Lekakis, L.J.; Oluwole
OO, Lin Y, Braunschweig I, Hill BT, Timmerman JM, et al. Five-year
follow-up of ZUMA-1 supports the curative potential of axicabtagene
ciloleucel in refractory large B-cell lymphoma. Blood 2023; 141(19):
2307-2315. https://doi.org/10.1182/blood.2022018893 PMid:36821768
- Neelapu
SS, Locke FL, Barlett NL, Lekakis LJ, Reagan PM, Miklos DB, Jacobson
CA, Braunschweig I, Oluwole OO, Siddiqi T, et al. Comparison of 2-year
outcomes with CAR T cells (ZUMA-1) vs salvage chemotherapy in
refractory large B-bell lymphoma. Blood Adv 2021; 5: 4149-4155. https://doi.org/10.1182/bloodadvances.2020003848 PMid:34478487 PMCid:PMC8945634
- Nastoupil
L.J, Jain MD, Feng L, Spiegel JY, Ghobadi A, Lin Y, Dahiya S, Lunning
M, Lekakis L, Reagan P, et al. Standard-of-care axicabtagene ciloleucel
for relapsed or refractory large B-cell lymphoma: results from the US
lymphoma CAR T consortium. J Clin Oncol 2020; 38(27): 3119-3128. https://doi.org/10.1200/JCO.19.02104 PMid:32401634 PMCid:PMC7499611
- Dean
EA, Mhaskar RS, Lu H, Mousa, MS, Krivenko GS, Lazaryan A, Bachmeier CA,
Chavez JC, Nishihori T, Davila Ml, et al. High metabolic tumor volume
is associated with decreased efficacy of axicabtagene ciloleucel in
large B-cell lymphoma. Blood Adv 2020; 4: 3268-3274. https://doi.org/10.1182/bloodadvances.2020001900 PMid:32702097 PMCid:PMC7391155
- Locke
FL, Rossi JM, Neelapu SS, Jacobson CA, Miklos DB, Ghobadi A, Oluwole
OO, Reagan PM, Lekakis LJ, Lin Y, et al. Tumor burden, inflammation,
and product attributes determine outcomes of axicabtagene ciloleucel in
large B-cell lymphoma. Blood Adv 2020; 4: 4898-4908. https://doi.org/10.1182/bloodadvances.2020002394 PMid:33035333 PMCid:PMC7556133
- Jain
MD, Zhao H, Wang X, Atkins R, Menges M, Reid K, Spitler K, Faramand R,
Bachmeier C, Dean EA, Cao B, et al. Tumor interferon signaling and
suppressive myeloid cells are associated with CAR T-cell failure in
large B-cell lymphoma. Blood 2023; 137(19): 2621-2633. https://doi.org/10.1182/blood.2020007445 PMid:33512407 PMCid:PMC8120145
- Scholler
N, Perbopst R, Locke FL, Jain MD, Turcan S, Danan C, Chang EC, Neelapu
SS, Miklos DB, Jacobson CA, et al. Tumor immune contexture is a
determinant of anti-CD19 CAR T cell efficacy in large B cell lymphoma.
Nat Med 2022; 28(9): 1872-1882. https://doi.org/10.1038/s41591-022-01916-x PMid:36038629 PMCid:PMC9499856
- Abramson
JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Amason J, Mehta A, Purev
E, Maloney DG, et al. Lisocabtagene maraleucel for patients with
relapsed or refractory large B-cell lymphomas (TRASCEND NHL 001): a
multicentre seamless design study. Lancet 2020; 396(10254): 839-852. https://doi.org/10.1016/S0140-6736(20)31366-0 PMid:32888407
- Kamdar
M, Solomon SR, Arnason J, Johnston PB, Glass B, Bachanova V, Ibrahimi
S, Mielke S, Mutsaer P. Lisocabtagene maraleucel versus standard of
care with salvage chemotherapy followed by autologous stem cell
transplantation as second-line treatment in patients with relapsed or
refractory large B-cell lymphoma (TRANSFORM): results from an interim
analysis of an open-label, randomized, phase 3 trial. Lancet 2022;
399(10343): 2294-2308. https://doi.org/10.1016/S0140-6736(22)00662-6 PMid:35717989
- Abramson
JS, Solomon SR, Arnason J, Johnston PB, Glass B, Bachanova V, Ibrahimi
S, Mielke S, Mulsaers P. Lisocabtagene maraleucel as second-line
therapy for large B-cell lymphoma: primary analysis of the phase 3
TRANSFORM study. Blood 2023; 141(14): 1675-1684. https://doi.org/10.1182/blood.2022018730 PMid:36542826
- Abramson
JS, Johnston PB, Kamdar M, Ibrahimi S, Isutzu K, Arnason J, Glass B,
Mutsaers P, Lunning M, Braverman J. Health-related quality of life with
licabtagene maraleucel vs standard of care in relapsed or refractory
LBCL. Blood Adv 2022, 6(23): 5969-5979. https://doi.org/10.1182/bloodadvances.2022008106 PMid:36149968 PMCid:PMC9713278
- Olson
NE, Ragan SP, Reiss DJ, Thorpe J, Kim Y, Abramson JS, McCoy C, Newwali
KJ, Fox BA. Exploration of tumor biopsy gene signatures to understand
the role of the tumor microenvironment in outcomes to lisocabtaegne
maraleucel. Mol Cancer Ther 2023; 22: 406-418. https://doi.org/10.1158/1535-7163.MCT-21-0506 PMid:36595660 PMCid:PMC9978882
- Lenz
G, Wright G, Dave SS, Xiao W, Powell J, Zhao H, Xu W, Tan B,
Goldschmidt N, Iqbal J, et al. Stromal gene signatures in large-B-cell
lymphomas. N Engl J Med 2008; 359(22): 2313-2323. https://doi.org/10.1056/NEJMoa0802885 PMid:19038878 PMCid:PMC9103713
- Schuster
SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuink JP, et al.
Tisagenleceucel in adult relapse or refractory diffuse large B-cell
lymphoma. N Engl J Med 2019; 380: 45-56. https://doi.org/10.1056/NEJMoa1804980 PMid:30501490
- Schuster
SJ, Tam CS, Borchmann P, Worel N, McGuirk JP, Holte H, et al. Long-term
clinical outcomes of tisagenlecleucel in patients with relapsed or
refractory aggressive B-cell lymphoma (JULIET): a multicentre, open
label, single-arm, phase 2 study. Lancet Oncol 2021; 22(10): 1403-1415.
https://doi.org/10.1016/S1470-2045(21)00375-2 PMid:34516954
- Maziarz
RT, Zhang J, Yang H, Chai X, Yuan C, Schwartz E, Jakovach M,
Martinez-Prieto M, Agarwal A, Degtyarev E, et al. Indirect comparison
of tisagenlecleucel and historical treatments for relapsed/refractory
diffuse large B-cell lymphoma. Blood Adv 2022; 6: 2536-2542. https://doi.org/10.1182/bloodadvances.2021006280 PMid:35030634 PMCid:PMC9043930
- Bishop
MR, Dickinson M, Purtill D, Barba P, Santoro A, Hamad N, Kato K, Sureda
A, Greil R, Thieblemont C. Second-line tisagenlecleucel or standard
care in aggressive B-cell lymphoma. N Engl J Med 2022, 386(7): 629-639.
https://doi.org/10.1056/NEJMoa2116596 PMid:34904798
- Bachy
E, Le Gouill S, Di Blasi R, Sesques P, Manson G, Cartron G, Beauvais D,
Roulin L, Gros FX, et al. A real-world comparison of tisagenlecleucel
and axicabtagene ciloleucel CAR T cells in relapsed or refractory
diffuse large B cell lymphoma. Nature Med 2022; 28: 2145-2154. https://doi.org/10.1038/s41591-022-01969-y PMid:36138152 PMCid:PMC9556323
- Jaeger
U, Tam CS, Borchmann P, McGuirk JP, Johansen M, Waller EK, Jaglowski S,
Andreadis C, Foley SR, Westing JR, et al. Long-term safety for patients
with tisagenlecleucel-treated relapsed/refractory diffuse large B-cell
lymphoma. Blood Adv 2022; 6(16): 4816-4820. https://doi.org/10.1182/bloodadvances.2021006193 PMid:35687492 PMCid:PMC9631665
- Jaeger
U, Worel N, McGuirk JP, Riedell PA, Fleury I, Du Y, Han X, Pearson D,
Redondo S, Waller EK. Safety and efficacy of tisagenleucleucel plus
pembrolizumab in patients with r/r DLBCL: phase 1b PORTIA study
results. Blood Adv 2023; 7(11): 2283-2286. https://doi.org/10.1182/bloodadvances.2022007779 PMid:36044388 PMCid:PMC10225880
- Neelapu
SS, Dickinson M, Munoz J, Ulrickson ML, Thieblemont C, Oluwole OO,
Herrera AF, Ujjani CS, Lin Y, Riedell PA, et al. Axicabtagene
ciloleucel as first-line therapy in high-risk large B-cell lymphoma:
the phase 2 ZUMA-12 trial: Nat Med 2022; 29: 735-742. https://doi.org/10.1038/s41591-022-01731-4 PMid:35314842 PMCid:PMC9018426
- Westin
J, Jacobson CA, Chavez JC, Sureda A, Morschhauser F, Glab B, Dickinson
M,Davies A, Flinn IW, Maloney DG, Chamuleau M, et al. ZUMA-23: a
global, phase 3, randomized controlled study of axicabtagene ciloleucel
versus standard of care as first-line therapy in patients with
high-risk large B-cell lymphoma. J Clin Oncol 2023; 142 (suppl 16): TPS
7578. https://doi.org/10.1097/01.HS9.0000975996.32786.92 PMCid:PMC10429721
- Roddie
C, Lekakis LJ, Marzolini M, Ramakrishnan A, Zhang Y, Hu Y,
Peddoreddigari V, Khokhafr N, Chen R, Basilico S, et al. Dual targeting
of CD19 and CD22 with bistronic CAR-T cells in patients with
relapsed/refractory large B-cell lymphoma. Blood 2023; 141: 2470-2482. https://doi.org/10.1182/blood.2022018598 PMid:36821767
- Tong
C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, Han X, Ti D, Dai H, Wang C, et
al. Optimized tandem CD19/CD20 CAR-engineered T cells in
refractory/relapsed B-cell lymphoma. Blood 2020; 136(14): 1632-1644. https://doi.org/10.1182/blood.2020005278 PMid:32556247 PMCid:PMC7596761
- Zhang
Y, Wang Y, Liu Y, Tong C, Wang C, Guo Y, Ti D, Yang Q, Qiao S, Wu Z,
Han W. Long-term activity of tandem CD19/CD20 CART Therapy in relapsed,
refractory B-cell lymphoma: a single-arm, phase 1-2 trial. Leukemia
2022; 36(11): 189-196. https://doi.org/10.1038/s41375-021-01345-8 PMid:34272481 PMCid:PMC8727291
- Interim
analysis of a phase II study of administered fresh bispecific
anti-CD20/CD19 CAR T-cell therapy-Zamtocabtagene Autocel (zamto-cel)
for relapsed/refractory (R/R) DLBCL (DAY II USA NCT04792489).
- Bhaskar
ST, Dholaria BR, Sengsayadeth SM, Savani BN, Oluwole OO. Role of
bridging therapy during chimeric antigen receptor T cell therapy.
eJHaem 2022; 3(suppl1): 39-45. https://doi.org/10.1002/jha2.335 PMid:35844303 PMCid:PMC9175845
- Roddie
C, Neill L, Osborne W, Lyengar S, Thilouli E, Irvine D, Chaganti S,
Besley C, Bloor A, Joens C, et al. Effective bridging therapy can
improve CD19 CAR-T outcomes while maintaining safety in patients with
large B-cell lymphoma. Blood Adv 2023; 7: 2872-2880. https://doi.org/10.1182/bloodadvances.2022009019 PMid:36724512 PMCid:PMC10300297
- Hubbeling
H, Silverman EA, Michaud L, Tomas AA, Shouval R, Flynn J, Devlin S,
Wiejtunga NA, Tringale KR, Batlevi C, et al. Bridging radiation rapidly
and effectively cytoreduces high-risk relapsed/refractory aggressive B
cell lymphomas prior to chimeric antigen receptor T cell therapy.
Tranplant Cell Ther 2023; https://doi.org/10.1016/j.jtct.2022.12.021 PMid:36587744
- Mangaonkar
AA, Patnaik MM. Clonal hematopoiesis of indetermeninate potential and
clonal cytopenias of undeterminated significance: 2023 update on
clinical associations and management recommendations. Am J Hematol
2023; 98(6): 951-964. https://doi.org/10.1002/ajh.26915 PMid:36938794
- Saini
NY, Swoboda DM, Greenbaum U, Ma J, Patel RD, Devashish K, Das K, Tanner
MR, Starti P, Nair R, et al. Clonal hematopoiesis is associated with
increased risk of severe neurotoxicity in axicabtagene ciloleucel
therapy of large B-cell lymphoma. Blood Cnecr Discov 2022; 3: 385-393. https://doi.org/10.1158/2643-3230.BCD-21-0177 PMid:35533245 PMCid:PMC9445749
- Eder
LN, Mrtinovic D, Mazzeo P, Ganster C, Hasenkamp J, Thomson J, Trummer
A, Haase D, Wulf G. Fata progression of muated TP53-associated clonal
hematopoiesis following anti-CD19 CAR-T cell therapy. Curr Oncol 2023;
30: 1146-1150. https://doi.org/10.3390/curroncol30010087 PMid:36661736 PMCid:PMC9858310
- Accorsi
Buttini E, Farina M, Lorenzi L, Polverelli N, Radici V, Morello E,
Colnaghi F, Almici C, Ferrari E, et al. High risk-myelodysplastic
syndrome following CAR T-cell therapy in a patient with relapsed
diffuse large B cell lymphoma: a care report and literature review.
Front Oncol 2023; 13: 1036455. https://doi.org/10.3389/fonc.2023.1036455 PMid:36741006 PMCid:PMC9897055
- Shah
NN, Ahn KW, Litovich C, He Y, Sauter C, Fenske TS, Hamadani M. Is
autologous transplant in relapsed DLBCL patients achieving only a PET+
PR appropriate in the CAR T-cell era? Blood 2021; 137: 1416-1423. https://doi.org/10.1182/blood.2020007939 PMid:33120429 PMCid:PMC7955408
- Shadman
M, Pasquini M, Ahn KW, Chen Y, Turtle CJ, Hematti P, Chen JB, Khimani
F, Ganguly S, Merrymar RW, et al. Autologous transplant vs chimeric
antigen receptor T-cell therapy for relapsed DLBCL in partial
remission. Blood 2022; 139: 1330-1339. https://doi.org/10.1182/blood.2021013289 PMid:34570879 PMCid:PMC8900276
- Akhtar
OS, Cao B, Wang X, Torka P, Al-Jamayli M, Locke FL, Freeman CL. CAR
T-cell therapy has comparable efficacy to autologous transplantation in
older adults with DLBCL in partial response. Blood Adv 2013; in press.
- Hamadani
M, Gopal AK, Pasquini M, Kim S, Qiu X, Ahmed S, Lazaryan A, Raj Bhatt
V, Daly A, Lulla P, et al. Allogeneic transplant and CAR-T therapy
after autologous transplant failure in DLBCL: a noncomparative cohort
analysis. Blood Adv 6(2): 486-494. https://doi.org/10.1182/bloodadvances.2021005788 PMid:34673903 PMCid:PMC8791562
- Zurko
J, Ramdial J, Shadman M, Ahmed S, Szabo A, Iovino L, Tomas AA, Sauter
C, Perales MA, Shah NN, et al. Allogeneic transplant following
CARt-cell therapy for large B-cell lymphoma. Haematologica 2023;
108(1): 98-109. https://doi.org/10.3324/haematol.2022.281242 PMid:35833303 PMCid:PMC9827150
- Tomas
AA, Fein JA, Fried S, Flynn JR, Devlin SM, Fingrut WB, Anagfnostou T,
Alperovich A, Shah N, Fraint E, et al. Outcomes of first therapy after
CD19-CAR-T treatment failure in large B-cell lymphoma. Leukemia 2023;
37(1): 154-163. https://doi.org/10.1038/s41375-022-01739-2 PMid:36335261 PMCid:PMC9892211
- Vercellino
L, Di Blasi R, Kanoun S, Tessoulin B, Rossi C, D'Aveny-Piney M, Obéric
L, Bodet-Milin C, Bories P, Olivier P, et al. Predictive factors of
early progression after CAR T-cell therapy in relapsed/refractory
diffuse large B-cell lymphoma. Blood Adv 2020; 5(22): 5607-5676. https://doi.org/10.1182/bloodadvances.2020003001 PMid:33180899 PMCid:PMC7686887
- Hirayama
AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, Li D, Cherian S,
Chen X, Pender BS, et al. The response to lymphodepletion impacts PFS
in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR
T cells. Blood 2019; 133(17): 1876-1887. https://doi.org/10.1182/blood-2018-11-887067 PMid:30782611 PMCid:PMC6484391
- Kittai
AS, Huang Y, Gordon M, Denlinger N, Mian A, Fritzgerald L, Bishop J,
Nagle S, Stephens DB, Jaglowski S, et al. Comorbidities predict
inferior survival in aptients receiving chimeric antigen recptor T cell
therapy for diffuse large B cell lymphoma : a mlticenter analysis.
Transplant Cell Ther 2021; 27: 46-52. https://doi.org/10.1016/j.bbmt.2020.09.028 PMid:33002640
- Garcia-Recio
M, Wudhikarn K, Pennisi M, Alonso-Trillo R, Flynn J, Shouval R, Afuye
AO, Silberberg ML, Batlevi CW, Dahi et al. The international prognostic
index is associated with outcomes in diffuse large B cell lymphoma
after chimeric antigen receptor T cell therapy. Transplant Cell Ther
2021; 27: 233-240. https://doi.org/10.1016/j.jtct.2020.10.022 PMid:33781518 PMCid:PMC8010220
- Shouse
G, Kaempf A, Gordon MJ, Artz A, Yashar D, Sigmung AM, Smilnak G, Bair
S, Mian A, Fitzgerald L, et al. A validated comorbidity index predicts
outcomes of CAR T-cell therapy in patients with diffuse large B cell
lymphoma. Blood Adv 2023; in press. https://doi.org/10.1182/bloodadvances.2022009309 PMid:36735393 PMCid:PMC10362276
- Plaks
V, Rossi JM, Chou J, Wang L, Poddar S, Han G, Wang Z, Kuang SQ, Chu F,
Davis RE, et al. CD19 target evasion as a mechanism of relapse in large
B-cell lymphoma treated with axicabtagene ciloleucel. Blood 2021;
138(12): 1081-1085. https://doi.org/10.1182/blood.2021010930 PMid:34041526 PMCid:PMC8462361
- Shouval
R, Tomas AA, Fein JA, Flynn JR, Markovits E, Mayer S, Afuye AO,
Alperovich A, Anagnostou T, Besser MJ, et al. Impact of TP53 genomic
alterations il large B-cell lymphoma treated with CD19-chimeric antigen
receptor T-cell therapy. J Clin Oncol 2021; 40: 369-381. https://doi.org/10.1200/JCO.21.02143 PMid:34860572 PMCid:PMC8797602
- Jain
MD, Zicheddu B, Coughlin CA, Faramand R, Griswold AJ, Reid KM, Menges
M, Zhang Y, Cen L, Wang X, et al. Whole-genome sequencing reveals
complex genomic features underlying anti-CD19 CAR T-cell treatment
failures in lymphoma. Blood 2022; 140(5): 491-503. https://doi.org/10.1182/blood.2021015008 PMid:35476848 PMCid:PMC9353150
- Cherng
HJ, Sun R, Sugg B, Irwin R, Yang H, Le CC, Deng Q, Fayad L, Fowler NH,
Parmar S, et al. Risk assessment with low-pass whole-genome sequencing
of cell-free DNA before CD19 CAR T-cell therapy for large B-cell
lymphoma. Blood 2022; 140(5): 504-515. https://doi.org/10.1182/blood.2022015601 PMid:35512184 PMCid:PMC9353148
- Kurtz
DM, Green MR, Bratman SV, Scherer F, Liu CL, Kunder CA, Takahashi K,
Glover C, Keane K, Kihira S, et al. Noninvasive monitoring of diffuse
large B-cell lymphoma by immunoglobulin high-throughput sequencing.
Blood 125(24): 3679-3687. https://doi.org/10.1182/blood-2015-03-635169 PMid:25887775 PMCid:PMC4463733
- Frank
MJ, Hossain NM, Bukhari A, Dean E, Spiegel JY, Claire GK, Kirsc I,
Jacob AP, Mullins CD, Lee LW, et al. Monitoring of circulating tumor
DNA improves early relapse detection after axicabtagene ciloleucel
infusion in large B-cell lymphoma: results of a prospective
multi-institutional trial. J Clin Oncol 2021; 39: 3034-3043. https://doi.org/10.1200/JCO.21.00377 PMid:34133196 PMCid:PMC10166351
- Sworder
BJ, Kurtz DM, Alig SK, Frank MJ, Shukla N, Garofalo A, Maculay CW.
Esfahani MS, Olsen MNB, Hamilton J, et al. Detrminants of resistance to
engineered T-cell therapies targeting CD19 in large B cell lymphomas.
Cancer 2023; 41(1): 210-225. https://doi.org/10.1016/j.ccell.2022.12.005 PMid:36584673
- Kotlov
N, Bagaev A, Revuelta MV, Phillip JM, Cacciapuoti MT, Antysheva Z,
Svekollin V, Tikhonova E, Miheecheva N, Kuzkina N, et al. Clinical and
biological subtypes of B-cell lymphoma revealed by microenvironmental
signatures. Cancer Discov 2021; 11(6):1468-1489. https://doi.org/10.1158/2159-8290.CD-20-0839 PMid:33541860 PMCid:PMC8178179
- Steen
CB, Luca BA, Esfahani MS, Azizi A, Sworder BJ, Nabet BY, Kurtz DM, Liu
CL, Khameneh F, Advani RH, et al. The landscape of tumor cell states
and ecosystems in diffuse large B cell lymphoma. Cancer 2021; 39(19):
1422-1437. https://doi.org/10.1016/j.ccell.2021.08.011 PMid:34597589 PMCid:PMC9205168
- Batlevi
CL, Nos G, Kotlov N, Sorokina M, Okwali M, Bagaev A, Michaud L, Shah
GL, Shouval R, Tomas AA, et al. Correlative biomarkers for CART19
response in patients with relapsed or refractory diffuse large B-cell
lymphoma. Blood 2022; 140 (suppl 1): 9228-9230. https://doi.org/10.1182/blood-2022-163333
- Dybkaer
K, Bogsted M, Falgreen S, Bodekr JS, Kjeldsen MK, Schmitz A, Bilgrau
AE, Xu-Monette ZY, Li L, Bergkvist KS, et al. Diffuse large B-cell
lymphoma classification system that associates normal B-cell subset
phenotypes with prognosis. J Clin Oncol 2015; 33(12): 1379-1388. https://doi.org/10.1200/JCO.2014.57.7080 PMid:25800755 PMCid:PMC4397280
- Haradhvala
NJ, Leich MB, Maurer K, Gohil SH, Larson RC, Yao N, Gallagher K, Katsis
K, F Rigault M, Southard J, et al. Distinct cellular dynamics
associated with response to CAR-T therapy for refractory. B-cell
lymphoma. Nat Med 2022; 28(9): 1848-1859. https://doi.org/10.1038/s41591-022-01959-0 PMid:36097221 PMCid:PMC9509487
- Deng
Q, Han G, Puebla-Orosio N, Ma MCJ, Strati P, Chasen B, Dai E, Dang M,
Jain N, Yang H, et al. Characteristics of anti-CD19 CAR T-cell infusion
products associates with efficacy and toxicity in patients with large
B-cell lymphomas. Nat Med 2020; 26(12): 1878-1888. https://doi.org/10.1038/s41591-020-1061-7 PMid:33020644 PMCid:PMC8446909
- Rejeski
K, Perez A, Sesques P, Hoster E, Berger C, Jentzsch L, Mougiakakos D,
Frolich L, Ackermann J, Bucklein V, et al. CAR-HEMATOTOX: a model for
CAR T-cell-cell related hematologic toxicity in relapsed/refractory
large B-cell lymphoma. Blood 2021; 138(24): 2499-2508. https://doi.org/10.1182/blood.2020010543 PMid:34166502 PMCid:PMC8893508
- Kampouri
E, Little JS, Rejeski K, Manuel O, Hammond SP, Hill JA. Infections
after chimeric antigen receptor (CAR)-T-cell therapy for hematologic
malignancies. Transpl Infect Dis. 2023 Oct 3:e14157 https://doi.org/10.1111/tid.14157 PMid:37787373
- Morris
EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine release syndrome and
associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. 2022
Feb;22(2):85-96. doi: 10.1038/s41577-021-00547 https://doi.org/10.1038/s41577-021-00547-6 PMid:34002066 PMCid:PMC8127450
- Makos
OL, D'Angelo CR. The shifting roles and toxicities of cellular
therapies in B-cell malignancies. Transpl Infect Dis. 2023 :e14145. https://doi.org/10.1111/tid.14145 PMid:37676749
- Rubin
DB, Danish HH, Ali AB, et al. Neurological toxicities associated with
chimeric antigen receptor T-cell therapy. Brain.2019;142(5):1334-1348. https://doi.org/10.1093/brain/awz053 PMid:30891590
- Ganatra
S, Dani SS, Yang EH, Zaha VG, Nohria A. Cardiotoxicity of T-Cell
Antineoplastic Therapies: JACC: CardioOncology Primer. JACC
CardioOncol. 2022 Dec 20;4(5):616-623. https://doi.org/10.1016/j.jaccao.2022.07.014 PMid:36636447 PMCid:PMC9830211
- Brammer
JE, Braunstein Z, Katapadi A, Porter K, Biersmith M, Guha A, Vasu S,
Yildiz VO, Smith SA, Buck B, Haddad D, Gumina R, William BM, Penza S,
Saad A, Denlinger N, Vallakati A, Baliga R, Benza R, Binkley P, Wei L,
Mocarski M, Devine SM, Jaglowski S, Addison D. Early toxicity and
clinical outcomes after chimeric antigen receptor T-cell (CAR-T)
therapy for lymphoma. J Immunother Cancer. 2021; 9:e002303 https://doi.org/10.1136/jitc-2020-002303 PMid:34429331 PMCid:PMC8386216
- Wudhikarn
K, Tomas AA, Flynn JR, Devlin SM, Brower J, Bachanova V, Nastoupil LJ,
McGuirk JP, Maziarz RT, Oluwole OO, Schuster SJ, Porter DL, Bishop MR,
Riedell PA, Perales MA. Low toxicity and excellent outcomes in patients
with DLBCL without residual lymphoma at the time of CD19 CAR T-cell
therapy. Blood Adv. 2023; 7: 3192-3198 https://doi.org/10.1182/bloodadvances.2022008294 PMid:36355838 PMCid:PMC10338201
- Oluwole
OO, Bouabdallah K, Muñoz J, De Guibert S, Vose JM, Bartlett NL, Lin Y,
Deol A, McSweeney PA, Goy AH, Kersten MJ, Jacobson CA, Farooq U,
Minnema MC, Thieblemont C, Timmerman JM, Stiff P, Avivi I, Tzachanis D,
Kim JJ, Bashir Z, McLeroy J, Zheng Y, Rossi JM, Johnson L, Goyal L, van
Meerten T. Prophylactic corticosteroid use in patients receiving
axicabtagene ciloleucel for large B-cell lymphoma. Br J Haematol. 2021
Aug;194(4):690-700. https://doi.org/10.1111/bjh.17527 PMid:34296427 PMCid:PMC8457222
- Mucha
SR, Rajendram P. Management and Prevention of Cellular-Therapy-Related
Toxicity: Early and Late Complications. Curr Oncol. 2023 May
15;30(5):5003-502 https://doi.org/10.3390/curroncol30050378 PMid:37232836 PMCid:PMC10217595
- Jain MD, Smith M, Shah NN. How I treat refractory CRS and ICANS after CAR T-cell therapy. Blood. 2023 ;141:2430-2442. https://doi.org/10.1182/blood.2022017414 PMid:36989488 PMCid:PMC10329191
- Gauthier
J, Gazeau N, Hirayama AV, et al. Impact of CD19 CAR T-cell product type
on outcomes in relapsed or refractory aggressive B-NHL. Blood.
2022;139(26):3722-3731. https://doi.org/10.1182/blood.2021014497 PMid:35439295 PMCid:PMC9247364
- Roschewski
M, Longo DL, Wilson WH. CAR T-Cell Therapy for Large B-Cell Lymphoma -
Who, When, and How? N Engl J Med. 2022 Feb 17;386(7):692-696. https://doi.org/10.1056/NEJMe2118899 PMid:34904797 PMCid:PMC9295142
- Parker
C, Liu FF, Deger KA, Franco-Villalobos C, Proskorovsky I, Keating SJ,
Sorensen S. Cost-Effectiveness of Lisocabtagene Maraleucel Versus
Axicabtagene Ciloleucel and Tisagenlecleucel in the Third-Line or Later
Treatment Setting for Relapsed or Refractory Large B-cell Lymphoma in
the United States. Adv Ther. 2023 May;40(5):2355-2374. https://doi.org/10.1007/s12325-023-02444-x PMid:36947328 PMCid:PMC10129927
- Foglia,
E.; Garagiola, E.; Ladisa, V.; Rambaldi, A.; Cairoli, R.; Sammassimo,
S.; Salè, E.O.; Zinzani, P.L.; Esposti, M.; Alberti, L.; et al.
Multidimensional Results and Reflections on CAR-T: The Italian
Evidence. Int. J. Environ. Res. Public Health 2023, 20, 3830. https://doi.org/10.3390/ijerph20053830 PMid:36900841 PMCid:PMC10001656