Michele
Bibas.
Department of Clinical Research,
Hematology. National Institute for Infectious Diseases “Lazzaro
Spallanzani” I.R.C.S.S.
Correspondence to:
Michele Bibas MD, Department of Clinical Research, Hematology. National
Institute for Infectious Diseases “Lazzaro Spallanzani” I.R.C.S.S. Via
Portuense 292 00148 Rome Italy.
Michele.bibas@inmi.it
Published: January 01, 2024
Received: October 09, 2023
Accepted: December 12, 2023
Mediterr J Hematol Infect Dis 2024, 16(1): e2024007 DOI
10.4084/MJHID.2024.007
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
This
two-part review aims to present a current and comprehensive
understanding of the diagnosis and management of plasmablastic
lymphoma. The first section, as presented in this paper, reviews
epidemiology, etiology, clinicopathological characteristics,
differential diagnosis, prognostic variables, and the impact of
plasmablastic lymphoma on specific populations.
Plasmablastic
lymphoma (PBL) is a rare and aggressive form of lymphoma. Previous and
modern studies have demonstrated a significant association between the
human immunodeficiency virus (HIV) and the development of the disease.
The limited occurrence of PBL contributes to a need for a more
comprehensive understanding of the molecular mechanisms involved in its
etiology. Consequently, the diagnostic procedure for PBL poses a
significant difficulty. Among the group of CD20-negative large B-cell
lymphomas, PBL can be correctly diagnosed by identifying its exact
clinical characteristics, anatomical location, and morphological
characteristics. PBL cells do not express CD20 or PAX5 but possess
plasmacytic differentiation markers such as CD38, CD138, MUM1/IRF4,
Blimp1, and XBP1. PBL must be distinguished from other B-cell
malignancies that lack the CD20 marker, including primary effusion
lymphoma, anaplastic lymphoma kinase-positive large B-cell lymphoma,
and large B-cell lymphoma (LBCL). This condition is frequently
associated with infections caused by the Epstein-Barr virus and genetic
alterations involving the MYC gene. Despite advances in our
comprehension of this disease, the prognosis remains dismal, resulting
in a low overall survival rate, although recent reports suggest an
apparent tendency towards substantial improvement.
|
Article highlights
• The 5th
edition of the World Health Organization classification for
hematological and lymphoid cancers recognizes plasmablastic lymphoma
(PBL) as a unique subtype. This classification includes PBL as "large
B-cell lymphomas".
• Fewer than 1100 cases of PBL have been reported in the medical literature.
•
HIV, immunodeficiencies, persistent immunological activation, and
oncogenic herpesviruses like EBV are suspected causes of this condition.
•
The disease is aggressive and destructive, primarily affecting the oral
cavity, as seen in its first clinical presentation. It is well known
that it also affects lymph nodes and areas outside the mouth.
• A
biopsy of the tissue mass or lymph nodes is necessary for diagnosis.
Core needle or small needle biopsy is often limited to inaccessible
areas.
• Plasmablasts, activated B cells undergoing somatic
hypermutation and class-switching recombination, are believed to be the
cells of origin of PBL.
• The immunophenotype often lacks CD45,
CD20, and CD79a expression, but it is positive for CD38, CD138, and
MUM1. Additionally, EBER and KI67 expression exceeds 80%.
• PBL is
distinct from other B-cell malignancies lacking CD20 marker, such as
primary effusion lymphoma, anaplastic lymphoma kinase-positive large
B-cell lymphoma, and HHV8+ large B-cell lymphoma (LBCL).
• It is
important to recognize that distinguishing plasmablastic myeloma from
plasmablastic lymphoma can be challenging and complex.
•
Historically, it has been observed that PBL has generally exhibited a
less favorable prognosis, as seen by a median overall survival (OS)
ranging from 8 to 15 months. Survival estimates of more recent data
ranged from 32 months to 62 months.
Definition
According
to the fifth edition of the World Health Organization Classification of
Hematololymphoid Tumors (WHO-HAEM5), PBL is classified as a specific
subtype within the category of "large B-cell lymphomas".[3] The nomenclature about PBL and other entities has undergone revision during the transition from the 4th edition of the World Health Organization Classification of Hematololymphoid Tumors (WHO-HAEM4)[12] to the 5th
edition (WHO-HAEM5), with the aim to promote consistency. The phrase
"diffuse large B-cell lymphoma" has been modified to "large B-cell
lymphoma".
History
In
the 1992 edition of "Neoplastic Hematopathology", Stein's article
started and provided a comprehensive description of PBL as a novel and
original concept.[1] In 1997, Delecluse and Stein documented the first
case series of PBL. This investigation utilized consultation files from
the lymphoma reference center at Benjamin Franklin Hospital in Berlin.
The study consisted of a sample size of 16 participants from Germany,
with 15 having been diagnosed with HIV.[2]The
tumor demonstrates a preference for the oral cavity, particularly the
gingiva or palate; additional sites, such as the bone marrow, may be
observed as a late or infrequent characteristic.
Epidemiology
The
incidence and temporal distributions of PBL in people with either
HIV-positive or HIV-negative status are not well established due to
their rarity. The estimated incidence of PBL in the context of
HIV-related lymphomas is approximately 2 percent.[5] In addition to its
notable association with HIV infection, there have been recorded cases
of PBL in persons with other types of immunodeficiency, including
iatrogenic immunosuppression subsequent to solid organ transplantation
and the geriatric population.[11-17] Moreover, it is crucial to recognize
that PBL can also occur in patients who are HIV-negative and may be
associated with preexisting lymphoproliferative or autoimmune diseases.
A few cases have been documented among people with healthy immune
systems.[17-19] PBL has been observed in people across a broad spectrum
of ages, from 1 to 90 years, but there is a scarcity of reported cases
of young individuals. The examination of the gender distribution of PBL
cases reveals a notable predominance of males, accounting for around
75% of the overall population.[19-22] PBL is a rare disease with a
published number of less than 1100 cases up to date. Nevertheless,
there has been a recent increase in the number of reported cases in the
literature. There is a possibility that the observed increase in the
incidence of this disease is primarily attributable to improved
diagnostic techniques and heightened awareness rather than a genuine
rise in frequency. However, this information needs to be analyzed with
large-scale epidemiological studies.
Pathogenesis
The biology of the originating cell.
Plasmablasts, which experienced the germinal center response and are
currently in the process of differentiation into plasma cells, are
widely recognized as the precursor cells of PBL. The essential role of
the germinal center (GC) reaction is to generate B-cell clones that
possess the highest affinity for a specific antigen. B-cell clones
exhibit bidirectional migration between two distinct regions within the
germinal center (GC), namely the "light zone" and the "dark zone". This
mobility allows them to compete for antigen presentation by follicular
dendritic cells while receiving survival signals from helper T cells.
Acquiring somatic mutations plays a crucial role in promoting affinity
development within B-cell clones.Furthermore,
B-cell clones are involved with DNA class-switching recombination, a
process that leads to the production of immunoglobulin A (IgA),
immunoglobulin E (IgE), or immunoglobulin G (IgG). This mechanism
serves to expand the range of antibodies produced. There is a prevalent
assumption that clones of autoimmune or anergic B cells undergo
apoptosis. Apoptosis is expected to occur in a significant proportion
of B cells during the germinal center reaction. Various factors can
induce apoptosis in the germinal center, including the B-cell receptor
(BCR), T-cell growth factor beta (TGF-β), and Fas-mediated pathways.
Both the B-cell receptor (BCR) and transforming growth factor-beta
(TGF-β) signaling pathways induce apoptosis by interacting with
proapoptotic components of the BCL-2 family. The previously mentioned
signaling mechanism results in the overexpression of BH3-only proteins
and the downregulation of BCL-XL, ultimately leading to mitochondrial
depolarization and the initiation of intrinsic apoptosis. [23-26]
B-cells sometimes experience programmed cell death via the FAS pathway.
The FAS death induction signaling complex, which is accountable for
initiating cell death, generally remains quiescent. Nevertheless, its
activation occurs in the presence of an imbalance in survival signals
originating from T cells and follicular dendritic cells. This
activation process plays a role in the caspase-8 activation and
the subsequent initiation of extrinsic apoptosis.[23-26]
The final fate of a particular subset of B lymphocytes is to undergo
differentiation into either long-lived lymphocytes or plasma cells.
Without antigenic stimulation, a certain subset of lymphocytes
undergoes a stochastic transformation process, generating plasma cells.[23-26]
Signaling pathways help the first stage of plasma cell differentiation
by turning off the transcription factors PAX-5 and BCL-6. This block is
done by the transcription factor BLIMP-1, which is mostly found in
plasma cells.Regarding
morphology, centrocytes undergo a process of transformation in which
they undergo differentiation into plasmablasts before attaining
maturity as plasma cells. From a phenotypic perspective, the cells
display the presence of CD38, interferon regulatory factor 4/multiple
myeloma 1 (IRF-4/MUM-1), and undergo a downregulation of CD20
expression while retaining CD19 expression.[23-27] In
summary, the currently accepted assumption is that plasmablasts,
activated B cells that have undergone somatic hypermutation and
class-switching recombination, constitute the primary biological origin
of PBL. The plasmablast undergoes a differentiation process during this
phase, transforming it into a mature plasma cell. Plasmablasts are a
characteristic element of reactive phenomena observed in viral
infections, such as Epstein-Barr virus (EBV) and HIV.
Etiology
The
precise etiology of PBL remains unknown, although recent studies have
shed light on the importance of genetic rearrangements in the MYC gene
and its correlation with EBV infection as essential pathogenic
mechanisms. Furthermore, the involvement of IRF4, JAK-STAT, Notch, and
RAS-RAF signaling pathways will be carefully highlighted.
The influence of Epstein-Barr virus (EBV).
The Epstein-Barr virus is classified as a DNA virus demonstrating a
preference for infecting B, T, natural killer, and epithelial cells.
The prevalence of seropositivity to EBV is estimated to be around 90%
among the global population. Around 80% of cases of PBL exhibit a
correlation with EBV infection. Moreover, the prevalence of EBV is
observed to be higher in PBL cases among individuals with human
immunodeficiency virus (HIV) in comparison to those without HIV.[28-29]
After the first infection with EBV, the virus establishes a latent
state among memory B cells and maintains its presence by avoiding the
host's immune system. Viral latency and persistence are promoted by
creating certain viral gene products, such as the Epstein-Barr nuclear
antigen (EBNA-1) and EBER. The Epstein-Barr virus (EBV) disrupts the
function of proapoptotic proteins, therefore promoting the survival of
host cells by activating intracellular signaling pathways such as NF-kB
and NOTCH signaling pathways.[27-29] The potential
outcome of an Epstein-Barr virus (EBV) infection is the acquisition of
oncogenic mutations, which may subsequently trigger the transformation
of B cells and ultimately contribute to the development of cancer.
There is an increased incidence of B-cell lymphomas associated with
Epstein-Barr virus (EBV) in populations affected by HIV infection,
individuals undergoing immunosuppressive therapy, and the elderly.[29]
Despite the utilization of combination antiretroviral therapy (cART)
and the attainment of viral suppression, individuals who are infected
with the human immunodeficiency virus (HIV) still experience an
increased vulnerability to the occurrence of cancers associated with
the Epstein-Barr virus (EBV). The phenomenon in question could
potentially be ascribed to a convergence of various mechanisms,
including immune evasion, prolonged inflammation characterized by a
modified cytokine profile, and immunological senescence. Significantly,
these characteristics continue to exist in individuals who are
HIV-positive even after receiving combination antiretroviral therapy
(cART) and maintaining virological suppression.[29-31] The
immunological senescence and evasion reported in cases of Epstein-Barr
virus (EBV)-positive PBL can be mostly related to the expression of
programmed death ligand 1 (PD-L1) by PBL cells and tumor-associated
macrophages (TAM). The primary function of this PD-L1 production is to
inhibit the cellular responses directed toward tumor suppression.[30]
The evasion of the immune system is made possible by the reduced
expression of major histocompatibility complex (MHC) class II proteins
in PBL infected with Epstein-Barr virus (EBV) and by the secretion of
immunosuppressive agents such as interleukin 10, transforming growth
factor beta (TGF-B), and other substances by tumor-associated
macrophages (TAM) and regulatory T cells.[31-32]
Histological analysis is employed to confirm the existence of
Epstein-Barr virus (EBV) infection in PBL, with a particular focus on
detecting Epstein-Barr-encoded small RNA (EBER) through in situ
hybridization (ISH) within the tumor cells. The EBV latency type I
program has been associated with the occurrence of PBL. Nevertheless,
it is important to acknowledge that latency types II and III have been
documented in cases associated with post-transplant and HIV-related
illnesses.[33-34]
The role of the MYC gene.
The oncogenic transcription factor MYC is a molecular entity that plays
a critical role in the development and progression of cancer. The MYC
protein is known to significantly impact the regulation of key
biological processes, including cell proliferation, metabolism,
apoptosis, and cell differentiation. The gene expression analysis has
revealed the consistent activation of MYC signaling in PBL.[35]
Immunohistochemical labeling was utilized to evaluate the expression of
MYC protein in most of the examined cases, demonstrating a notable
predominance of upregulated expression. In different studies, it has
been reported that MYC translocations were identified in roughly 50% of
PBL cases. This discovery implies that MYC translocation does not only
account for the upregulation of MYC at the protein level.[35-36]
Approximately two-thirds of PBL cases include translocations between
the MYC gene and the heavy chain immunoglobulin gene (IgH), while the
remaining one-third of cases exhibit translocations with non-IgH
partners. Patients with PBL who tested positive for MYC translocation
displayed a significantly elevated Ki67 proliferation score. However,
there is an ongoing debate questioning the influence of MYC
translocation on the survival outcomes of patients diagnosed with PBL.[37]
Around 10% of primary PBL cases demonstrate observable MYC mutations
associated with translocations. The current understanding of the
biological effects of these MYC mutations is still unknown. A
correlation was observed between the occurrence of MYC mutations and
the concurrent prevalence of MYC translocations.[38]
The
presence of MYC mutations can be linked to the activation-induced
cytidine deaminase (AID) facilitated abnormal somatic hypermutation
mechanism, as demonstrated by the relocation of the MYC gene to the IgH
locus in most cases.[39] This mechanism could explain
a higher frequency of silent mutations and the predominance of
subclonal MYC mutations. In addition to modifications in passenger
genes, specific mutations in the MYC gene can affect important
functional regions, thereby increasing the carcinogenic capacity of
MYC. Prior research provided empirical support suggesting that the
occurrence of MYC T58A and P57S mutations, which are frequently
detected in Burkitt lymphoma, plays a role in lymphoma progression by
promoting cellular proliferation and inhibiting the proapoptotic
characteristics generally associated with MYC.[38-40]
Additionally, it is important to acknowledge that MYC's transactivation
domain (TAD) undergoes ubiquitination, resulting in subsequent
proteolytic degradation.[40] Modifications to the
topologically associated domain (TAD) can impede the proteolysis
mechanism, enhancing the MYC protein's stability. Approximately 50% of
the MYC mutations identified in PBL were located within the
transactivation domain (TAD) of exon 2. Despite the absence of a
designated mutational hotspot in the study, additional functional
examinations are required to definitively determine the precise role of
MYC mutations in the development of PBL.[41-43] The
observation that several cancer-promoting pathways can activate MYC in
diverse cell types underscores the significance of MYC-mediated
transcriptional regulation in the biology of PBL. The gene MYC assumes
a pivotal function as a target gene within the JAK-STAT signaling
pathway; its upregulation is facilitated by the direct contact
between the activated STAT3 protein and the promoter region of the
gene. Moreover, there have been reports suggesting that the activation
of the RAS-RAF signaling pathway results in the enhancement of MYC
production by stabilizing the MYC protein and impeding its degradation
by the proteasome. MYC activation as a downstream target through NOTCH1
signaling has been documented in T-cell acute lymphoblastic
leukemia/lymphoma cases.[42-47] The higher levels of
MYC protein seen in PBL cells without MYC translocation may be because
MYC is activated through various pathways that have been altered.[48]
The Impact of the IRF4 gene.
The expression of IRF4 is restricted to hematopoietic systems, and it
is widely recognized as a pivotal regulator of plasmacytic
differentiation. Centrocytes induce plasmacytic differentiation by
downregulating the expression of BCL6, a crucial regulator involved in
the formation and maintenance of the germinal center. Concurrently,
they upregulate the expression of transcription factors such as IRF4.[49]
The depletion of IRF4 under specific conditions in live experiments led
to the complete absence of plasma cells, thereby emphasizing the
essential function of IRF4.[50] Additionally, it has
been reported that there is a notable increase in the expression of
IRF4 in several forms of lymphoid malignancies. Subsequent functional
analyses have revealed that the signaling path of the IRF4 network is
of paramount importance in the viability and proliferation of PCM, ABC,
DLBCL, and ALCL cells.[51] The immunohistochemical
staining results revealed a substantial increase in the expression of
IRF4 in PBL samples. An underlying mechanism was identified whereby
approximately 33% of primary PBL patients exhibited a unique and
localized amplification of 6p25.3.[52,53] The
reported amplification proved to include a restricted group of genes,
one of which was identified as IRF4. The molecular target IRF4 has been
recognized in various lymphoma forms, including Hodgkin lymphoma,
specific subtypes of T-cell lymphoma, and plasma cell myeloma (PCM).
However, there is currently no data indicating the presence of
recurring focal amplifications of 6p25.3. The lack of detected
amplifications suggests that these genetic abnormalities may function
as a unique genetic anomaly in the context of PBL. The prevalence of
IRF4 mutations was seen in approximately 4% of the first PBL samples.[50-54]
The role of the JAK-STAT pathway.
The JAK-STAT signaling pathway is of paramount importance in modulating
cellular differentiation, proliferation, viability, and the immune
response. As a result, it has been linked to the carcinogenic
mechanisms associated with a wide array of cancers. Cytokines, like
interleukin-6 (IL-6), engage in interactions with receptor tyrosine
kinases (RTKs) that are linked to cytosolic domain proteins belonging
to the Janus kinase (JAK) family, thereby initiating the JAK-STAT
signaling cascade.[55-56] JAK transactivation occurs
when JAKs become activated, resulting in the phosphorylation of
tyrosine residues on receptor tyrosine kinases (RTKs). The
phosphorylated residues act as binding sites for Janus kinases (JAKs),
which turn on several effector proteins. In order to facilitate the
regulation of gene expression, STAT proteins are recruited, undergo
dimerization upon phosphorylation, and subsequently translocate to the
nucleus. Mutations affecting the constituents of JAK-STAT signaling
have been identified in many subtypes of T-cell lymphoma, such as
T-cell large granulocytic lymphocytic leukemia (T-LGL) and conditions
involving natural killer cells. However, these mutations are rarely
reported in aggressive B-cell lymphomas.[57-60] The
presence of somatic mutations in PBL has been observed to affect many
genes responsible for encoding elements of the JAK-STAT signaling
pathway. This finding highlights the importance of this pathway in the
disease's pathogenesis. A total of 25% of the PBL patients examined
displayed STAT3 mutations.[61] Notably, most of these
observed mutations were situated inside the SH2 domain, which plays a
crucial role in dimerization and activation mechanisms. It is
noteworthy that a significant link exists between mutations in the
STAT3 gene and the presence of concomitant HIV infection.[61]
The prevalence of STAT3 mutations in PBL derived from HIV-positive
patients was around 50%, but the occurrence of these mutations in PBL
derived from HIV-negative patients was lower than 10%.[61]
The discrepancy observed was less pronounced in individuals with
immunocompetent PBL than in those with any level of immunodeficiency.
This suggests that HIV infection may have a direct impact on the
development of lymphoma, independent of its immunosuppressive effects.[61] In
addition to STAT3 mutations, PBL has a high frequency of mutations in
other genes that code for parts of the JAK-STAT pathway. Mutations in
the JAK1 gene were observed in 14% of individuals with HIV, with a
significant clustering of these mutations observed at the G1097 codon.[62] mutations at the G1097 codon have also been previously recorded in specific subgroups of T-cell lymphoma.[62]
Several cases of PBL demonstrated concomitant alterations in various
constituents of the JAK-STAT pathway, such as STAT3 and SOCS1/3. A
considerable percentage of cases of PBL in individuals without human
immunodeficiency virus (HIV) infection, specifically more than
one-third, as well as a huge majority of PBL cases associated with HIV
infection, specifically over sixty percent, had alterations within the
Janus kinase-signal transducer and activator of transcription
(JAK-STAT) pathway.[4,61,62] In the
context of PBL, it is relevant to recognize that the JAK-STAT pathway
can influence not just mutations but also recurrent somatic copy-number
alterations (SCNAs). The research findings indicated a high occurrence
of recurring focal amplifications in the 1q21.3 area, detected in 52%
of PBLs.[4,61,62] The change impacts
the IL-6 receptor (IL-6R) gene and the MCL1 gene linked to
antiapoptotic activity. The gene MCL1 is prone to direct activation by
the JAK-STAT signaling pathway, whereas the IL-6R protein plays a vital
role in initiating the activation of the JAK-STAT system at a higher
level.[62-64]
Role of NOTCH in promoting the development of PBL.
The activation of NOTCH signaling starts when ligands cut apart
receptors into shorter pieces. Mammals have an overall total of four
unique NOTCH receptors, specifically identified as NOTCH1, NOTCH2,
NOTCH3, and NOTCH4.[65] The transmembrane receptor
NOTCH undergoes proteolytic cleavage upon contact with specific ligands
provided by neighboring cells, leading to the release of the NOTCH
intracellular domain (NICD). The NICD molecule, which is unbound and
not associated with any other molecules, can penetrate the cellular
nucleus and performs the function of controlling the process of gene
transcription.[66] The NOTCH pathway is a highly
conserved process that regulates cellular differentiation, survival,
and proliferation with the specific consequences being dependent on the
cellular context. The scope of NOTCH signaling goes beyond
intracellular mechanisms, embracing intercellular communication and its
relevance in tumor formation, notably in enhancing interactions between
tumor cells and their surrounding environment.[65-67]
Approximately 25% of PBL cases exhibited mutations in genes responsible
for encoding subunits of the NOTCH signaling system. The genes Notch1,
Notch4, and SPEN, responsible for encoding a negative regulator of the
Notch signaling pathway, demonstrated the highest incidence of
mutations. Except for a small group of cases, NOTCH mutations were not
mostly found in either the HD or the PEST domain, as seen in T-ALL and
other B-cell cancers. The primary site of most alterations has been
identified as the extracellular epidermal growth factor (EGF)-like
domain. Nevertheless, the functional consequences of these mutations in
PBL pathogenesis have yet to be fully understood.[41,68]
The findings of a limited-scale investigation involving individuals
diagnosed with PBL revealed a consistent expression of NOTCH1,
predominantly localized inside the nuclear region, as indicated by the
staining pattern. The findings of this study underscore a substantial
prevalence of NOTCH activation.[69]
Role of the RAS-RAF pathway in promoting the development of PBL. The
RAS-RAF pathway, which regulates fundamental cellular processes such as
differentiation, proliferation, apoptosis, and migration, is frequently
altered in the context of cancer.[70] RAS proteins'
activation involves several cytokines and receptor tyrosine kinases,
which promote the transition of RAS proteins into their active state
while being bound to GTP. The activation signal subsequently promotes
the recruitment of RAF kinases to the cellular membrane, resulting in
additional downstream activation. Following this, RAF kinases undergo
the dimerization process and play a role in transmitting signals by
interacting with MEK and ERK proteins, thus activating various
transcription factors. Although the occurrence of RAS-RAF signaling
mutations in aggressive lymphomas is not prevalent, it is recognized as
one of the most frequently affected oncogenic pathways in the context
of cancer. Somatic mutations in the RAS gene are commonly seen in
primary cutaneous melanoma (PCM), with NRAS and KRAS mutations detected
in around 20% of PCM cases. The abovementioned mutations, which are
primarily subclonal, correlate with disease progression.[71-76]
The frequency of recurrent NRAS mutations in PBL cases was
approximately 30%, whereas KRAS mutations were detected in 10% of
cases, and BRAF mutations were identified in 6% of cases. The
investigators observed that a significant proportion of RAS mutations
were localized at the widely recognized mutational hotspot sites G13
and Q61.[41,71-75] The above
mutations are called gain-of-function mutations because they can keep
the pathway active by making RAS more stable when it is bound to GTP.[76-81]
Clinical Evaluation and Staging
Patients
suspected of having PBL should have their medical history reviewed,
focusing on B symptoms, including fever, night sweats, and weight loss
over 10% in the past six months. Additionally, inspect Waldeyer's ring,
the integumentary system, the hepatic region, and the splenic region,
which contain lymph nodes. Further, patient performance must be
assessed. A full blood count, chemical analysis with LDH determination,
beta-2 microglobulin measurement, immunoglobulin profile by
immunofixation, and plasma and urine kappa and lambda tests should be
performed in the lab. Serological testing is required for identifying
antibodies against hepatitis B and C viruses, cytomegalovirus,
Epstein-Barr virus, Toxoplasma, and varicella-zoster virus, quantifying
HIV loads in plasma, and analyzing CD4+ and CD8+ T-lymphocyte
subpopulations. Before chemotherapy, patients' renal, hepatic, and
cardiac functions must be assessed. Echocardiography is also indicated
for anthracycline-based chemotherapy patients to evaluate heart
function. If imaging discloses symptoms or lesions, gastrointestinal
endoscopy may be recommended. Women of reproductive age must have
pregnancy testing and fertility preservation counseling before
treatment. According to the current guidelines, PET/CT is the
recommended imaging modality for evaluating aggressive lymphoma.
Further imaging with CT or MRI may be performed if there is a clinical
indication. Up to 25% of PBL patients have bone marrow involvement,
requiring a biopsy and aspiration. Diagnostic lumbar punctures are
recommended for HIV and PBL patients due to the higher risk of CNS
involvement, particularly in the meninges. Flow cytometry, cytology,
and molecular tests are advised to detect leptomeningeal lymphoma in
cerebrospinal fluid. The most common staging approach is Ann Arbor. The
International Prognostic Index (IPI) is recommended for risk
stratification. Excisional and incisional biopsies are mandatory. FNA
biopsy cannot reliably diagnose lymphoma in the majority of cases. Core
needle biopsy may not be effective, but it can be used in some cases.
When an excisional or incisional lymph node is inaccessible, a core
biopsy (preferably many biopsies) and fine-needle aspiration (FNA)
biopsies are recommended. Enhancing these procedures with appropriate
ancillary techniques helps differentiate diagnoses. For this scope,
immunohistochemistry (IHC), flow cytometry, molecular analysis, and
cytogenetic methods like karyotyping or FISH are used to detect
immunoglobulin gene rearrangements and significant translocations. This
comprehensive technique may offer a lot of diagnostic data. In cases
where the material is undiagnostic, another biopsy is needed.[5]
Clinical Features and Presentation
As
previously outlined, PBL is defined by its propensity to affect the
oral cavity. Nevertheless, it is crucial to remember that a significant
percentage, about 45%, of reported cases have been observed in
different structures outside of the oral cavity. The anatomical
locations covered in this list consist of the gastrointestinal tract,
skin, soft tissue, heart, mediastinum, retroperitoneum, liver, lungs,
testes, vulva, parotid gland, breast, central nervous system (CNS),
lymph nodes, bone marrow. The lymphoma exhibits a male predominance
ratio of 4:1 and typically manifests at a median age of 40 years. In
around 5% of cases, this disease occurs as the primary manifestation of
HIV infection. The median age of diagnosis usually ranges within the
approximate range of 40 years. However, it is crucial to always keep in
mind that people with HIV tend to present at a younger median age of 40
years, in contrast to non-HIV patients whose median age exceeds 50
years. This finding offers evidence for the hypothesis that age-related
senescence might play a role in a distinct subgroup of people who are
not infected with HIV. Reported cases involving children have also been
documented.[8-10] A significant proportion of cases
are characterized by an accelerated disease progression, frequently
with destructive lesions. This condition is typically accompanied by an
increased concentration of lactate dehydrogenase (LDH) and the presence
of B symptoms.At
the presentation, it was noted that a majority of individuals confirmed
to have HIV (more than 65%), patients who received transplantation
(50%), and people with apparently normal immune systems (25%) exhibited
advanced disease, notably fitting into Ann Arbor stages III and IV.
However, clinical differences are still observed across patients with
different immunological conditions. The anatomical distribution of PBL
sites exhibits a higher degree of variability among persons who test
negative for HIV in comparison to those who test positive for HIV.
Furthermore, it is seen that bone marrow involvement and the presence
of B symptoms are less commonly observed in individuals who are
HIV-negative.[12-13] Although lymph node involvement
at the time of diagnosis is very rare, it has been observed in
approximately 30% of people who have had transplantation. The incidence
of bone marrow infiltration in patients with PBL has been reported to
exhibit variance among individuals with HIV infection and those without
HIV infection. A comprehensive examination was conducted on a large
cohort of 590 patients diagnosed with PBL. The findings of this study
indicate that a considerable proportion of both HIV-associated cases,
up to 40%, and HIV-negative cases, up to 25%, exhibited bone marrow
involvement.[5]
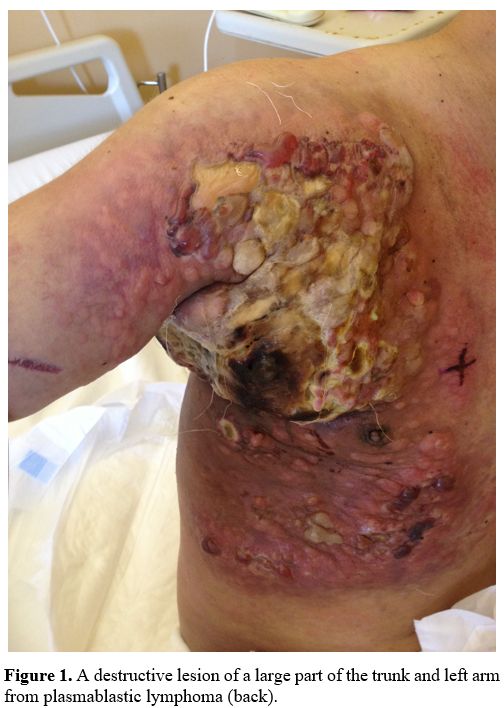 |
Figure 1. A destructive lesion of a large part of the trunk and left arm from plasmablastic lymphoma (back). |
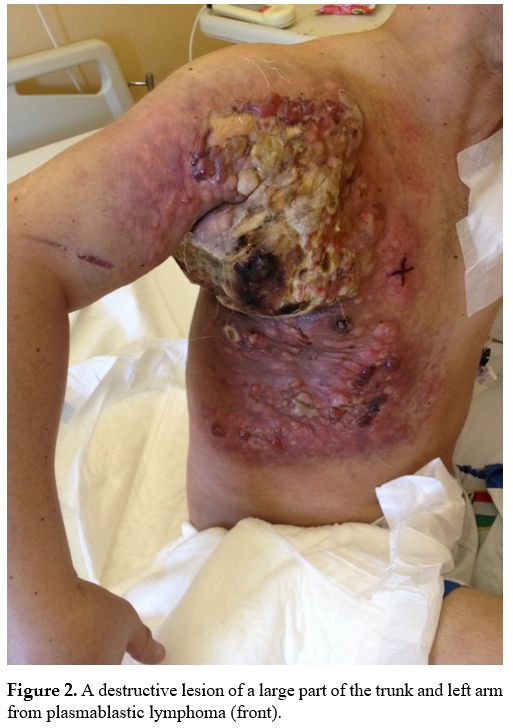 |
Figure 2. A destructive lesion of a large part of the trunk and left arm from plasmablastic lymphoma (front). |
 |
Figure 3. An extensive
lesion caused by plasmablastic lymphoma, which affects both the
integumentary system and the cranial bones.
|
Pathological Features
PBL
is a very aggressive malignancy characterized by large immunoblasts or
giant plasma cells that express plasma cell markers but do not express
B-cell markers.[2-5] The tumor demonstrates a diffuse
growth pattern, leading to impairment of the structural integrity of
both extranodal and nodal locations. The frequent observation of a
"starry-sky" pattern, defined by the abundance of tingible body
macrophages, is evident. The neoplastic cells exhibit features
reminiscent of large immunoblasts, including a significant cytoplasmic
volume and oval vesicular nuclei with prominent nucleoli. The presence
of aberrant cells displaying morphological characteristics resembling
larger centroblasts and/or immunoblasts may be a distinguishing
hallmark of PBL in HIV-positive individuals.On
the other hand, in those without HIV, the occurrence of plasma cell
differentiation is frequently seen at extranodal sites distinct from
the oral mucosa; its distinguishing features include the existence of
cytoplasm with a basophilic staining pattern, the presence of para
nuclear hof, and the presence of large nuclei positioned eccentrically.
Necrosis, karyorrhexis, and larger mitotic figures are commonly
encountered phenomena.[82-83]Specifically,
this disease has an immunophenotype similar to plasma cell neoplasms,
as evidenced by positive markers including CD79a, IRF-4/MUM-1, BLIMP-1,
CD38, and CD138. The neoplastic cells lack B-cell markers CD19, CD20,
and PAX-5 expression. Nevertheless, a subset of these cells may display
a little positive reaction to CD45. In specific cases, the expression
of T-cell markers such as CD2 or CD4 has been seen.[8]
The MIB-1 antibody is commonly employed in immunohistochemical
investigations to detect the Ki-67 proliferation marker, which is
typically shown to be expressed in malignant cells, if not universally.
The expression of the MYC gene is detected in approximately 50% of
cases and is frequently associated with MYC translocations or
amplification. EBV-encoded RNA (EBER) has been seen in around 80
percent of cases, making it the most sensitive method for identifying
EBV infection in malignant cells. According to the results of a recent
study, it was observed that the occurrence of Epstein-Barr virus (EBV)
infection, as evidenced by the production of EBV-encoded RNA (EBER),
exhibited a higher rate among HIV-positive individuals (80%), patients
who acquired post-transplantation primary PBL (67%), and
immunocompetent individuals (50%).[83] Some findings
suggest a predominantly unfavorable presence of EBV LMP-1, with the
typical latency pattern being type I. However, it is important to note
that persons who have HIV infection and posttransplant PBL may display
latency pattern type III. Based on molecular genetic testing, it has
been observed that over 66% of cases present MYC rearrangements, with a
lower proportion exhibiting MYC amplification. The comparative genomic
hybridization analysis results indicate that PBL displays a greater
genetic similarity to diffuse large B-cell lymphoma (DLBCL) compared to
multiple myeloma.[84-86] It is of utmost importance
to remember that distinguishing between plasmablastic myeloma and
lymphomas with plasmablastic features may present difficulties in
correctly identifying tumor cells, hence introducing complexities to
the diagnostic procedure.
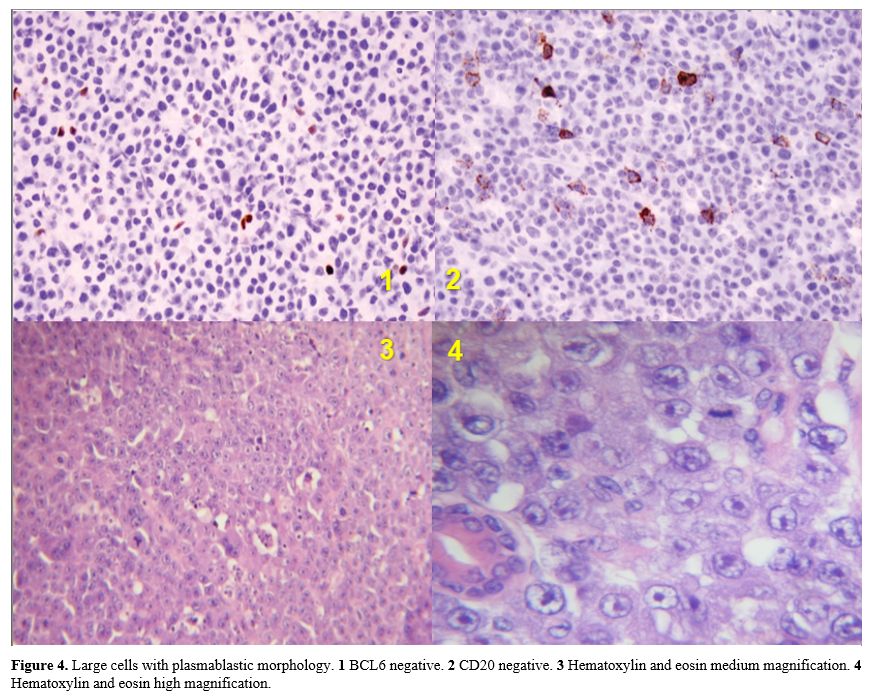 |
Figure 4. Large cells with
plasmablastic morphology. 1 BCL6 negative. 2 CD20 negative. 3
Hematoxylin and eosin medium magnification. 4 Hematoxylin and eosin
high magnification. |
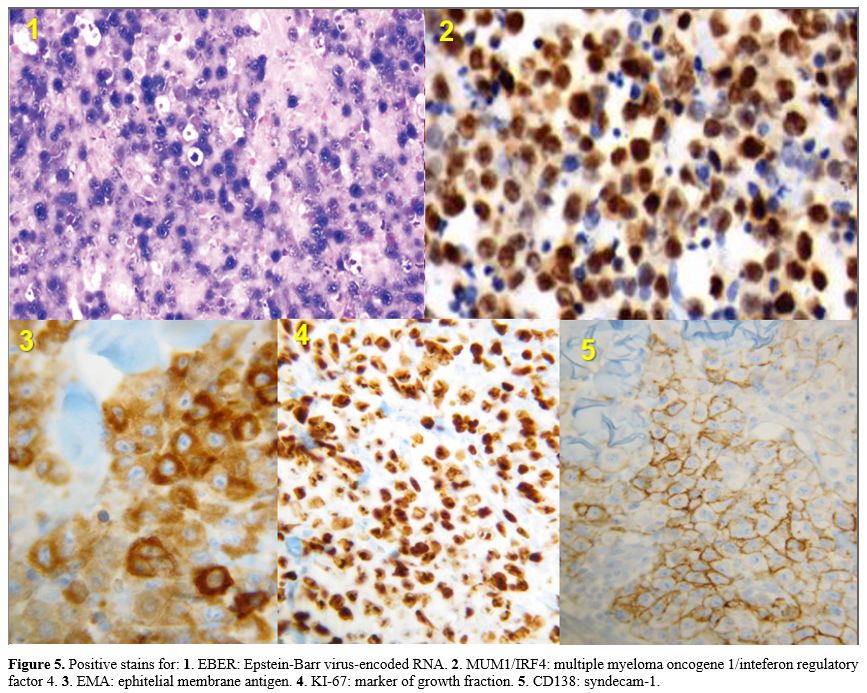 |
Figure 5. Positive
stains for: 1. EBER: Epstein-Barr virus-encoded RNA. 2. MUM1/IRF4:
multiple myeloma oncogene 1/inteferon regulatory factor 4. 3. EMA:
ephitelial membrane antigen. 4. KI-67: marker of growth fraction. 5.
CD138: syndecam-1.
|
Differential Diagnosis
Plasmablastic
lymphoma (PBL), extramedullary plasma cell tumor/plasmablastic myeloma
(EMPCT/PBM), primary effusion lymphoma (PEL), HHV8+ diffuse large B
cell lymphoma (DLBCL) not otherwise specified, and ALK+ large B cell
lymphoma (LBCL) belong to a category of lymphoproliferative neoplasms
that share a common characteristic of exhibiting plasmablastic
morphology. These neoplasms commonly display a tendency towards
aggressive behavior and frequently correlate to a poor prognosis.
Identifying each distinct entity frequently poses challenges due to the
diseases' rarity and histopathologic traits overlapping with each
other.
Except for cavity-based primary effusion lymphoma (PEL),
which displays the presence of plasmablasts and immunoblasts within the
effusion, all other neoplasms mentioned in this context exhibit a
layout of plasmablasts and immunoblasts in a pattern of sheets. In
immunophenotypic analysis, it was found that most of these tumors do
not have the typical expression of mature B-cell antigens like CD19,
CD20, PAX5, and CD79a. However, they demonstrate the presence of plasma
cell markers, including CD138, VS38c, and MUM-1.
HHV8-positive
diffuse large B-cell lymphoma (DLBCL) is a distinct entity that
exhibits different levels of B-cell antigen expression and reduced
expression of plasma cell antigens. EBER in situ hybridization and
immunohistochemical analysis of HHV-8, LANA1, and ALK can usually be
used to make a correct diagnosis. PBL is frequently distinguished by
the presence of Epstein-Barr virus-encoded small RNA (EBER) positivity,
human herpesvirus 8 (HHV8) negativity, and anaplastic lymphoma kinase
(ALK) negativity. In contrast, PBM is distinguished by the lack of
Epstein-Barr virus-encoded small RNA (EBER), human herpesvirus 8
(HHV8), and anaplastic lymphoma kinase (ALK). Both primary effusion
lymphoma (PEL) and extra-cavitary PEL demonstrate frequently
Epstein-Barr virus-encoded small RNA (EBER) positivity, human
herpesvirus 8 (HHV8) positivity, and anaplastic lymphoma kinase (ALK)
negativity. Diffuse large B-cell lymphoma (DLBCL) that is positive for
human herpesvirus 8 (HHV8) is distinguished nearly always lack of
Epstein-Barr virus-encoded RNA (EBER), presence of HHV8, and absence of
anaplastic lymphoma kinase (ALK) expression.[5,87] To summarize all of
those points, ALK-positive large B-cell lymphoma (LBCL) is
distinguished by the lack of Epstein-Barr virus-encoded small RNA
(EBER), human herpesvirus 8 (HHV8) negativity, and the presence of
anaplastic lymphoma kinase (ALK) positivity. The diagnosis of ALK+ LBCL
is rather straightforward due to the specific presentation of the ALK
protein, which sets it apart from the other five recognized
classifications.[5,87] Furthermore, unlike PBL, PEL, and HHV8+ DLBCL,
ALK+ LBCL does not demonstrate a preference for HIV+ or
immunocompromised people. The differentiation between ALK+ LBCL and
ALK+ anaplastic large cell lymphoma, as well as ALK+ non-hematopoietic
malignancies, must be made thanks to the existence of ALK expression.
The distinction between ALK+ LBCL and other ALK+ malignancies can be
established centered on the differential expression of BOB-1 and OCT2
and the absence of CD30 expression. Distinguishing between PBL and PBM
is crucial within a therapeutic context, given the significant
disparities in treating these two types of neoplasms. Plasmablastic
lymphoma and PBM demonstrate notable resemblances within the domain of
morphology. Based on an analysis of immunophenotypic characteristics,
it is apparent that these two organisms exhibit notable similarities.[86]
Both neoplasms demonstrate the presence of plasma cell-associated
antigens, specifically MUM1, CD138, CD38, and PRDM1, but lose the
expression of B-cell antigens such as CD19, CD20, and PAX-5. The rate
of elevated EBER expression is greater in most cases of PBL, especially
in cases where the patient is HIV positive. Conversely, EBER expression
is rarely observed in cases of PBM. Cyclin D1 expression has been
detected in a distinct subset of PBM. In contrast, PBL lacks the
expression of cyclin D1. There have been documented cases of excessive
expression of CD117 in select cases of PBM, while a lack of CD117
expression has been observed in cases of PBL. Therefore, in the context
of plasmablastic morphology, detecting EBER expression may often be
regarded as diagnostically meaningful for identifying plasmablastic
lymphoma (PBL). In contrast, the presence of cyclin D1 or CD117
expression suggests a probable diagnosis of plasmablastic myeloma
(PBM). The immunohistochemical analyses suggest that both PBL and PBM
exhibit MYC gene expression. Interphase fluorescence in situ
hybridization (FISH) is a more common way to find MYC translocations in
PBL, especially when the translocations involve immunoglobulin
genes.[81-88] Based on the small number of next-generation sequencing
studies conducted on PBL and PBM, it can be concluded that gene
mutational analysis does not appear to play a substantial role in
distinguishing between these two neoplasms. Instead, differentiation is
primarily achieved by considering clinical presentation and laboratory
findings, particularly when markers such as EBER, cyclin D1, and CD117
exhibit negative results.[81-88] The probability of detecting PBL is
higher in people with compromised immune systems when there is
significant extramedullary involvement or lymphadenopathy present.
Conversely, the likelihood of diagnosing PBM increases when the disease
predominantly impacts the bone marrow or when there are signs that
fulfill the CRAB criteria, including elevated levels of M protein. When
a definitive distinction cannot be determined via thorough clinical and
histopathologic evaluation, the designation of plasmablastic malignancy
may be given, with plasmablastic lymphoma (PBL) and plasmablastic
myeloma (PBM) being regarded as potential alternative diagnoses.[81-88]
In certain cases, there can be difficulties in differentiating between
HHV8+ DLBCL and PEL, especially in the context of extracavitary PEL.
Human herpesvirus 8 (HHV8) and Epstein-Barr virus-encoded small RNA
(EBER) are often found together in primary effusion lymphoma (PEL).
However, HHV8-positive diffuse large B-cell lymphoma (DLBCL) generally
does not exhibit EBER expression despite its HHV8 positivity. That
said, it is important to know that some types of primary effusion
lymphoma (PEL), which are only found in people who do not have HIV, do
not show expression of Epstein-Barr virus-encoded small RNA
(EBER).[81-88] Patients diagnosed with primary effusion lymphoma (PEL)
exhibit a clinical characteristic known as concurrent body cavity
involvement. However, this characteristic is not observed in
individuals diagnosed with human herpesvirus 8-positive diffuse large
B-cell lymphoma (HHV8+ DLBCL). On the other hand, it is important to
keep in mind that HHV8-positive diffuse large B-cell lymphoma (DLBCL)
can arise from a lower-grade HHV8-associated lymphoproliferative
disorder. Accordingly, certain signs that point to HHV8+ multicentric
Castleman disease or germinotropic lymphoproliferative disease in the
patient's medical history, whether from the past or the present, are
used to support the diagnosis of HHV8+ DLBCL. HHV8-positive diffuse
large B-cell lymphoma (DLBCL) often has different levels of pan-B-cell
markers and less consistent levels of CD138 and CD38 expression within
the immunophenotype. The presence of IgM and cytoplasmic k light-chain
expression has been found in tumor cells. In contrast, prior studies
have shown that PEL tends to demonstrate negative expression for
pan-B-cell markers but positive expression for CD138 and CD38.[5,81-88]
The PEL lymphoma cells have an impairment in the expression of both
immunoglobulin heavy and light chains. The prevailing hypothesis
suggests that the lymphoma cells observed in HHV8+ DLBCL exhibit the
characteristics of naive B cells, specifically those that have not
undergone somatic hypermutation and are in a pre-germinal center state.
Further, the PEL lymphoma cells have the characteristics of B cells
that have undergone terminal differentiation subsequent to the germinal
center stage, and they possess somatic mutations in their
immunoglobulin genes.[87-88] Taking advantage of molecular analysis, when
available, appears to offer benefits in determining a definitive
diagnosis for complex cases by determining the presence or absence of
somatic mutations. Although there have been sporadic mentions in
research papers regarding the presence of EBER positivity in HHV8+
DLBCL cases, it is crucial to contemplate the practical implications.
The coexistence of Epstein-Barr virus-encoded small RNA (EBER) and
human herpesvirus 8 latency-associated nuclear antigen 1 (HHV8 LANA1),
in conjunction with plasmablastic and immunoblastic proliferation,
serves as a robust indication for the diagnosis of PEL. Nevertheless,
it is imperative to rule out any possibility for transformation from
previous HHV8-associated lymphoproliferative disease or the existence
of only nodal or splenic disease.[86-89]
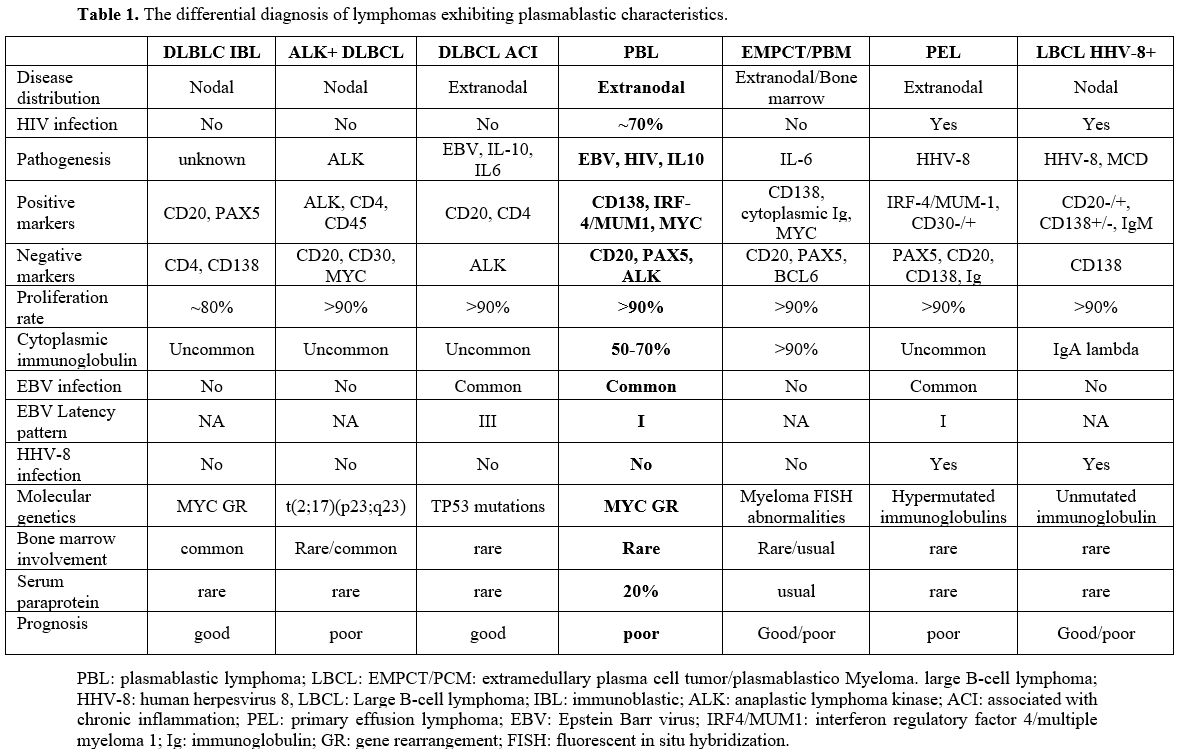 |
- Table 1. The differential diagnosis of lymphomas exhibiting plasmablastic characteristics.
|
Prognostic Factors and Survival
In
earlier research investigations, it was reported that the median
overall survival ranged from 8 to 15 months. Individuals whose
condition was not treated experienced a significantly low median
overall survival (OS) that corresponds to previously reported results,
surviving for an average of 1.9 months.[91] Presented
below are a few illustrative examples. In a study of 112 HIV-positive
PBL patients, the median overall survival (OS) was 15 months, and the
3-year OS rate was 25%.[90] In another study of 76 PBL patients with negative HIV tests, the median OS was 9 months, and the 2-year OS rate was 10%.[91]
In a large study of 300 PBL patients, the median overall survival (OS)
was 8 months. Three patient groups were compared for median overall
survival (OS). HIV-positive individuals had a median OS of 10 months,
while HIV-negative immunocompetent patients had 11 months. After
transplantation, PBL patients had a smaller median OS of 7 months.[8]
Another academic study of 50 HIV-positive people using cART found
comparable results with a median 11-month survival length and a 24%
5-year survival rate.[6] An early investigation in
Germany comprised 18 HIV-positive PBL patients diagnosed after 2005.
The study of 30 medical centers found a median OS of 5 months.[9]
The AIDS Malignancy Consortium abstracted data from nine locations,
including 19 HIV patients who received medication after 1999. The
estimated one-year survival rate was 67%.[92]
In
recent years, there have been reports of enhanced survival rates. The
Lymphoma Study Association (LYSA) examined 135 people with PBL for the
study. Among these patients, 80% received chemotherapy. The study found
that the median overall survival (OS) was 32 months.[23]
The investigation conducted on 248 treated patients in the SEER
database revealed a median overall survival (OS) of 47 months.[24]
Very recently, in small-scale research including patients with PBL, the
administration of bortezomib in combination with dose-adjusted
etoposide, prednisone, vincristine, cyclophosphamide, and doxorubicin
(EPOCH) resulted in a median survival duration of 62 months.[142] Overall, several comparison studies demonstrate that HIV does not significantly affect PBL outcomes.[5-10]
However, several data points suggest a link between immunosuppression and worse outcomes in HIV-negative patients.[93]
The International Prognostic Index (IPI) scoring system is widely used
to classify aggressive lymphomas. PBL patients' International
Predictive Index (IPI) scores are predictive, according to several
retrospective investigations. However, in PBL, the International
Prognostic Index (IPI) score appears to be most indicative of a poor
prognosis when advanced disease stages and reduced functioning status
are present.[94] An independent study linked age to LDH levels and unfavorable outcomes.[9]
The prognostic consequences of Epstein-Barr virus (EBV)-related antigen
expression in PBL remain unclear. Many studies have found no
correlation between Epstein-Barr virus (EBV) expression and
HIV-associated PBL prognosis.[5-9] However, Epstein-Barr virus (EBV) has been linked to a better prognosis in immunocompetent PBL patients.[94]
It is crucial to note that the production of EBER by malignant cells
frequently affects Epstein-Barr virus (EBV) expression. Recent research
has linked MYC gene rearrangements to a shorter life expectancy in PBL
patients. The results showed that patients with MYC increases or
translocations had significantly lower overall survival (OS) than those
with normal MYC status.[5-11] MYC gene rearrangements were also linked to death from any cause in HIV-positive PBL patients, six times greater.[5-11]
It is unclear if CD20 or CD45 expression levels affect PBL patients'
clinical results. However, other studies have associated Ki-67
expression levels above 80% with a poor prognosis.[5-11]
Overall survival was not significantly related to low CD4 counts in
HIV-associated PBL, but lower CD4 levels were linked to shorter
progression-free survival.[5-11]
HIV-negative Patients with PBL. The Moffitt Cancer Center studied nine consecutive PBL patients without HIV between 1999 and 2010.[93]
The median age at diagnosis was 58, ranging from 46 to 67. Five of nine
patients (55%) had aaIPI values greater than two. Seven of nine
patients (78% of the sample) underwent CHOP. Rituximab was given to
four patients. Two patients (22% of the nine-patient sample) received
hyper-CVAD therapy. Seven chemotherapy patients (78% of the sample)
achieved complete remission. One patient had a partial response
sufficient for autologous hematopoietic cell transplantation (AHCT)
consolidation, while another needed further treatment. 89% of
individuals responded. After CR1, four patients (44% of the sample)
with aaIPI 2 underwent AHCT. At data collection, the median overall
survival (OS) was unknown.[93] The authors also
reviewed the literature on 70 PBL patients who tested negative for
HIV.93 CHOP, or a comparable treatment, was given to 60% of patients.
Two patients with primary refractory illnesses survived 6 and 12 months
after autologous hematopoietic cell transplantation (AHCT). Overall
survival (OS) was 9 months. The researchers found that advanced age,
extranodal disease, and immunosuppression are risk factors for
unfavorable outcomes. This single-institution study found better
results than earlier ones due to the fast adoption of autologous
hematopoietic cell transplantation (AHCT) in high-risk patients.[92]
Differences between HIV-positive and negative PBLs.
Despite limited data on HIV-negative PBL patients, few differences have
been found. HIV-negative PBL is more common in women and older adults.
Regarding the clinical onset, HIV-negative PBL exhibits more extra-oral
symptoms, indicating greater heterogeneity. Around 50% of HIV-negative
PBL cases are linked to immunosuppression.[94] The literature study shows that HIV-negative PBL patients had a worse outcome than HIV-positive patients.[5-11]
HIV-negative PBL patients have a median survival of nine months.
Interestingly, a complete remission after induction chemotherapy is the
single predictor of better outcomes.[93] Multiple
studies have shown that HIV-associated PBL patients respond better to
chemotherapy than HIV-negative PBL patients. Antiretroviral therapy
combination (cART) in HIV-positive people who never received cART may
explain this. That treatment method improves immune surveillance and
restores immunological function, contributing to the reported
improvement. Patients with HIV-associated PBL who test positive for
Epstein-Barr virus (EBV) have a better prognosis than those who test
negative.[30-33] Results vary because cART reduces viral replication.[30-32]
In addition, HIV-negative older adults often develop PBL. This group
has inferior performance status and physiological reserve, making them
less able to tolerate higher-dose chemotherapy.[94] HIV-associated PBL patients receiving cART and those not receiving cART have similar rates of opportunistic infections.[31]
Approximately 77% of HIV-associated PBL patients who received
chemotherapy, specifically the CHOP regimen or more aggressive
protocols, responded.[94] Overall survival was poor
in this trial, with a median survival period of 14 months. No evidence
suggests that more intensive treatment regimens enhance survival.[22,93-95]
Oral versus extra-oral plasmablastic lymphoma.
PBLs have been classified into two separate categories, respectively
oral and extra-oral, as indicated in different research investigations.
This classification implies that there is heterogeneity in PBL based on
the particular location.[96,97] This issue was
analyzed in a cohort of 101 cases of oral and extraoral PBL from a
specific institution located in a location with a high prevalence of
HIV.[98] The results of this study contradict the
assumption that oral and extra-oral PBL should be considered distinct
and unrelated entities, proposing instead that they are part of a
continuum of the same disease.[98] These results
suggest that regardless of the location and presence of HIV and EBV,
PBLs displayed comparable clinicopathologic, immunophenotypic, and
behavioral features. Analysis of molecular characteristics in oral and
extra-oral PBL failed to identify any noticeable differences associated
with the location of the tumor.[98] The explanation
behind the increased incidence of oral manifestations of PBL in
comparison to extraoral manifestations remains unknown. Nevertheless, a
similar trend has been noted in instances of HIV-associated Kaposi
sarcoma and Burkitt lymphoma. In conclusion, the extra-oral PBL cases
demonstrated comparable attributes to their oral counterparts with
regards to gender, age distribution, HIV status, morphological
appearance, immunophenotypic profile, and Epstein-Barr virus-encoded
small RNA (EBER) status.[99]
CD138 negative Plasmablastic Lymphoma.
Approximately 10% of PBL cases have an unusual immunophenotypic
profile, which is defined by the lack of CD138 expression. The
diagnosis procedure is made much more difficult by this specific
feature. When compared to CD138+ PBL, which mostly manifests as oral
lesions, CD138-PBL is characterized by extraoral lesions, the most
common of which is lymphadenopathy, which is then followed by
gastrointestinal lesions.[100-102] Compared to
patients who are CD138+, these patients had a reduced documented
incidence of HIV and EBV infection. The MYC gene has reportedly a
significant impact on disease development, especially when the
Epstein-Barr virus (EBV) is present. There were no noticeable
variations in survival between patients with PBL who were
CD138-positive and CD138-negative.[100-105]
CD 20 positive Plasmablastic Lymphoma. The presence of CD20 expression has been detected in 10% of patients who are HIV-positive and have PBL.[5-11]
Nevertheless, it is generally recognized that a significant proportion
of PBL patients have a lack of CD20 expression, hence limiting the
possible benefit of the anti-CD20 monoclonal antibody Rituximab.
However, there have been recorded cases of CD20+ HIV-negative PBL.[106]
As is well known, Rituximab has shown significant improvements in the
curative treatment of diffuse large B-cell lymphoma (DLBCL) patients
who are positive for CD20.[106] Therefore, it is
recommended to include the anti-CD20 monoclonal antibody Rituximab in
the treatment strategy for CD20+ PBL after doing an in-depth evaluation
of the patient's histological characteristics.
International prognostic Indices.
In a recent study, the performance of three scoring systems, namely the
International Prognostic Index (IPI), Revised International Prognostic
Index (R-IPI), and National Comprehensive Cancer Network International
Prognostic Index (NCCN-IPI), were examined in the context of
plasmablastic lymphoma.[107-110] The results of this
study provide evidence to endorse the utilization of the International
Prognostic Index (IPI) and National Comprehensive Cancer Network (NCCN)
IPI scores in PBL settings.[110] Nevertheless, it is
imperative to have a novel prognostic tool that can effectively
identify subgroups at risk of early relapse or refractory disease, as
well as late relapses. The incorporation of molecular characterization
and cART therapy is a promising approach to potentially achieving this
outcome within the patient population.[110]
Special Popolation PBL
Pediatric cases.
Pediatric lymphoma is a rather rare phenomenon. Lymphoma accounts for
around 8% of cancers observed in the pediatric and teenage populations.[111]
In particular, the incidence of non-Hodgkin lymphoma among pediatric
cancer patients is estimated to be around 5%, while Hodgkin lymphoma is
detected in approximately 3% of cases. According to currently published
research, there is evidence to support the assumption that people who
are younger than 14 years old display an increased likelihood of
developing non-Hodgkin lymphoma.[112] The presence
of PBL has been observed in pediatric patients with HIV infection, as
well as in patients with different immunological deficiencies and
immunocompetent young people, despite the higher prevalence in adult
patients. The literature review resulted in the identification of a
total of 35 cases.[5-10] According to the study, the
condition was observed in individuals at a median age of ten years,
with a range ranging from 0 to 17 years. Furthermore, it is significant
that more than 80% of the patients had been classified into the
advanced stage at their initial presentation.[5] In
addition, it was observed that the jaw and oral cavity exhibited the
highest incidence of disease presentation. Still, there have been
reported cases of extranodal manifestations in various locations, such
as the skin, vulva, spine, and head. The overall prognosis is typically
detrimental, as seen by the scarcity of known long-term survivors.
Specifically, there have been only two recorded examples of individuals
who have lived for 3.5 and eight years, respectively. A total of 25
cases showed tests positive for HIV, while 10 of them were negative for
HIV.[10,112-118] Limited-stage plasmablastic lymphoma (LS PBL).
Due to the limited patient population and lack of randomized studies,
developing definite treatment guidelines for limited-stage
plasmablastic lymphoma (LS-PBL) is difficult. Thus, guidelines largely
use retroactive evidence. Due to the aggressive nature of the disease,
greater chances of relapse, bad outcomes, and insufficient
evidence-based outcomes for limited-stage PBL patients, intensive
treatment regimens are often suggested. Various studies on LS and ES
patients have indicated that intensive chemotherapy and consolidation
therapy with autologous stem cell transplantation improves survival.[119]
However, other studies have found little advantage in intense therapy.
Thus, healthcare practitioners face ambiguity when suggesting
treatments, especially for LS patients.[119,121] A recent study on LS PBL described it in detail and reported mostly positive outcomes.[121]
The study calculated the three-year probability of progression-free
survival (PFS) and overall survival (OS) at 72% (95% CI 62–83) and 79%
(95% CI 69–89). HIV-positive patients had no trend toward worse
progression-free survival (PFS) or overall survival (OS). Instead,
HIV-positive people had better progression-free survival (PFS) and
overall survival (OS) in the multivariate analysis. When frontline
treatment was considered, the multivariate regression analysis showed a
statistically significant link between high levels of lactate
dehydrogenase (LDH) at diagnosis and a higher risk ratio (HR) for
progression-free survival (PFS). Looking to clinical characteristics
like gender, age, stage, and EBV expression did not significantly
affect progression-free survival (PFS) or overall survival (OS).
Compared to CHOP- or EPOCH-based first-line cytotoxic treatments,
aggressive first-line treatments like hyper-CVAD or modified hyper-CVAD
did not improve progression-free survival (PFS) or overall survival
(OS). The study found that radiation therapy (RT) consolidation
improved progression-free survival (PFS) and overall survival (OS).
Statistically significant (p < 0.05) data from 108 patients showed
this improvement.[120] It appears that PBL-LS
patients have good results, especially when they receive
disease-specific treatment. The multivariate regression analysis
demonstrates that EPOCH-based regimens improved progression-free
survival (PFS) more than CHOP-based regimens as the first therapy
choice. Radiation consolidation after the first chemotherapy improved
results, although the improvements were not statistically significant.[121]Post-transplant PBL (PT-PBL).
Plasmablastic lymphoma is a type of lymphoma that may occur following
transplantation. Post-transplant lymphoproliferative disorder (PTLD) is
a rare and aggressive tumor that mostly affects patients who have
received solid organ transplants.[102] PT-PBL
usually develops at a later stage following transplantation, with a
median onset occurring at 96 months post-transplantation and a range
encompassing from 2 to 360 months.[5-11] This
condition exhibits a higher prevalence in males and in recipients of
heart and kidney allografts. The clinical presentation is characterized
by a predominance of skin and lymph node involvement and digestive
diseases.[5,10,101]
The characterization of PT-PBL currently needs to be improved in
clarity, and available evidence is scarce regarding the clinical and
genetic alterations that underlie this condition. Although
there are similarities in the genetic changes observed in HIV-related
PBL and PT-PBL, there are also discernible distinctions between the
two. A significant proportion, approximately 50%, of the cases
examined, encompassing both EBV-positive (EBV+) and EBV-negative (EBV-)
cases, had small foci characterized by plasmacytic differentiation.
These observations accord with the conclusions drawn by previous
researchers in this area.[5-10] Similar to other
B-cell post-transplant lymphoproliferative disorders (PTLDs), the
majority (64%) of PT-PBL exhibited germinal center transit. While the
B-cell program is often suppressed in PBL, certain cases exhibit the
presence of B-cell antigens. A study revealed that 27% of (PT-PBL)
displayed partial CD20 expression, a percentage that is similar to
other forms of PBL (20%). More than 50% of the PT-PBL samples exhibited
positivity for PAX5 and/or CD79a.[122,123] The expression of CD79a has been seen in 45% of PBL associated with HIV infection as well as in 68% of PT-PBL.[3]
Additionally, PAX5 expression has been detected in 22–26% of mostly
HIV-related PBL. The presence of PAX5+ was observed in a significant
proportion (60%) of Epstein-Barr virus-positive (EBV+) PT-PBL. However,
no disparities were observed in the functional groupings of mutations
between cases that expressed B-cell antigens and those that did not.[124]
There is a significant variation observed in the Ki-67 proliferation
indices within PT-PBL, with Ki-67 labeling ranging from 25% to 100%. The
presence of PD-L1 expression, infiltration of PD1+ T cells into tumors,
and elevation of immune escape genes have been documented in cases of
Epstein-Barr virus-positive PBL (EBV+ PBL) and post-transplant
lymphoproliferative disorder (PTLD).[124-125] The
presence of PD-L1 expression was seen in specific subsets of
EBV-positive and EBV-negative (PT-PBL). The EBV-negative PT-PBL
fraction had additional alterations in immune evasion genes, namely FAS
and CD58.[124] PT-PBL demonstrates infrequent
manifestations of morphologic or immunophenotypic indications of marrow
infiltration or the presence of bone (lytic) lesions on imaging, as
well as myeloma-associated laboratory irregularities, such as
monoclonal gammopathy and hypercalcemia. However, a small percentage of
cases can manifest these alterations.[123-125]
Moreover, several genetic abnormalities identified in PT-PBL exhibited
similarities with those observed in MM. Notably, these abnormalities
were detected in both EBV-positive and EBV-negative PT-PBL cases, as
well as in PBL cases associated with HIV infection. The overall
quantity of modifications exhibited variation in comparison to MM.[67,125]
The prognosis of PT-PBL is determined by various factors, including
age, stage of the disease, and nodal involvement. However, it has been
previously documented that certain PT-PBL patients have exhibited
long-term survival.[98] The primary objectives of
post-transplant lymphoproliferative disorder (PTLD) care encompass the
elimination of PTLD and the preservation of allograft function. These
aims often entail conflicting treatment approaches, and often, one
objective will be given priority over the other based on the individual
patient's specific requirements. So, the primary strategy employed in
the eradication of post-transplant lymphoproliferative disorder (PTLD)
is the lowering of immunosuppression. However, it is important to note
that this approach has the potential danger of graft rejection and
failure. The care strategies for post-transplant lymphoproliferative
disorder (PTLD) might vary across different institutions. However, a
common method involves administering lymphoma-directed drugs, which
often consist of conventional chemotherapy and/or radiotherapy, to the
majority of patients. A comprehensive examination of existing academic
literature revealed the presence of 37 cases of PBL in individuals who
had undergone organ transplantation.[5-10] Among these
cases, it was observed that 28 (76%) were male, and the median age at
which the disorder manifested was 62 years. A total of 38% of the
observed cases occurred subsequent to a heart transplant, while 27%
were reported following a kidney transplant.[5]
Additionally, 14% of the cases were observed after a hematopoietic stem
cell transplant, 11% after a lung transplant, 8% after a liver
transplant, and 3% after a pancreas transplant. Interestingly, the
lymph nodes were found to be the most frequently affected region,
accounting for 30% of cases, with the skin being the second most
regularly implicated site at 22%. Approximately half of the patients
diagnosed with posttransplant PBL exhibit advanced clinical stages.[5-10]Spontaneous regression.
A considerable amount of empirical data exists, consisting of several
reported cases and comprehensive analyses, that supports the occurrence
of spontaneous regression in low-grade lymphomas, but it is
infrequently observed in aggressive lymphomas.[126]
In particular circumstances, there have been documented cases where
aggressive PBL has shown spontaneous regression following the
introduction of antiretroviral therapy (ART).[126-132]
This therapy has been observed to contribute to the restoration of
immune function in individuals infected with HIV and the subsequent
activation of immune surveillance against the lymphoma, resulting in
its regression.Moreover,
in some cases, the cessation of methotrexate treatment without the use
of additional anti-neoplastic therapy could contribute to the
regression of PBL.[126-132]Transformed PBL.
The evolution of indolent lymphomas into aggressive histologies is a
critical phenomenon in the management of patients, requiring a
modification in their treatment approach. Despite a declining trend in
the general occurrence of transformation, this condition continues to
pose a significant problem. It is associated with a less favorable
prognosis when compared to patients who do not experience
transformation.[133] Based on a comprehensive
analysis of existing research articles, it has been reported that there
are 30 cases in which PBL has originated from a preexisting
hematological disorder.[5] Typically arising as a
consequence of a transformation from chronic lymphocytic leukemia or
low-grade follicular lymphoma. A total of 10 cases have been documented
in which double-hit follicular lymphoma, or DLBCL, has transformed PBL
among individuals who are HIV-negative.[133-138] In
cases where a transformation is suspected, it is imperative to conduct
a biopsy in order to confirm the diagnosis and acquire tissue for
genomic analysis. Because the transformation could mean either a change
from the original hematological disease or the appearance of a new
primary lymphoma, this is very important because it affects the
prognosis and the treatment options. In order to establish the clonal
link between the primary tumor and the plasmablastic neoplasm, it is
possible to perform a PCR and FISH investigation targeting the
immunoglobulin heavy chain and BCL2 genes.[138-141]
When compared to clonally related T-PBL cases, cases that are
genetically and immunologically different have a better response to
chemoimmunotherapy. Finally, the remaining patients must enroll in the
clinical trial.[138-141]
Conclusions
This
first part of the state-of-the-art review provided an overview of the
epidemiology, etiology, clinicopathologic characteristics, differential
diagnosis, prognostic variables, and special populations associated
with plasmablastic lymphoma. In the second part of this article, our
focus will be on the treatment of plasmablastic lymphoma, specifically
examining both the conventional, consolidated approach and the novel
therapeutic strategy.
References
- Stein H. &
Dallenbach F. in Neoplastic
Hematopathology (ed. Knowles, D.M.) 675-714 (Williams &
Wilkins,
Baltimore, MA, 1992).
- Delecluse
HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of
the oral cavity: a new entity associated with the human
immunodeficiency virus infection. Blood. 1997;89:1413-1420. https://doi.org/10.1182/blood.V89.4.1413
PMid:9028965
- Alaggio
R, Amador C, Anagnostopoulos I, et al.: The 5th
edition of the World Health Organization Classification of
Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. 2022
Jul;36(7):1720-1748. doi: 10.1038/s41375-022-01620-2. Epub 2022 Jun 22.
PMID:35732829; PMCID:PMC9214472.
- Campo
E, Jaffe ES, Cook JR, et al.: The International Consensus
Classification of Mature Lymphoid Neoplasms: a report from the Clinical
Advisory Committee. Blood. 2022 Sep 15;140(11):1229-1253. doi:
10.1182/blood.2022015851. https://doi.org/10.1182/blood.2022015851
PMid:35653592 PMCid:PMC9479027
- Castillo
JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic
lymphoma. Blood. 2015 Apr 9;125(15):2323-30. doi:
10.1182/blood-2014-10-567479. Epub 2015 Jan 30. https://doi.org/10.1182/blood-2014-10-567479
PMid:25636338
- Castillo
JJ, Furman M, Beltran BE, et al. Human immunodeficiency
virus-associated plasmablastic lymphoma: poor prognosis in the era of
highly active antiretroviral therapy. Cancer. 2012; 118(21):5270-5277. https://doi.org/10.1002/cncr.27551
PMid:22510767
- Colomo
L, Loong F, Rives S, et al. Diffuse large B-cell lymphomas with
plasmablastic differentiation represent a heterogeneous group of
disease entities. Am J Surg Pathol. 2004;28(6): 736-747. https://doi.org/10.1097/01.pas.0000126781.87158.e3
PMid:15166665
- Morscio
J, Dierickx D, Nijs J, et al. Clinicopathologic comparison of
plasmablastic lymphoma in HIV-positive, immunocompetent, and
posttransplant patients: single-center series of 25 cases and
meta-analysis of 277 reported cases. Am J Surg Pathol.
2014;38(7):875-886. https://doi.org/10.1097/PAS.0000000000000234
PMid:24832164
- Schommers
P, Wyen C, Hentrich M, et al. Poor outcome of HIV-infected patients
with plasmablastic lymphoma: results from the German AIDS-related
lymphoma cohort study. AIDS. 2013;27(5):842-845. 7. https://doi.org/10.1097/QAD.0b013e32835e069d
PMid:23574794
- Teruya-Feldstein
J, Chiao E, Filippa DA, et al. CD20-negative large-cell lymphoma with
plasmablastic features: a clinically heterogenous spectrum in both
HIV-positive and -negative patients. Ann Oncol. 2004;15(11):1673-1679. https://doi.org/10.1093/annonc/mdh399
PMid:15520070
- Bibas
M, Castillo JJ. Current knowledge on HIVassociated Plasmablastic
Lymphoma. Mediterr J Hematol Infect Dis. 2014;6(1):e2014064. https://doi.org/10.4084/mjhid.2014.064
PMid:25408850 PMCid:PMC4235470
- Swerdlow
SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J (Eds.):
World Health Organization classification of Tumours of Haematopoietic
and Lymphoid Tissues. Revised 4th
ed. Lyon: IARC; 2017.
- Bailly
J, Jenkins N, Chetty D, Mohamed Z, Verburgh ER, Opie JJ. Plasmablastic
lymphoma: An update. Int J Lab Hematol. 2022 Sep;44 Suppl 1(Suppl
1):54-63. doi: 10.1111/ijlh.13863. Erratum in: Int J Lab Hematol. 2022
Dec;44(6):1121. https://doi.org/10.1111/ijlh.13863
PMid:36074710 PMCid:PMC9545967
- Carbone
A, Vaccher E, Gloghini A. Hematologic cancers in individuals infected
by HIV. Blood. 2022 Feb 17;139(7):995-1012. doi:
10.1182/blood.2020005469. https://doi.org/10.1182/blood.2020005469
PMid:34469512
- Re
A, Cattaneo C, Montoto S. Treatment management of haematological
malignancies in people living with HIV. Lancet Haematol. 2020
Sep;7(9):e679-e689. doi: 10.1016/S2352-3026(20)30115-0. Epub 2020 Aug
10. https://doi.org/10.1016/S2352-3026(20)30115-0
PMid:32791044
- Rapiti
N, Peer N, Abdelatif N, Rapiti P, Moosa Y. HIV-associated plasmablastic
lymphoma: A single-centre 12-year experience in Kwa-Zulu Natal, South
Africa. HIV Med. 2022 Sep;23(8):837-848. doi: 10.1111/hiv.13266. Epub
2022 Mar 1. https://doi.org/10.1111/hiv.13266
PMid:35229978
- Jessa
R, Chien N, Villa D, Freeman CL, Slack GW, Savage KJ, Scott DW, Sehn
LH, Song KW, Gerrie AS. Clinicopathological characteristics and
long-term outcomes of plasmablastic lymphoma in British Columbia. Br J
Haematol. 2022 Oct;199(2):230-238. doi: 10.1111/bjh.18399. Epub 2022
Aug 12. PMID: 35961783. https://doi.org/10.1111/bjh.18399
PMid:35961783
- Makady
NF, Ramzy D, Ghaly R, Abdel-Malek RR, Shohdy KS. The Emerging Treatment
Options of Plasmablastic Lymphoma: Analysis of 173 Individual Patient
Outcomes. Clin Lymphoma Myeloma Leuk. 2021 Mar;21(3):e255-e263. doi:
10.1016/j.clml.2020.11.025. Epub 2020 Dec 3. https://doi.org/10.1016/j.clml.2020.11.025
PMid:33419717
- Mai
B, Wang W, Lin M, Hu S, Wang XI, Chen L, Wahed A, Nguyen A, Ma HY,
Medeiros LJ, Hu Z. HIV-associated plasmablastic lymphoma in the era of
HAART: a single-center experience of 21 patients. AIDS. 2020 Oct
1;34(12):1735-1743. doi: 10.1097/QAD.0000000000002590. Erratum in:
AIDS. 2020 Nov 15;34(14):2159. https://doi.org/10.1097/QAD.0000000000002590
PMid:32889849
- Li
YJ, Li JW, Chen KL, Li J, Zhong MZ, Liu XL, Yi PY, Zhou H. HIV-negative
plasmablastic lymphoma: report of 8 cases and a comprehensive review of
394 published cases. Blood Res. 2020 Mar;55(1):49-56. doi:
10.5045/br.2020.55.1.49. Epub 2020 Mar 30. https://doi.org/10.5045/br.2020.55.1.49
PMid:32269975 PMCid:PMC7106118
- Castelli
R, Schiavon R, Preti C, Ferraris L. HIV-Related Lymphoproliferative
Diseases in the Era of Combination Antiretroviral Therapy. Cardiovasc
Hematol Disord Drug Targets. 2020;20(3):175-180. doi:
10.2174/1871529X20666200415121009. https://doi.org/10.2174/1871529X20666200415121009
PMid:32294049 PMCid:PMC8226149
- Lopez
A, Abrisqueta P. Plasmablastic lymphoma: current perspectives. Blood
Lymphat Cancer. 2018 Oct 4;8:63-70. doi:
10.2147/BLCTT.S142814. https://doi.org/10.2147/BLCTT.S142814
PMid:31360094 PMCid:PMC6467349
- Tchernonog
E, Faurie P, Coppo P, Monjanel H, Bonnet A, Algarte Génin M, Mercier M,
Dupuis J, Bijou F, Herbaux C, Delmer A, Fabiani B, Besson C, Le Gouill
S, Gyan E, Laurent C, Ghesquieres H, Cartron G. Clinical
characteristics and prognostic factors of plasmablastic lymphoma
patients: analysis of 135 patients from the LYSA group. Ann Oncol. 2017
Apr 1;28(4):843-848. doi: 10.1093/annonc/mdw684. https://doi.org/10.1093/annonc/mdw684
PMid:28031174
- Florindez
JA, Alderuccio JP, Reis IM, Lossos IS. Survival analysis in treated
plasmablastic lymphoma patients: a population-based study. Am J
Hematol. 2020 Nov;95(11):1344-1351. doi: 10.1002/ajh.25955. Epub 2020
Aug 26. https://doi.org/10.1002/ajh.25955
PMid:32777103
- Gars
E, Butzmann A, Ohgami R, Balakrishna JP, O'Malley DP. The life and
death of the germinal center. Ann Diagn Pathol. 2020 Feb;44:151421.
doi: 10.1016/j.anndiagpath.2019.151421. Epub 2019 Nov 13. https://doi.org/10.1016/j.anndiagpath.2019.151421
PMid:31751845
- Huang
C. Germinal Center Reaction. Adv Exp Med Biol. 2020;1254:47-53. doi:
10.1007/978-981-15-3532-1_4. https://doi.org/10.1007/978-981-15-3532-1_4
PMid:32323268
- Elsner
RA, Shlomchik MJ. Germinal Center and Extrafollicular B Cell Responses
in Vaccination, Immunity, and Autoimmunity. Immunity. 2020 Dec
15;53(6):1136-1150. doi: 10.1016/j.immuni.2020.11.006. https://doi.org/10.1016/j.immuni.2020.11.006
PMid:33326765 PMCid:PMC7748291
- Shindiapina
P, Ahmed EH, Mozhenkova A, Abebe T, Baiocchi RA. Immunology of
EBV-related lymphoproliferative disease in HIV-positive individuals.
Front Oncol. 2020; 10: 1723. https://doi.org/10.3389/fonc.2020.01723
PMid:33102204 PMCid:PMC7556212
- Marques-Piubelli
ML, Salas YI, Pachas C, Becker-Hecker R, Vega F, Miranda RN.
Epstein-Barr virus-associated B-cell lymphoproliferative disorders and
lymphomas: a review. Pathology. 2020; 52(1): 40- 52. https://doi.org/10.1016/j.pathol.2019.09.006
PMid:31706670
- El-Sharkawy
A, Al Zaidan L, Malki A. Epstein-Barr virus-associated malignancies:
roles of viral Oncoproteins in carcinogenesis. Front Oncol. 2018; 8:
265. https://doi.org/10.3389/fonc.2018.00265
PMid:30116721 PMCid:PMC6082928
- Gravelle
P, Pericart S, Tosolini M, et al. EBV infection determines the immune
hallmarks of plasmablastic lymphoma. Onco Targets Ther. 2018;
7(10):e1486950. https://doi.org/10.1080/2162402X.2018.1486950
PMid:30288350 PMCid:PMC6169584
- Vaughan
J, Perner Y, Mayne E, Wiggill T. Plasmablastic lymphoma in
Johannesburg, South Africa, in the era of widescale antiretroviral
therapy use. HIV Med. 2021; 22(3): 225- 230. https://doi.org/10.1111/hiv.12965
PMid:33022825
- Schmelz
M, Montes-Moreno S, Piris M, Wilkinson ST, Rimsza LM. Lack and/or
aberrant localization of major histocompatibility class II (MHCII)
protein in plasmablastic lymphoma. Haematologica. 2012; 97(10): 1614-
1616. https://doi.org/10.3324/haematol.2011.060186
PMid:22689685 PMCid:PMC3487566
- Ramburan
A, Kriel R, Govender D. Plasmablastic lymphomas show restricted EBV
latency profile and MYC gene aberrations. Leuk Lymphoma. 2022;
63(2):370-376. https://doi.org/10.1080/10428194.2021.1986218
PMid:34612761
- Nguyen
L, Papenhausen P, Shao H. The role of c-MYC in B-Cell lymphomas:
diagnostic and molecular aspects. Genes. 2017;8(4):116 https://doi.org/10.3390/genes8040116
PMid:28379189 PMCid:PMC5406863
- Chapman
J, Gentles AJ, Sujoy V, et al. Gene expression analysis of
plasmablastic lymphoma identifies downregulation of B-cell receptor
signaling and additional unique transcriptional programs. Leukemia.
2015;29(11):2270-2273 https://doi.org/10.1038/leu.2015.109
PMid:25921246
- Montes-Moreno
S, Martinez-Magunacelaya N, Zecchini-Barrese T, et al. Plasmablastic
lymphoma phenotype is determined by genetic alterations in MYC and
PRDM1. Mod Pathol. 2017;30(1):85-94 https://doi.org/10.1038/modpathol.2016.162
PMid:27687004
- Pasqualucci
L, Neumeister P, Goossens T, et al. Hypermutation of multiple
proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature.
2001;412(6844): 341-346. https://doi.org/10.1038/35085588
PMid:11460166
- Walker
BA, Wardell CP, Murison A, et al. APOBEC family mutational signatures
are associated with poor prognosis translocations in multiple myeloma.
Nat Commun. 2015;6:6997. https://doi.org/10.1038/ncomms7997
PMid:25904160 PMCid:PMC4568299
- Hemann
MT, Bric A, Teruya-Feldstein J, et al. Evasion of the p53 tumour
surveillance network by tumourderived MYC mutants. Nature.
2005;436(7052): 807-811 https://doi.org/10.1038/nature03845
PMid:16094360 PMCid:PMC4599579
- Salghetti
SE, Kim SY, Tansey WP. Destruction of myc by ubiquitin-mediated
proteolysis: cancer-associated and transforming mutations stabilize
myc. EMBO J. 1999;18(3):717-72 https://doi.org/10.1093/emboj/18.3.717
PMid:9927431 PMCid:PMC1171164
- Frontzek
F, Staiger AM, Zapukhlyak M, et al. Molecular and functional profiling
identifies therapeutically targetable vulnerabilities in plasmablastic
lymphoma. Nat Commun. 2021;12(1):5183. https://doi.org/10.1038/s41467-021-25405-w
PMid:34465776 PMCid:PMC8408158
- Li
WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol.
2008;18(11):545-551. https://doi.org/10.1016/j.tcb.2008.08.008
PMid:18848449 PMCid:PMC3082280
- Kiuchi
N, Nakajima K, Ichiba M, et al. STAT3 is required for the
gp130-mediated full activation of the c-myc gene. J Exp Med.
1999;189(1):63-73. https://doi.org/10.1084/jem.189.1.63
PMid:9874564 PMCid:PMC1887683
- Kerkhoff
E, Houben R, Loffler S, et al. Regulation of cmyc expression by ras/raf
signalling. Oncogene. 1998; 16(2):211-216. https://doi.org/10.1038/sj.onc.1201520
PMid:9464539
- Sears
R, Leone G, DeGregori J, et al. Ras enhances myc protein stability. Mol
Cell. 1999;3(2):169-179. https://doi.org/10.1016/S1097-2765(00)80308-1
PMid:10078200
- Weng
AP, Millholland JM, Yashiro-Ohtani Y, et al. cMyc is an important
direct target of Notch1 in T-cell acute lymphoblastic leukemi
a/lymphoma. Genes Dev. 2006;20(15):2096-2109. https://doi.org/10.1101/gad.1450406
PMid:16847353 PMCid:PMC1536060
- Dent
AL, Shaffer AL, Yu X, et al. Control of inflammation, cytokine
expression, and germinal center formation by BCL-6. Science.
1997;276(5312):589-592. https://doi.org/10.1126/science.276.5312.589
PMid:9110977
- Falini
B, Fizzotti M, Pucciarini A, et al. A monoclonal antibody (MUM1p)
detects expression of the MUM1/IRF4 protein in a subset of germinal
center B cells, plasma cells, and activated T cells. Blood.
2000;95(6):2084-2092. https://doi.org/10.1182/blood.V95.6.2084
PMid:10706878
- Klein
U, Casola S, Cattoretti G, et al. Transcription factor IRF4 controls
plasma cell differentiation and class switch recombination. Nat
Immunol. 2006;7(7):773-782. https://doi.org/10.1038/ni1357
PMid:16767092
- Shaffer
AL, Emre NC, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature.
2008;454(7201):226-231. https://doi.org/10.1038/nature07064
PMid:18568025 PMCid:PMC2542904
- Mondala
PK, Vora AA, Zhou T, et al. Selective antisense oligonucleotide
inhibition of human IRF4 prevents malignant myeloma regeneration via
cell cycle disruption. Cell Stem Cell. 2021;28(4):623-636.e9. https://doi.org/10.1016/j.stem.2020.12.017
PMid:33476575 PMCid:PMC8026723
- Weilemann
A, Grau M, Erdmann T, et al. Essential role of IRF4 and MYC signaling
for survival of anaplastic large cell lymphoma. Blood.
2015;125(1):124-132. https://doi.org/10.1182/blood-2014-08-594507
PMid:25359993
- Aldinucci
D, Rapana B, Olivo K, et al. IRF4 is modulated by CD40L and by
apoptotic and anti-proliferative signals in Hodgkin lymphoma. Br J
Haematol. 2010;148(1):115-118 https://doi.org/10.1111/j.1365-2141.2009.07945.x
PMid:19821826
- Rawlings
JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. J Cell Sci.
2004;117(Pt 8): 1281-1283. https://doi.org/10.1242/jcs.00963
PMid:15020666
- Vainchenker
W, Constantinescu SN. JAK/STAT signaling in hematological malignancies.
Oncogene. 2013; 32(21):2601-2613 https://doi.org/10.1038/onc.2012.347
PMid:22869151
- Darnell
JE Jr. STATs and gene regulation. Science. 1997;277(5332):1630-1635 https://doi.org/10.1126/science.277.5332.1630
PMid:9287210
- Crescenzo
R, Abate F, Lasorsa E, et al. Convergent mutations and kinase fusions
lead to oncogenic STAT3 activation in anaplastic large cell lymphoma.
Cancer Cell. 2015;27(4):516-532. https://doi.org/10.1016/j.ccell.2015.03.006
PMid:25873174 PMCid:PMC5898430
- Koskela
HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large
granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913. https://doi.org/10.1056/NEJMoa1114885
PMid:22591296 PMCid:PMC3693860
- Jerez
A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the
pathogenesis of chronic lymphoproliferative disorders of NK cells and
T-cell large granular lymphocyte leukemia. Blood. 2012;120(15):
3048-3057. https://doi.org/10.1182/blood-2012-06-435297
PMid:22859607 PMCid:PMC3471515
- Chapuy
B, Stewart C, Dunford AJ, et al. Molecular subtypes of diffuse large B
cell lymphoma are associated with distinct pathogenic mechanisms and
outcomes. Nat Med. 2018;24(5):679-690. [23] Schmitz R, Wright GW, Huang
DW, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma.
N Engl J Med. 2018;378(15):1396-1407 https://doi.org/10.1056/NEJMoa1801445
PMid:29641966 PMCid:PMC6010183
- Dolcetti
R, Gloghini A, Caruso A, et al. A lymphomagenic role for HIV beyond
immune suppression? Blood. 2016;127(11):1403-1409 https://doi.org/10.1182/blood-2015-11-681411
PMid:26773045 PMCid:PMC4826146
- Nicolae
A, Xi L, Pham TH, et al. Mutations in the JAK/ STAT and RAS signaling
pathways are common in intestinal T-cell lymphomas. Leukemia.
2016;30(11): 2245-2247. https://doi.org/10.1038/leu.2016.178
PMid:27389054 PMCid:PMC5093023
- Wenzel
SS, Grau M, Mavis C, et al. MCL1 is deregulated in subgroups of diffuse
large B-cell lymphoma. Leukemia. 2013;27(6):1381-1390. https://doi.org/10.1038/leu.2012.367
PMid:23257783
- Bray
SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol
Cell Biol. 2006;7(9):678-689. https://doi.org/10.1038/nrm2009
PMid:16921404
- Kovall
RA, Gebelein B, Sprinzak D, et al. The canonical notch signaling
pathway: structural and biochemical insights into shape, sugar, and
force. Dev Cell. 2017; 41(3):228-241. https://doi.org/10.1016/j.devcel.2017.04.001
PMid:28486129 PMCid:PMC5492985
- Meurette
O, Mehlen P. Notch signaling in the tumor microenvironment. Cancer
Cell. 2018;34(4):536-548 https://doi.org/10.1016/j.ccell.2018.07.009
PMid:30146333
- Liu
Z, Filip I, Gomez K, et al. Genomic characterization of HIV-associated
plasmablastic lymphoma identifies pervasive mutations in the JAK-STAT
pathway. Blood Cancer Discov. 2020;1(1):112-125 https://doi.org/10.1158/2643-3230.BCD-20-0051
PMid:33225311 PMCid:PMC7679070
- Seegmiller
AC, Wang HY, Hladik C, et al. Uniform expression of Notch1, suppressor
of B-cell-specific gene expression, in plasmablastic lymphoma. Arch
Pathol Lab Med. 2011;135(6):770-775 https://doi.org/10.5858/2009-0691-OA.1
PMid:21631271
- Santarpia
L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling
pathway in cancer therapy. Expert Opin Ther Targets.
2012;16(1):103-119. https://doi.org/10.1517/14728222.2011.645805
PMid:22239440 PMCid:PMC3457779
- Lowenstein
EJ, Daly RJ, Batzer AG, et al. The SH2 and SH3 domain-containing
protein GRB2 links receptor tyrosine kinases to ras signaling. Cell.
1992;70(3): 431-442. https://doi.org/10.1016/0092-8674(92)90167-B
PMid:1322798
- Bourne
HR, Sanders DA, McCormick F. The GTPase superfamily: a conserved switch
for diverse cell functions. Nature. 1990;348(6297):125-132. https://doi.org/10.1038/348125a0
PMid:2122258
- Leevers
SJ, Paterson HF, Marshall CJ. Requirement for ras in raf activation is
overcome by targeting raf to the plasma membrane. Nature.
1994;369(6479): 411-414. https://doi.org/10.1038/369411a0
PMid:8196769
- Schmitz
R, Young RM, Ceribelli M, et al. Burkitt lymphoma pathogenesis and
therapeutic targets from structural and functional genomics. Nature.
2012; 490(7418):116-120. https://doi.org/10.1038/nature11378
PMid:22885699 PMCid:PMC3609867
- Lohr
JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in
multiple myeloma: implications for targeted therapy. Cancer Cell.
2014;25(1):91-101. https://doi.org/10.1016/j.ccr.2013.12.015
PMid:24434212 PMCid:PMC4241387
- Walker
BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number
changes, and outcome: results of a sequencing study of patients with
newly diagnosed myeloma. J Clin Oncol. 2015;33(33): 3911-3920. https://doi.org/10.1200/JCO.2014.59.1503
PMid:26282654 PMCid:PMC6485456
- Hobbs
GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a
glance. J Cell Sci. 2016; 129(7):1287-1292. https://doi.org/10.1242/jcs.182873
PMid:26985062 PMCid:PMC4869631
- Biankin
AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal
aberrations in axon guidance pathway genes. Nature.
2012;491(7424):399-405. https://doi.org/10.1038/nature11547
PMid:23103869 PMCid:PMC3530898
- Brose
MS, Volpe P, Feldman M, et al. BRAF and RAS mutations in human lung
cancer and melanoma. Cancer Res. 2002;62(23):6997-7000.
- Trahey
M, McCormick F. A cytoplasmic protein stimulates normal N-ras p21
GTPase, but does not affect oncogenic mutants. Science.
1987;238(4826):542-545. https://doi.org/10.1126/science.2821624
PMid:2821624
- Scheffzek
K, Ahmadian MR, Kabsch W, et al. The RasRasGAP complex: structural
basis for GTPase activation and its loss in oncogenic ras mutants.
Science. 1997;277(5324):333-338. https://doi.org/10.1126/science.277.5324.333
PMid:9219684
- Vega
F, Chang CC, Medeiros LJ, et al. Plasmablastic lymphomas and
plasmablastic plasma cell myelomas have nearly identical
immunophenotypic profiles. Mod Pathol. 2005; 18(6):806-815.
https://doi.org/10.1038/modpathol.3800355 PMid:15578069
- Carbone
A. AIDS-related non-Hodgkin's lymphomas: from pathology and molecular
pathogenesis to treatment. Hum Pathol. 2002; 33(4):392-404
https://doi.org/10.1053/hupa.2002.124723 PMid:12055673
- Bogusz
AM, Seegmiller AC, Garcia R, Shang P, Ashfaq R, Chen W. Plasmablastic
lymphomas with MYC/IgH rearrangement: report of three cases and review
of the literature. Am J Clin Pathol. 2009;132(4):597-605.
https://doi.org/10.1309/AJCPFUR1BK0UODTS PMid:19762538
- Valera
A, Balagu 'e O, Colomo L, et al. IG/MYC rearrangements are the main
cytogenetic alteration in plasmablastic lymphomas. Am J Surg Pathol.
2010;34(11):1686-1694. https://doi.org/10.1097/PAS.0b013e3181f3e29f
PMid:20962620 PMCid:PMC2982261
- Chang
CC, Zhou X, Taylor JJ, et al. Genomic profiling of plasmablastic
lymphoma using array comparative genomic hybridization (aCGH):
revealing significant overlapping genomic lesions with diffuse large
B-cell lymphoma. J Hematol Oncol. 2009;2:47.
https://doi.org/10.1186/1756-8722-2-47 PMid:19909553 PMCid:PMC2789747
- Zhou
J, Nassiri M. Lymphoproliferative Neoplasms With Plasmablastic
Morphology. Arch Pathol Lab Med. 2022 Apr 1;146(4):407-414. doi:
10.5858/arpa.2021-0117-RA. https://doi.org/10.5858/arpa.2021-0117-RA
PMid:34559873
- Ahn
JS, Okal R, Vos JA, Smolkin M, Kanate AS, Rosado FG. Plasmablastic
lymphoma versus plasmablastic myeloma: an ongoing diagnostic dilemma. J
Clin Pathol. 2017 Sep;70(9):775-780. doi:
10.1136/jclinpath-2016-204294. Epub 2017 Mar 1.
https://doi.org/10.1136/jclinpath-2016-204294 PMid:28249941
- Wang
W, Kanagal-Shamanna R, Medeiros LJ. Lymphoproliferative disorders with
concurrent HHV8 and EBV infection: beyond primary effusion lymphoma and
germinotropic lymphoproliferative disorder. Histopathology. 2018
Apr;72(5):855-861. doi: 10.1111/his.13428. Epub 2018 Jan 3. PMID:
29105119. https://doi.org/10.1111/his.13428 PMid:29105119
- Castillo
J, Pantanowitz L, Dezube BJ. HIVassociated plasmablastic lymphoma:
lessons learned from 112 published cases. Am J Hematol.
2008;83(10):804-809. https://doi.org/10.1002/ajh.21250
PMid:18756521
- Castillo
JJ, Winer ES, Stachurski D, et al. HIVnegative plasmablastic lymphoma:
not in the mouth. Clin Lymphoma Myeloma Leuk. 2011; 11(2):185-189 https://doi.org/10.1016/j.clml.2011.03.008
PMid:21575922
- Noy
A, Chadburn A, Lensing SY. Moore P. Plasmablastic Lymphoma Is Curable
The HAART Era. A 10 Year Retrospective By The AIDS Malignancy
Consortium (AMC) [abstract]. Blood. 2013;122(21). Abstract 1801. https://doi.org/10.1182/blood.V122.21.1801.1801
- Liu
JJ, Zhang L, Ayala E, et al. Human immunodeficiency virus
(HIV)-negative plasmablastic lymphoma: a single institutional
experience and literature review. Leuk Res. 2011; 35(12):1571-1577. https://doi.org/10.1016/j.leukres.2011.06.023
PMid:21752466
- Castillo
JJ, Winer ES, Stachurski D, et al. Prognostic factors in
chemotherapy-treated patients with HIV-associated Plasmablastic
lymphoma. Oncologist. 2010;15(3):293-299. https://doi.org/10.1634/theoncologist.2009-0304
PMid:20167839 PMCid:PMC3227958
- Li
YJ, Li JW, Chen KL, Li J, Zhong MZ, Liu XL, Yi PY, Zhou H. HIV-negative
plasmablastic lymphoma: report of 8 cases and a comprehensive review of
394 published cases. Blood Res. 2020 Mar;55(1):49-56. doi:
10.5045/br.2020.55.1.49. Epub 2020 Mar 30. https://doi.org/10.5045/br.2020.55.1.49
PMid:32269975 PMCid:PMC7106118
- Meer
S, Perner Y, McAlpine ED, Willem P. Extraoral plasmablastic lymphoma in
a high human immunodeficiency virus endemic area. Histopathology.
2020;76:212-221. https://doi.org/10.1111/his.13964
PMid:31361906
- Alli
N, Meer S. Head and neck lymphomas: a 20-year review in an Oral
Pathology Unit, Johannesburg, South Africa, a country with the highest
global incidence of HIV/AIDS. Oral Oncol. 2017;67:17-23. https://doi.org/10.1016/j.oraloncology.2017.01.011
PMid:28351573
- Hansra
D, Montague N, Stefanovic A, et al. Oral and extraoral plasmablastic
lymphoma: similarities and differences in clinicopathologic
characteristics. Am J Clin Pathol. 2010;134:710-719. https://doi.org/10.1309/AJCPJH6KEUSECQLU
PMid:20959653
- Loghavi
S, Alayed K, Aladily TN, et al. Stage, age, and EBV status impact
outcomes of plasmablastic lymphoma patients: a clinicopathologic
analysis of 61 patients. J Hematol Oncol. 2015;8(1):65. https://doi.org/10.1186/s13045-015-0163-z
PMid:26055271 PMCid:PMC4472407
- Marks
E, Shi Y, Wang Y. CD117 (KIT) is a useful marker in the diagnosis of
plasmablastic plasma cell myeloma. Histopathology. 2017;71(1):81-88. https://doi.org/10.1111/his.13196
PMid:28226184
- Dittus
C, Sarosiek S. A case of HIV-negative plasmablastic lymphoma of the
bone marrow with a unique immunophenotype. Clin Case Rep.
2017;5(6):902- 904. https://doi.org/10.1002/ccr3.878
PMid:28588836 PMCid:PMC5458007
- Zimmermann
H, Oschlies I, Fink S, et al. Plasmablastic posttransplant lymphoma:
cytogenetic aberrations and lack of Epstein-Barr virus association
linked with poor outcome in the prospective German Posttransplant
Lymphoproliferative Disorder Registry. Transplantation.
2012;93(5):543-550. https://doi.org/10.1097/TP.0b013e318242162d
PMid:22234349
- Scheper
MA, Nikitakis NG, Fernandes R, Gocke CD, Ord RA, Sauk JJ. Oral
plasmablastic lymphoma in an HIV-negative patient: a case report and
review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol.
2005;100(2):198-206. https://doi.org/10.1016/j.tripleo.2004.11.050
PMid:16037778
- Huang
X, Zhang Y, Gao Z. Plasmablastic lymphoma of the stomach with CMYC
rearrangement in an immunocompetent young adult: a case report.
Medicine (Baltimore). 2015;94(4):e470 https://doi.org/10.1097/MD.0000000000000470
PMid:25634193 PMCid:PMC4602980
- Choudhuri
J, Pan Z, Yuan J, Chen M, Wu X, Zheng G, Zhao C, Yuan Y, Agarwal B, Liu
J, Ma MY, Wang Y, Shi Y. CD138- Plasmablastic Lymphoma. Arch Pathol Lab
Med. 2022 Sep 26. doi: 10.5858/arpa.2021-0462-OA. Epub ahead of print. https://doi.org/10.5858/arpa.2021-0462-OA
PMid:36161544
- Yan
M, Dong Z, Zhao F, Chauncey T, Deauna-Limayo D, Wang-Rodriguez J, Liu
D, Wang HY, Pilz R. CD20-positive plasmablastic lymphoma with excellent
response to bortezomib combined with rituximab. Eur J Haematol. 2014
Jul;93(1):77-80. doi: 10.1111/ejh.12286. Epub 2014 Apr 1. https://doi.org/10.1111/ejh.12286
PMid:24528507
- International
Non-Hodgkin's Lymphoma Prognostic Factors P A predictive model for
aggressive non-Hodgkin's lymphoma. N Engl J Med. 1993;329:987-994. doi:
10.1056/NEJM199309303291402. https://doi.org/10.1056/NEJM199309303291402
PMid:8141877
- Sehn
LH, et al. The revised international prognostic index (R-IPI) is a
better predictor of outcome than the standard IPI for patients with
diffuse large B-cell lymphoma treated with R-CHOP. Blood.
2007;109:1857-1861. doi: 10.1182/blood-2006-08-038257. https://doi.org/10.1182/blood-2006-08-038257
PMid:17105812
- Zhou
Z, et al. An enhanced International Prognostic Index (NCCN-IPI) for
patients with diffuse large B-cell lymphoma treated in the rituximab
era. Blood. 2014;123:837-842. doi: 10.1182/blood-2013-09-524108. https://doi.org/10.1182/blood-2013-09-524108
PMid:24264230 PMCid:PMC5527396
- Hertel
N, Merz H, Bernd HW, Bernard V, Künstner A, Busch H, von Bubnoff N,
Feller AC, Witte HM, Gebauer N. Performance of international prognostic
indices in plasmablastic lymphoma: a comparative evaluation. J Cancer
Res Clin Oncol. 2021 Oct;147(10):3043-3050. doi:
10.1007/s00432-021-03580-z. Epub 2021 Mar 3. https://doi.org/10.1007/s00432-021-03580-z
PMid:33660007 PMCid:PMC8397630
- Gross
TG, Kamdar KY, Bollard CM. Chapter 19: Malignant Non-Hodgkin Lymphomas
in Children. In: Blaney SM, Adamson PC, Helman LJ, eds. Pizzo and
Poplack's Principles and Practice of Pediatric Oncology. 8th ed.
Philadelphia Pa: Lippincott Williams & Wilkins; 2021.
- Chabay
P, De Matteo E, Lorenzetti M, Gutierrez M, Narbaitz M, Aversa L,
Preciado MV. Vulvar plasmablastic lymphoma in a HIV-positive child: a
novel extraoral localisation. J Clin Pathol. 2009 Jul;62(7):644-6. doi:
10.1136/jcp.2009.064758. https://doi.org/10.1136/jcp.2009.064758
PMid:19561233
- Mala
A, Khan YN, Ahmed A, Khayyam N, Aftab K. Tibial Plasmablastic Lymphoma
in a HIV-Negative Child: A Novel Extraoral Localisation. Case Rep
Hematol. 2022 Jun 8;2022:4353830. doi: 10.1155/2022/4353830. https://doi.org/10.1155/2022/4353830
PMid:35721803 PMCid:PMC9200553
- Hernandez
C, Cetner AS, Wiley EL. Cutaneous presentation of plasmablastic
post-transplant lymphoproliferative disorder in a 14-month-old. Pediatr
Dermatol. 2009 Nov-Dec;26(6):713-6. doi:
10.1111/j.1525-1470.2009.01019.x. https://doi.org/10.1111/j.1525-1470.2009.01019.x
PMid:20199447
- Vaubell
JI, Sing Y, Ramburan A, Sewram V, Thejpal R, Rapiti N, Ramdial PK.
Pediatric plasmablastic lymphoma: a clinicopathologic study. Int J Surg
Pathol. 2014 Oct;22(7):607-16. doi: 10.1177/1066896914531815. Epub 2014
Apr 25. https://doi.org/10.1177/1066896914531815
PMid:24771257
- Zhang
Y, Mariani R, Gong M, Kirschmann D, Caparelli E, Wallace N, Turner R,
Lu X, Gao J, Yap KL, Gong S. Distinctive Clinicopathologic Features of
Monomorphic B-cell Post-transplant Lymphoproliferative Disorders in
Children. Pediatr Dev Pathol. 2021 Jul-Aug;24(4):318-326. doi:
10.1177/10935266211007254. Epub 2021 Apr 19. https://doi.org/10.1177/10935266211007254
PMid:33872109
- Pather
S, MacKinnon D, Padayachee RS. Plasmablastic lymphoma in pediatric
patients: clinicopathologic study of three cases. Ann Diagn Pathol.
2013 Feb;17(1):80-4. doi: 10.1016/j.anndiagpath.2012.08.005. Epub 2012
Oct 2. https://doi.org/10.1016/j.anndiagpath.2012.08.005
PMid:23036261
- Modi
N, Khurana U, Mukhopadhyay S, Joshi D, Soni D, Chaudhary N, Malik S,
Shet T. An unusual case of paediatric plasmablastic lymphoma presenting
with malignant effusion and the challenges in its diagnosis. Diagn
Cytopathol. 2021 Oct;49(10):E389-E394. doi: 10.1002/dc.24829. Epub 2021
Jul 23. https://doi.org/10.1002/dc.24829
PMid:34296830
- Al-Malki
MM, Castillo JJ, Sloan JM, Re A. Hematopoietic cell transplantation for
plasmablastic lymphoma: a review. Biol Blood Marrow Transplant. 2014
Dec;20(12):1877-84. doi: 10.1016/j.bbmt.2014.06.009. Epub 2014 Jun 16. https://doi.org/10.1016/j.bbmt.2014.06.009
PMid:24946718
- Cattaneo
C, Finel H, McQuaker G, Vandenberghe E, Rossi G, Dreger P. Autologous
hematopoietic stem cell transplantation for plasmablastic lymphoma: the
European Society for Blood and Marrow Transplantation experience. Biol
Blood Marrow Transplant. 2015 Jun;21(6):1146-7. doi:
10.1016/j.bbmt.2015.03.008. Epub 2015 Mar 14. https://doi.org/10.1016/j.bbmt.2015.03.008
PMid:25783635
- Hess
BT, Giri A, Park Y, Patel KK, Link BK, Nowakowski GS, Maliske SM,
Fortin S, Chavez JC, Saeed H, Hill BT, Mejia Garcia AV, Maddocks KJ,
Hanel W, Wagner-Johnston ND, Messmer MR, Kahl BS, Watkins M, Alderuccio
JP, Lossos IS, Nathan S, Orellana-Noia VM, Portell CA, Landsburg DJ,
Ayers EC, Castillo JJ. Outcomes of patients with limited-stage
plasmablastic lymphoma: A multi-institutional retrospective study. Am J
Hematol. 2023 Feb;98(2):300-308. doi: 10.1002/ajh.26784. Epub 2023 Jan
1. https://doi.org/10.1002/ajh.26784
PMid:36588409 PMCid:PMC10107934
- Vakiani
E, Basso K, Klein U, Mansukhani MM, Narayan G, Smith PM, Murty VV,
Dalla-Favera R, Pasqualucci L, Bhagat G. Genetic and phenotypic
analysis of B-cell post-transplant lymphoproliferative disorders
provides insights into disease biology. Hematol Oncol. 2008
Dec;26(4):199-211. doi: 10.1002/hon.859. https://doi.org/10.1002/hon.859
PMid:18457340
- Leeman-Neill
RJ, Soderquist CR, Montanari F, Raciti P, Park D, Radeski D, Mansukhani
MM, Murty VV, Hsiao S, Alobeid B, Bhagat G. Phenogenomic heterogeneity
of post-transplant plasmablastic lymphomas. Haematologica. 2022 Jan
1;107(1):201-210. doi: 10.3324/haematol.2020.267294. https://doi.org/10.3324/haematol.2020.267294
PMid:33297669 PMCid:PMC8719101
- Green
MR, Rodig S, Juszczynski P, Ouyang J, Sinha P, O'Donnell E, Neuberg D,
Shipp MA. Constitutive AP-1 activity and EBV infection induce PD-L1 in
Hodgkin lymphomas and posttransplant lymphoproliferative disorders:
implications for targeted therapy. Clin Cancer Res. 2012 Mar
15;18(6):1611-8. doi: 10.1158/1078-0432.CCR-11-1942. Epub 2012 Jan 23.
Erratum in: Clin Cancer Res. 2012 Apr 1;18(7):2117. https://doi.org/10.1158/1078-0432.CCR-11-1942
PMid:22271878 PMCid:PMC3321508
- Garcia-Reyero
J, Martinez Magunacelaya N, Gonzalez de Villambrosia S, Loghavi S,
Gomez Mediavilla A, Tonda R, Beltran S, Gut M, Pereña Gonzalez A,
d'Ámore E, Visco C, Khoury JD, Montes-Moreno S. Genetic lesions in MYC
and STAT3 drive oncogenic transcription factor overexpression in
plasmablastic lymphoma. Haematologica. 2021 Apr 1;106(4):1120-1128.
doi: 10.3324/haematol.2020.251579. https://doi.org/10.3324/haematol.2020.251579
PMid:32273478 PMCid:PMC8018103
- Armstrong
R, Bradrick J, Liu YC. Spontaneous regression of an HIV-associated
plasmablastic lymphoma in the oral cavity: a case report. J Oral
Maxillofac Surg. Jul 2007;65(7):1361-4. doi:10.1016/j.joms. 2005.12.039
https://doi.org/10.1016/j.joms.2005.12.039
PMid:17577503
- Nasta
SD, Carrum GM, Shahab I, Hanania NA, Udden MM. Regression of a
plasmablastic lymphoma in a patient with HIV on highly active
antiretroviral therapy. Leuk Lymphoma. Feb 2002;43(2):423-6.
doi:10.1080/10428190290006260 https://doi.org/10.1080/10428190290006260
PMid:11999580
- Lee
KT, Misron NA, Abdul Aziz N, Wong CH, Liew HK. Spontaneous Regression
of Plasmablastic Lymphoma in an Immunocompetent Patient: Case Report
and Review of the Literature. Case Rep Hematol. 2022 May
30;2022:1142049. doi: 10.1155/2022/1142049. https://doi.org/10.1155/2022/1142049
PMid:35677531 PMCid:PMC9170507
- Flatow-Trujillo
L, Win K, Jencks A, Andritsos L, Arana Yi C. Spontaneous resolution of
untreated diffuse large B-cell lymphoma of maxillary bone after
incisional biopsy. Clin Case Rep. 2019 Sep 27;7(11):2082-2086. doi:
10.1002/ccr3.2408. https://doi.org/10.1002/ccr3.2408
PMid:31788256 PMCid:PMC6878081
- García-Noblejas
A, Velasco A, Cannata-Ortiz J, Arranz R. Remisión espontánea de un
linfoma no Hodgkin plasmablástico asociado a metotrexato tras
disminución de la dosis del fármaco [Spontaneous regression of
immunodeficiency associated plasmablastic lymphoma related to
methotrexate after decrease of dosage]. Med Clin (Barc). 2013 Jun
18;140(12):569-70. Spanish. doi: 10.1016/j.medcli.2012.10.001. Epub
2012 Nov 21. https://doi.org/10.1016/j.medcli.2012.10.001
PMid:23177304
- Igawa
T, Sato Y, Kawai H, Kondo E, Takeuchi M, Miyata-Takata T, Takata K,
Yoshino T. Spontaneous regression of plasmablastic lymphoma in an
elderly human immunodeficiency virus (HIV)-negative patient. Diagn
Pathol. 2015 Oct 6;10:183. doi: 10.1186/s13000-015-0421-y. https://doi.org/10.1186/s13000-015-0421-y
PMid:26445485 PMCid:PMC4596369
- Yordanova
K, Stilgenbauer S, Bohle RM, Lesan V, Thurner L, Kaddu-Mulindwa D,
Bittenbring JT, Scharberger M, Aßmann G, Bewarder M. Spontaneous
regression of a plasmablastic lymphoma with MYC rearrangement. Br J
Haematol. 2019 Sep;186(6):e203-e207. doi: 10.1111/bjh.16082. Epub 2019
Jul 1. https://doi.org/10.1111/bjh.16082
PMid:31257571
- Smith
S. Transformed lymphoma: what should I do now? Hematology Am Soc
Hematol Educ Program. 2020 Dec 4;2020(1):306-311. doi:
10.1182/hematology.2020000115. https://doi.org/10.1182/hematology.2020000115
PMid:33275671 PMCid:PMC7727564
- Marini
C, Baldaia H, Trigo F, Castillo JJ. Transformation of a previously
diagnosed diffuse large B-cell lymphoma to plasmablastic lymphoma. Am J
Hematol. 2016 Aug;91(8):E324. doi: 10.1002/ajh.24375. Epub 2016 May
2. https://doi.org/10.1002/ajh.24375
PMid:27012155
- Castillo
JJ, LaMacchia J, Flynn CA, Sarosiek S, Pozdnyakova O, Treon SP.
Plasmablastic lymphoma transformation in a patient with Waldenström
macroglobulinemia treated with ibrutinib. Br J Haematol. 2021
Nov;195(3):466-468. doi: 10.1111/bjh.17759. Epub 2021 Aug 6. https://doi.org/10.1111/bjh.17759
PMid:34355802
- Marvyin
K, Tjønnfjord EB, Breland UM, Tjønnfjord GE. Transformation to
plasmablastic lymphoma in CLL upon ibrutinib treatment. BMJ Case Rep.
2020 Sep 29;13(9):e235816. doi: 10.1136/bcr-2020-235816. https://doi.org/10.1136/bcr-2020-235816
PMid:32994268 PMCid:PMC7526319
- Aqil
B, Kaur A, Ramos J, Sukhanova M, Ma S, Gao J, Lu X, Chen YH, Chen Q.
Richter transformation to aggressive plasmablastic neoplasm related to
selection of a BTK-mutated clone in a patient with CLL/SLL treated by
ibrutinib. Leuk Lymphoma. 2022 Nov 6:1-4. doi:
10.1080/10428194.2022.2140286. Epub ahead of print. https://doi.org/10.1080/10428194.2022.2140286
PMid:36336991
- Ise
M, Kageyama H, Ikebe D, Araki A, Kumagai K, Itami M. Transformation of
double-hit follicular lymphoma to plasmablastic lymphoma: a partial
role of MYC gene rearrangement. J Clin Exp Hematop. 2018 Sep
19;58(3):128-135. doi: 10.3960/jslrt.18003. Epub 2018 Jul 14. https://doi.org/10.3960/jslrt.18003
PMid:30012920 PMCid:PMC6408176
- Ouansafi
I, He B, Fraser C, Nie K, Mathew S, Bhanji R, Hoda R, Arabadjief M,
Knowles D, Cerutti A, Orazi A, Tam W. Transformation of follicular
lymphoma to plasmablastic lymphoma with c-myc gene rearrangement. Am J
Clin Pathol. 2010 Dec;134(6):972-81. doi: 10.1309/AJCPWY1SGJ9IEAOR. https://doi.org/10.1309/AJCPWY1SGJ9IEAOR
PMid:21088162
- Condoluci
A, Rossi D. Biology and Treatment of Richter Transformation. Front
Oncol. 2022 Mar 22;12:829983. doi: 10.3389/fonc.2022.829983. https://doi.org/10.3389/fonc.2022.829983
PMid:35392219 PMCid:PMC8980468
- Thompson
PA, Siddiqi T. Treatment of Richter's syndrome. Hematology Am Soc
Hematol Educ Program. 2022 Dec 9;2022(1):329-336. doi:
10.1182/hematology.2022000345. https://doi.org/10.1182/hematology.2022000345
PMid:36485138 PMCid:PMC9820569
- Castillo
JJ, Guerrero-Garcia T, Baldini F, Tchernonog E, Cartron G, Ninkovic S,
Cwynarski K, Dierickx D, Tousseyn T, Lansigan F, Linnik Y, Mogollon R,
Navarro JT, Olszewski AJ, Reagan JL, Fedele P, Gilbertson M,
Grigoriadis G, Bibas M. Bortezomib plus EPOCH is effective as frontline
treatment in patients with plasmablastic lymphoma. Br J Haematol. 2019
Feb;184(4):679-682. doi: 10.1111/bjh.15156. Epub 2018 Mar 12.
https://doi.org/10.1111/bjh.15156
PMid:29527667