Background: Erythrocytosis
is a relatively common condition; however, a large proportion of these
patients (70%) remain without a clear etiologic explanation.
Methods: We set up a targeted NGS panel for patients with
erythrocytosis, and 118 sporadic patients with idiopathic
erythrocytosis were studied.
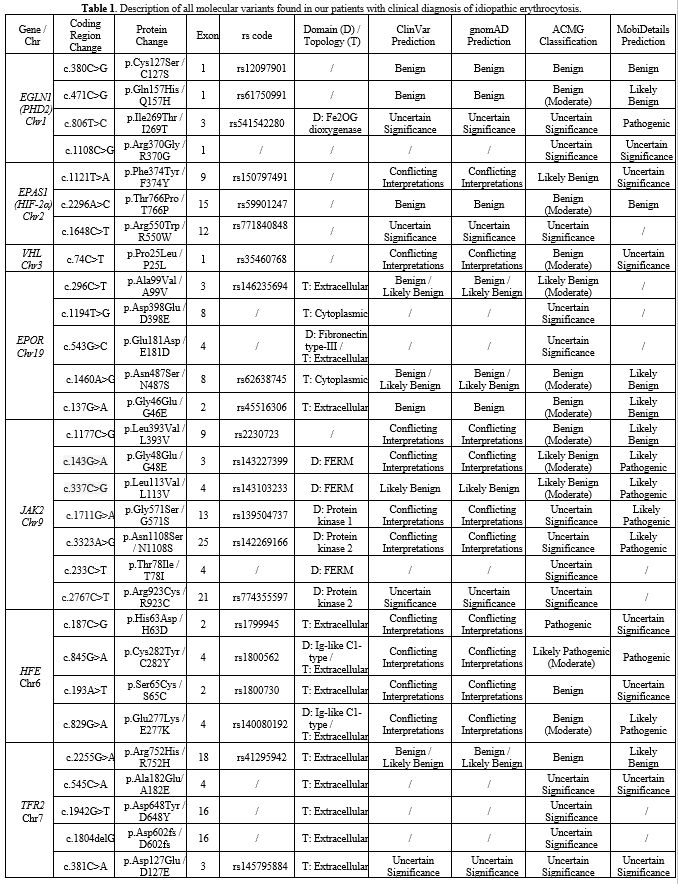
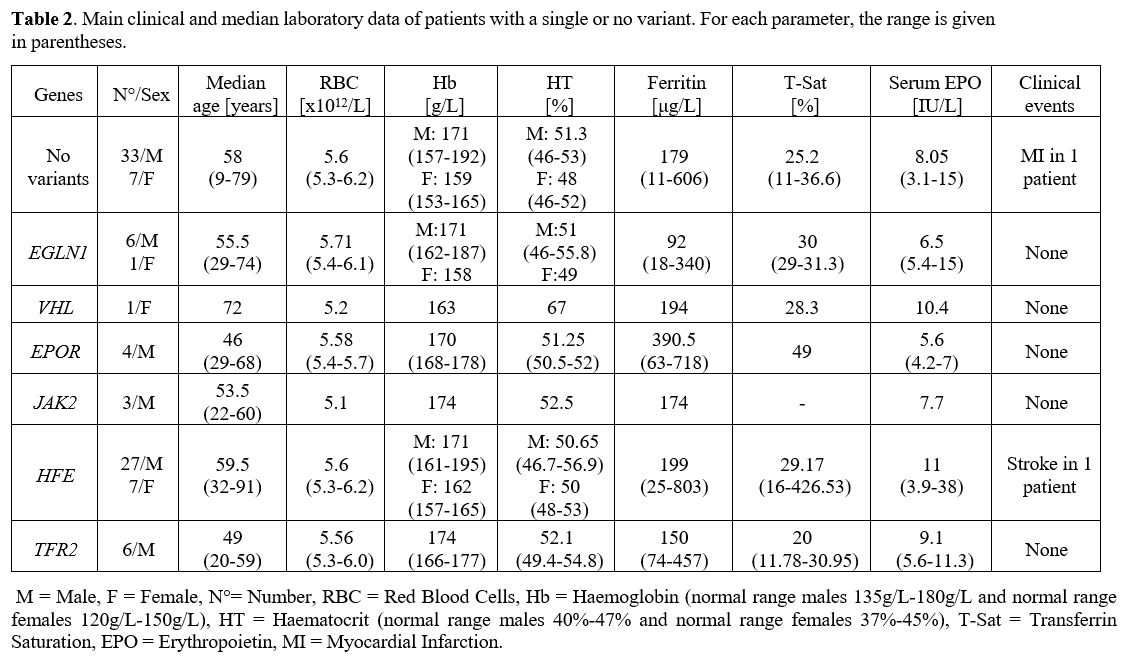
Results: In 40 (34%) patients, no variant was found, while in 78
(66%), we identified at least one germinal variant; 55 patients (70.5%)
had 1 altered gene, 18 (23%) had 2 alterations, and 5 (6.4%) had 3. An
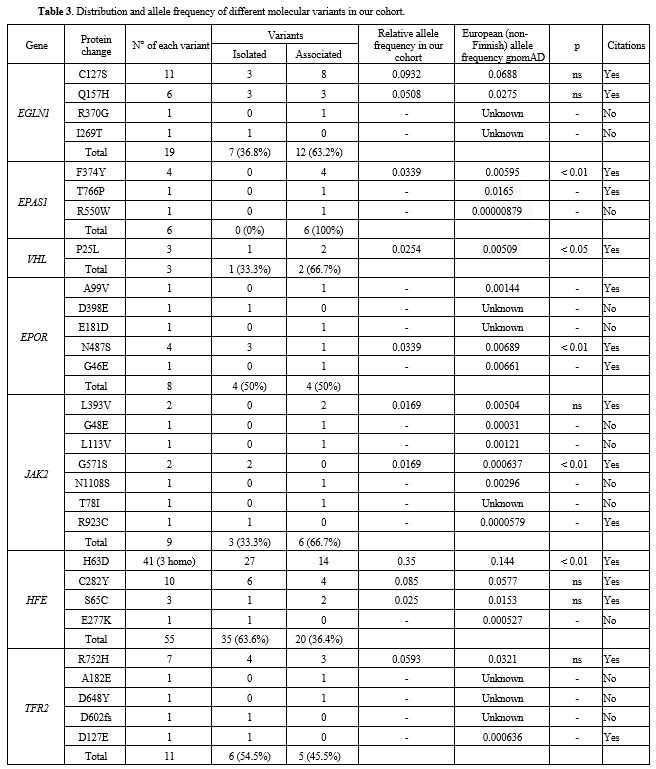
altered HFE gene was observed in 51 cases (57.1%), EGLN1 in 18 (22.6%)
and EPAS1, EPOR, JAK2, and TFR2 variants in 7.7%, 10.3%, 11.5%, and
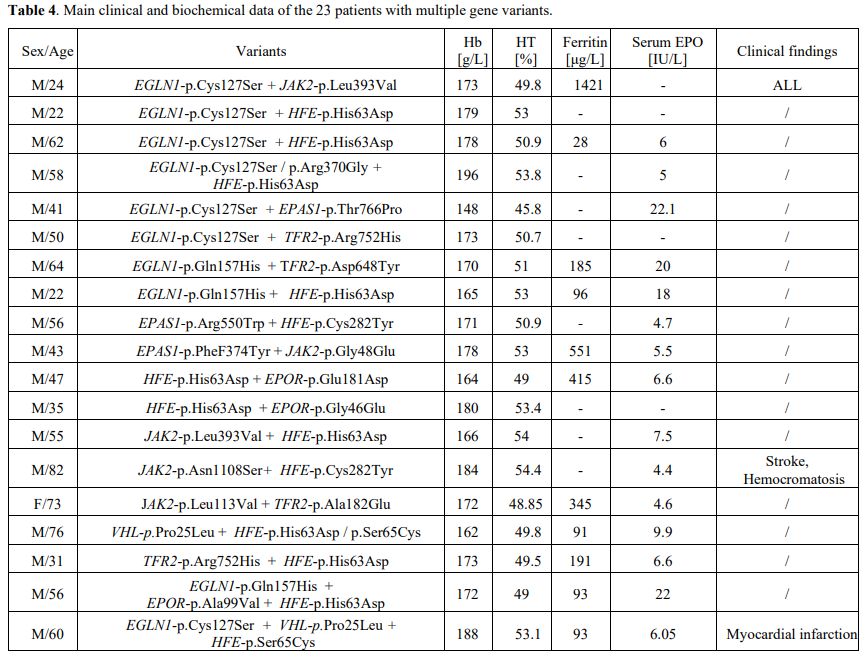
14.1% patients, respectively. In 23 patients (19.45%), more than 1
putative variant was found in multiple genes.
Conclusions: Genetic variants in patients with erythrocytosis
were detected in about 2/3 of our cohort. An NGS panel including more
candidate genes should reduce the number of cases diagnosed as
“idiopathic” erythrocytosis in which a cause cannot yet be identified.
It is known that HFE variants are common in idiopathic erythrocytosis.
TFR2 alterations support the existence of a relationship between genes
involved in iron metabolism and impaired erythropoiesis. Some novel
multiple variants were identified. Erythrocytosis appears to be often
multigenic.