Ugo
Testa1, Simona Sica2,3, Elvira
Pelosi1, Germana Castelli1
and Giuseppe Leone3.
1
Istituto Superiore di Sanità, Roma.
2
Dipartimento di Diagnostica per
Immagini,
Radioterapia Oncologica ed
Ematologia, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Roma,
Italy. Sezione di Ematologia.
3 Dipartimento d Scienze Radiologiche ed
Ematologiche, Università Cattolica del Sacro Cuore, Roma, Italy
Published: January 01, 2024
Received: November 16, 2023
Accepted: December 14, 2023
Mediterr J Hematol Infect Dis 2024, 16(1): e2024010 DOI
10.4084/MJHID.2024.010
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
|
Abstract
Treatment
of refractory and relapsed (R/R) B acute lymphoblastic leukemia (B-ALL)
is an unmet medical need in both children and adults. Studies carried
out in the last two decades have shown that autologous T cells
engineered to express a chimeric antigen receptor (CAR-T) represent an
effective technique for treating these patients. Antigens expressed on
B-cells, such as CD19, CD20, and CD22, represent targets suitable for
treating patients with R/R B-ALL. CD19 CAR-T cells induce a high rate
(80-90%) of complete remissions in both pediatric and adult R/R B-ALL
patients. However, despite this impressive rate of responses, about
half of responding patients relapse within 1-2 years after CAR-T cell
therapy. Allo-HSCT after CAR-T cell therapy might consolidate the
therapeutic efficacy of CAR-T and increase long-term outcomes; however,
not all the studies that have adopted allo-HSCT as a consolidative
treatment strategy have shown a benefit deriving from transplantation.
For
B-ALL patients who relapse early after allo-HSCT or those with
insufficient T-cell numbers for an autologous approach, using T cells
from the original stem cell donor offers the opportunity for the
successful generation of CAR-T cells and for an effective therapeutic
approach.
Finally, recent studies have introduced allogeneic CAR-T
cells generated from healthy donors or unmatched, which are opportunely
manipulated with gene editing to reduce the risk of immunological
incompatibility, with promising therapeutic effects.
|
Introduction
Chimeric
antigen receptor (CAR) T cells are engineered fusion proteins targeting
T lymphocytes to a specific membrane antigen expressed on the surface
of cancer cells, thus generating a specific antitumor immune response.[1]
CAR-T
cells targeting membrane antigens expressed on B-lymphoid cells, such
as CD19, CD20, or CD22, were shown to have significant therapeutic
activity in the treatment of patients with refractory and/or relapsed
B-cell malignancies, including B-acute lymphoblastic leukemia (B-ALL)
and B-cell lymphomas, such as mantle cell lymphoma, diffuse large
B-cell lymphoma and indolent B-cell lymphomas.[2]
Patients
with refractory/relapsed (R/R) B-ALL have a dismal prognosis (overall
survival of only 6 months), with a remission rate ranging from 20 to
40%. The introduction of new treatment strategies based on either
inotuzumab ozogamicin (a humanized anti-CD22 antibody conjugated with
calicheamicin) or blinatumomab (bispecific T cell antibody binding to
both CD3 on T lymphocytes and CD19 on B cells) have improved the
progression-free survival and overall survival of R/R B-ALL; however
durable remissions usually require allo-HSCT consolidation.[3-4]
CD19 CAR-T Cells
Two
CD19-targeted CAR-T cell products are currently approved by the FDA for
the treatment of R/R B-ALL patients: Tisagenlecleucel (Tisa-cel) and
Brexacabtagene autoileucel (Brexa-cel). These two products are
second-generation constructs, including an antigen-binding domain
(anti-CD19), hinge and transmembrane domains (derived from CD8a in
Tisa-cel and CD28 in Brexa-cel), co-stimulatory domain (derived from
4-1BB in Tisa-cel and from CD28 in Brexa-cel) and a T cell activation
domain (derived from CD3ζ). Another notable difference between these
two CAR-T cell products is that Brexa-cel is delivered using a
gammaretrovirus, while Tisa-cel is delivered using a lentivirus.
Tisagenlecleucel.
Tisa-cel (CTL019) was evaluated in B-ALL patients. An initial study
using Tisa-Cel evaluated 35 adult B-ALL patients with R/R disease, aged
20-70 years, treated with Tisa-Cel at three different doses: the
complete remission (CR) rate was 69% (90% in the patients treated at
the highest dose of Tisa-cel); in the whole-treated population the EFS
was 5.6 months; in the cohort of 20 patients treated with the highest
dose the 2-year OS was 73% and EFS 49.5%; 38% of patients achieving CR
were treated with allo-HSCT.[5]
Other studies
have evaluated the safety and efficacy of Tisa-Cel in young pediatric
B-ALL patients. The primary analysis of the phase II ELIANA trial,
involving 75 pediatric (children, adolescent) and young R/R B-ALL
patients, showed an ORR of 81% and an EFS of 76%, with 69% of responder
patients remaining relapse-free at 12 months.[6] This study led to FDA
approval of Tisa-Cel for pediatric/young R/R B-ALL patients. A more
recent study evaluated the safety profile and efficacy in 79 pediatric
and young adult patients with R/R B-ALL with a median follow-up of 38.8
months.[7] The overall remission rate was 82%, with an EFS of 24 months and the median overall survival not reached.[7] At a 3-year follow-up, the EFS was 44%, OS 63%, and RFS 52%.[7] Grade 3-4 adverse events were observed in 29% of patients.[7] 17% of patients in CR received consolidative allo-HSCT.[7] In 46 responding patients, a consistent improvement in quality of life was observed.[7]
Few
studies have evaluated the safety and efficacy of Tisa-Cel in children
and infant B-ALL patients. Ghorasian et al. reported the results of a
retrospective study based on the treatment of 35 children and infants
younger than 3 years with R/R B-ALL: 76% of these patients have
KMT2A-rearranged B-ALL and 66% relapsed after previous allo-HSCT.[8] The patients received a single infusion of Tisa-cel; 18% of patients previously received inotuzumb and 37% blinatumomab.[8] After a median follow-up of 14 months, the OS at 12 months was 84%, EFS 69%.[8] Adverse events grade 3 or more consisting of CRS were observed in 14% of cases; no severe neurotoxicity was observed.[8]
Makop
and coworkers retrospectively analyzed 14 infant R/R B-ALL patients who
received Tisa-Cel: 64% of these patients achieved MRD-negative
remission after CAR-T therapy, and 50% remained in remission at the
last follow-up (median duration of follow-up 231 days).[9] 86% of these patients had KMT2A rearrangements and 29% had prior HSCT.[9] The treatment was well tolerated, and 3/4 of patients displayed grade 3 cytokine release syndrome.[9] The estimated post-CAR-T 6-month EFS and OS were 48% and 71%, respectively.
Studies
performed in a real-world setting confirmed the efficacy of Tisa-Cel in
the treatment of R/R B-ALL patients. Thus, the Center for International
Blood and Marrow Transplant Research (CIBMTR) performed a retrospective
analysis of 255 pediatric patients with R/R B-ALL treated with
Tisa-Cel; after a median follow-up of 13.4 months, an initial CR rate
of 85.5% was observed, with a 12-month duration of response, EFS and OS
rates of 60.9%, 52.4%, and 77.2%, respectively.[10]
An updated analysis of the CIBMTR registry of real-world data,
presented at the 2021 ASH (American Society of Hematology) meeting,
displayed the outcomes of 451 R/R B-ALL children /young patients (≤25
years-old) treated with Tisa-Cel.[11] With a median
follow-up of 21 months, the ORR was 86.8%, the mDOR (median Duration of
Remission) was 23.9 months, mEFS was 14 months and mRFS was 23.9
months; 12-month EFS and RFS were 54.3% and 62.3%, respectively; mOS
was not reached.[11] Grade 3 CRS and neurotoxicity events were observed in 17.8% and 10% of patients, respectively.[11]
Fabrizio
et al. have retrospectively evaluated 184 R/R B-ALL patients treated in
the context of the Pediatric Real World CAR Consortium (PRWCC) with the
specific aim of assessing the efficacy of this therapy in patients with
extramedullary disease, subdivided into those with central nervous
system (CNS) and non-CNS involvement.[12] In patients with CNS disease,
88% achieved a CR, compared to 66% in those with non-CNS disease.[12]
The 24-months OS and 11-month RFS were similar in patients with CNS and
non-CNS disease and in those with bone marrow-only disease.[12]
Other
studies have explored some biomarkers or clinical parameters associated
with the clinical response to Tisa-Cel in R/R B-ALL patients. A
retrospective analysis carried out in a total of 200 R/R B-ALL patients
treated with Tisa-Cel, involving 15 USA institutions, reported a CR of
85%, with 12-month OS of 72%.[13] Univariate and multivariate analyses
showed an association between high disease burden (defined by ≥5% bone
marrow leukemic blasts and presence of extramedullary disease) with
inferior outcomes with a 12-month OS of 58% and EFS of 31%, compared
with patients with low-disease tumor burden, exhibiting OS of 95% and
EFS of 72%.[13] Furthermore, the high-burden tumor
was also associated with increased toxicity (grade 3 cytokine release
syndrome and neurotoxicity).[13]
Pulsipher and
coworkers have evaluated the predictive role of the persistence of
B-cell aplasia and detection of minimal residual disease (MRD) by NGS
in B-ALL patients undergoing treatment with Tisa-Cel in the context of
the ELIANA and ENSIGN trials.[14] In these studies, the large majority of relapses occurred within the first year after CAR-T cell infusion.[14]
B-cell aplasia was used as a pharmacodynamic marker of the persistence
of CAR-T functional activity. An association was observed between
B-cell recovery and shorter EFS; however, the measurement of B-cell
aplasia after CAR-T cell treatment by itself is not sufficient to
predict relapse since CD19-negative relapse can occur early and at
higher frequency in patients with persistence of B-cell aplasia.[14]
The study of MRD by NGS was the most sensitive technique for defining
the risk of relapse after CAR-T cell therapy. The evaluation of MRD at
day 28 post-infusion showed that a small percentage of patients who
have NGS MRD positivity display long-term responses, thus suggesting
that in these patients at day 28, the response is not complete and is
continuing.[14] Therefore,
repeated NGS-MRD evaluations are required for an adequate prediction of
the relapse risk in these patients. Importantly, clonality analyses
comparing MRD clones with the corresponding baseline clones showed no
differences in IgH rearrangements, thus suggesting that the relapsing
clones evolved from the original clones.[14]
CAR-T
cell expansion and/or persistence are major determinants of response to
CAR-T cell treatment. An appropriate lymphodepletion prior to CAR-T
cell infusion is required for optimal CAR-T cell expansion and
survival. Particularly, the addition of fludarabine to cyclophosphamide
as a lymphodepletion regimen significantly improved CAR-T cell
expansion and persistence in children and young adult B-ALL patients
receiving Tisa-Cel.[15] A retrospective analysis
carried out on 28 B-ALL patients treated with Tisa-Cel showed a
significantly improved probability of leukemia-free survival, a lower
incidence of CD19-positive relapsed, and a delayed B-cell recovery in
patients receiving high fludarabine regimens compared to those
receiving low-fludarabine regimens.[15]
Dourthe
and coworkers have explored the factors associated with outcome in 51
relapsed-refractory B-ALL patients undergoing treatment with Tisa-Cel:
49/51 patients achieved CR/Cri with an 18-month overall survival of
74%; 22/49 patients relapsed with a median time of 3.7 months: 12 had
CD19-positive relapse and 8 CD19-negative relapse.[16] Factors associated
with a high tumor burden, such as cytokine release syndrome and prior
blinotumumab therapy, were associated with an increased risk of relapse
and a reduced EFS and OS.[16] Pre-lymphodepletion
high disease burden and detectable MRD at day 28 correlated with an
increased risk of CD19-negative relapse, while low disease burden and
loss of B-cell aplasia predicted an increased risk of CD19-positive
relapses.[16] These observations supported an important role of prior therapy on patient outcomes.
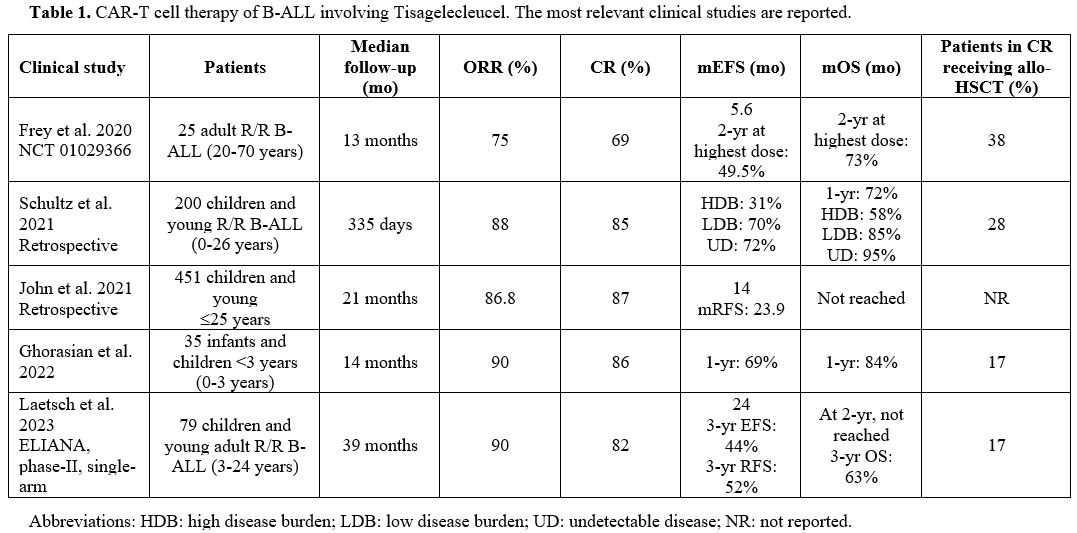 |
- Table 1. CAR-T cell therapy of B-ALL involving Tisagelecleucel. The most relevant clinical studies are reported.
|
Other CAR-T Cell Products with 4-1BB Co-Stimulatory Domain
Hay
et al. have explored the safety and the efficacy of CAR-T cells
manufactured using a CAR composed of a single-chain variable fragment
(scFv) derived from an anti-CD19 monoclonal antibody fused to an IgG4
hinge region, CD28 transmembrane domain, 4-1BB co-stimulatory domain,
and CD3ζ signaling sequence.[17]
CAR-T cells were infused in a defined CD4+:CD8+ ratio to improve
uniformity and maximize the potency of the infused product, according
to preclinical studies.[17]
Using these CAR-T cells, a phase I/II study was carried out, involving
the enrollment of 53 R/R adult B-ALL patients; at a follow-up of 31
months, 89% of patients achieved a CR/CRi response, with a mEFS of 6
months among patients achieving MRD-negative status.[17] 40% of patients who achieved a CR with MRD-negativity were processed for allo-HSCT.[17]
With a median follow-up of 28.4 months after allo-HSCT, the EFS and OS
rates were 61% and 72%, respectively; allo-HSCT after CAR-T cell
therapy was associated with longer EFS compared with no allogeneic
HSCT.[17] mEFS and mOS were significantly better among patients achieving CR with MRD-negativity compared to those with MRD-positivity.[17]
In patients achieving CR with MRD-negative condition, lower LDH levels,
higher platelet counts, incorporation of fludarabine in the
lymphodepletion regimen, and allo-HSCT after CAR-T cell therapy were
associated with improved EFS.[17]An
et al. reported the results of a multicentre phase II study involving
the treatment of 47 R/R children and adult B-ALL patients (aged 3-72
years) using Sino CD19 CAR-T cells obtained using a CAR composed of
scFv, IgG4 hinge, CD28 transmembrane domain and 4-1BB co-stimulatory
domains.[18]
81% of patients achieved a CR, and after a follow-up of 12 months, the
RFS was 10.5 months, and 26% of patients in CR received consolidative
allo-HSCT.[18] 19/28
of the patients achieving allo-HSCT relapsed after CAR-T cell therapy;
patients who underwent allo-HSCT after CAR-T cell therapy had a lower
risk of relapse and death, but not statistically significant when
compared with those who did not.[18]
Factors associated with poor outcome were the presence of high-risk
cytogenetic factors, the presence of extramedullary disease, and higher
levels of circulating T-reg cells.[18]
CAR-T Cells with CD28 Co-Stimulatory Domains
Several
studies involved CAR-T cell products based on hinge, transmembrane and
co-stimulatory domains derived from CD28; these CAR-T cell products
include the commercialized product Brexu-Cel. Pivotal studies carried
out by Hollyman and coworkers have described the development of CAR-T
cells obtained by the genetic manipulation of T cells with a
replicon-defective gamma retroviral vector derived from Maloney murine
leukemia virus encoding a CAR targeted to CD19 (19-28z), containing
mouse scFv, CD28 H/T and CD28 co-stimulatory domains.[19]
Using this CAR-T cell product, Park et al. have evaluated 53 adult R/R
B-ALL patients (23-74 years) heavily pretreated and reported their
evaluation at a median follow-up of 29 months: the median EFS was 6.1
months; the mOS was 12.9 months (Table 2).
Patients with a low disease burden prior to CAR-T cell therapy (<5%
BM blasts) displayed enhanced remission duration and survival, with a
mEFS of 10.6 months and a mOS of 20.1 months compared to 4 and 12
months, respectively, in those with high-disease burden (>5% BM
blasts or extramedullary disease).[20] Furthermore, patients with high disease burden showed a significantly increased incidence of CRS and neurotoxic events.[20] A parallel study evaluated the safety and efficacy of 19-28z CAR-T cells in 25 pediatric/young adult R/R B-ALL patients.[21]
The study of these patients supported the conclusion that the dose
intensity of conditioning chemotherapy and low pretreatment disease
burden displayed a positive impact on response without a negative
impact on treatment-associated toxicity.[21]
Finally, a retrospective study evaluated 38 adult B-ALL patients who
progressed after 19-28z CAR-T cell therapy; the median time to
progression after CAR-T cell therapy in these patients was 5.5 months,
with a mOS of 7.5 months.[22] A high pretreatment disease burden was associated with the risk of disease progression after CAR-T cell therapy.[22]
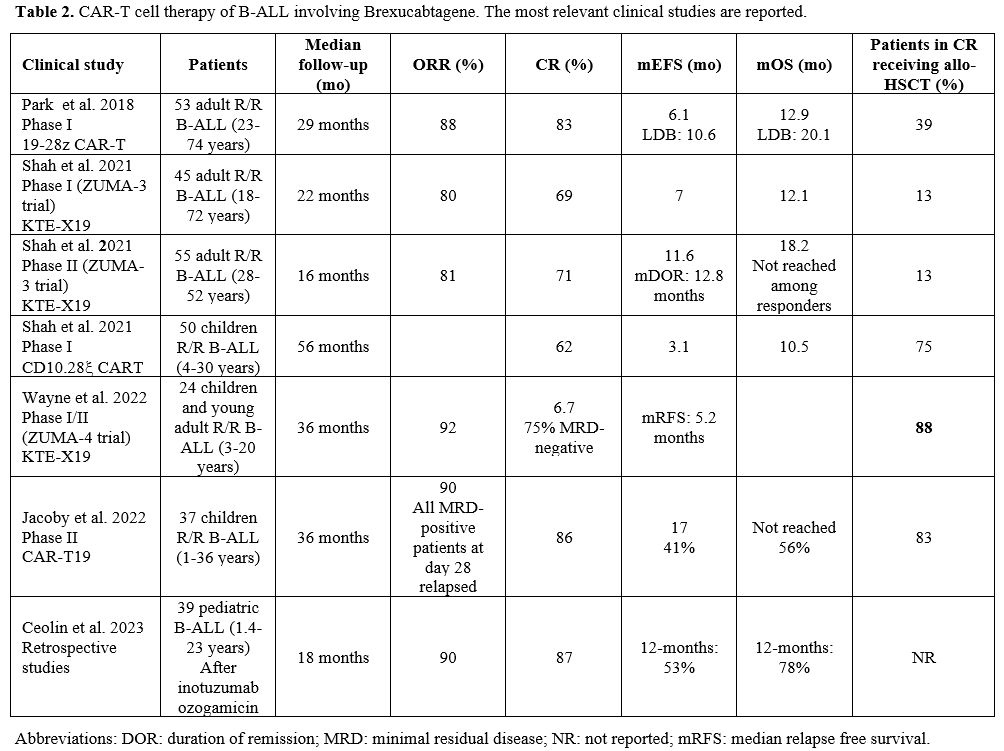 |
- Table 2. CAR-T cell therapy of B-ALL involving Brexucabtagene. The most relevant clinical studies are reported.
|
Several
studies have explored autologous CAR-T cells manufactured using the
product KTE-X19 (commercialized as Brexu-Cel). In a first phase I study
(ZUMA-3 study), 45 adult R/R B-ALL patients with age comprised between
18 and 77 years were treated with Brexu-Cel at a dosage of 2x106 cells per Kg (6 patients) or 1x106 (23 patients) or 0.5x106 (16 patients).[23] Grade ≥3 CRS was observed in 31% of patients and grade ≥3 neurologic events in 38% of cases.[23]
At a median follow-up of 22.1 months, the ORR was 69%, with 53% of
patients achieving CR and 16% achieving Cri; the median duration of
response for the patients achieving a CR/Cri was 14.5 months (18 months
for patients treated at 1x106 dose).[22] MRD was undetectable in all responding patients; 13% of patients received allo-HSCT.[23]The
ZUMA-3, phase II study involved the treatment of 55 adult (28-52 years
old) B-ALL patients with R/R B-ALL patients treated with Brexu-Cel; at
a median follow-up of 16.4 months, 71% of treated patients displayed a
CR, with a median duration remission of 12.8 months; mRFS was 11.6
months and mOS was 18.2 months.[24] Among responders, mOS was not reached. 18% of patients received allo-HSCT treatment after CAR-T cell therapy infusion.[24]
The most common adverse events were grade 3 or higher anemia (49%),
pyrexia (36%), and infections (25%); CRS or neurological events of
grade 3 or higher occurred in 24% and 25% of cases, respectively.[24]On
the basis of the results of the ZUMA-3 trial, Bruxa-Cel received
approval from the FDA for the treatment of adult patients with R/R
B-ALL.[25]A
longer follow-up confirmed the consistent therapeutic efficacy of
Brexu-Cel in R/R B-ALL patients, with a CR rate of 75%, median duration
of remission, and OS of 14.6 and 25.4 months, respectively.[26]
Furthermore, in the SCHOLAR-3 study, 49 patients treated with Brexu-Cel
in the context of the ZUMA-3 trial were matched with 40 treated
patients from historical clinical trials, with a comparative mOS of
25.4 and 5.5 months, respectively.[26] A detailed
comparative analysis of the results obtained in the SCHOLAR-3 study
showed that outcomes of patients treated in historical standard-of-care
trials were poor, irrespectively of the prior treatment
(blinatumomab/inotuzumab-treated or naïve) with a mOS of <60 months,
compared with a mOS of >25 months in matched patients enrolled in
the ZUMA-3 trial.[27]The
phase I ZUMA-4 trial explored the safety and the efficacy of Brexu-Cel
in 24 pediatric/adolescent R/R B-ALL patients; the overall CR rate was
67%, and MRD-negativity was 100% among responders.28 85% of responders
underwent subsequent allo-HSCT; the median duration of remission at
HSCT time and median OS were not reached.[28] Grade 3 CRS was 33%, and grade ≥3 neurologic events were observed in 11-27% of patients following the dose of CAR-T cells.[28]Other
studies have explored the efficacy of CAR-T cells manufactured using
vectors similar to those used in Brexu-Cel in infants/children patients
with R/R B-ALL. Thus, Jacoby et al. reported the long-term response of
37 infants/children and young adult patients (1-36 years) with R/R
B-ALL treated with CD19 CAR-T cells in the context of a phase II study;
the CR rate was 86%, with 71% of the responding patients achieving an
MRD-negative status; 83% of the patients in CR proceeded to allo-HSCT;
all MRD-positive patients at day 28 post-CAR-T cell infusion relapsed.[29] mEFS was 17 months, and mOS was not reached, with 56% of patients surviving at 3 years.[29]
A prior HSCT did not affect the response to CAR-T cell therapy, but a
consolidation with allo-HSCT after CAR-T cell therapy improved
long-term survival.[29]Shah
et al. reported the long-term results of a single-center phase I study
involving the treatment of 50 children and young adult (4-30 years)
patients with R/R B-ALL treated with CD19-28z CAR-T cells; at a
follow-up of 4.8 years, 62% of patients displayed a CR, with a mEFS of
3.1 months and a mOS of 10.1 months.[30] 56% of
patients displayed a CR with an MRD-negative status, and 75% of these
patients were processed for consolidative allo-HSCT.[30]Ceolin
et al. have retrospectively analyzed the response to CD19 CAR-T cell
therapy in 39 pediatric R/R B-ALL patients (1.4-23 years old)
previously treated with inotuzumab ozogamicin; at an 18-month
follow-up, an ORR of 53% and a mOS of 78% were observed.[31] These results were comparable to those previously reported for patients without prior inotuzumab ozogamicin exposure.[31]
CD19-Targeted CAR-T Cells Engineered with Lower Affinity Anti-CD19
Ghorashian
et al. have explored whether a lower CAR binding affinity could improve
CAR-T activity and reduce the toxic effects induced by CAR-T cell
therapy.[32] To this end, these authors have
generated a novel CD19 CAR (CAT) with a lower affinity than FMC63, the
high-affinity anti-CD19 scFv used in most CD19 CAR constructs.[32] CAT CAR-T cells exhibited increased proliferation and cytotoxicity in vitro and enhanced proliferative and in vivo antitumor activity compared with FMC63 CAR-T cells.[32]
The safety and the efficacy of CAT-CAR-T cells were explored in 17
pediatric patients with high-risk R/R CD19+ B-ALL; 14 patients received
an infusion of CAR-CAR-T cells, and 12 had a molecular complete
response after 3 months; the expansion of CAT-CAR-T cells observed in
these patients was about threefold higher than that reported for
Tisa-Cel at 28 days post-infusion.[32] At 1 year, the OS was 63%, and the event-free survival was 46%.[32] No patients required tocilizumab or intensive care support for grade ≥3 CRS.[32]A
subsequent study evaluated the safety and efficacy of CAT19-CAR-T cells
(AUTO1) in 20 patients with R/R adult B-ALLs; 65% of these patients
received prior allo-HSCT.[33] No patient experienced
grade ≥3 CRS, and 3 of 20 patients had grade neurotoxicity that
resolved within three days of treatment with steroids.[33]
85% of patients achieved a complete MRD-negative response at 1 and 3
months post-infusion; 17 patients underwent allo-HSCT while in
remission.[33] The event-free survival was 68% and 48%, respectively. A high level of CAR-T cell expansion was observed in 15 of 20 patients.[33] It is interesting to note that risk-adaptive and split-dosing were incorporated into the design of this study.Based
on these results, the pivotal FELIX study was proposed; this is a phase
Ib/II study enrolling R/R B-ALL patients with >5% of bone marrow
blasts (cohort A), MRD-positivity (cohort B), or extramedullary disease
(cohort C): The patients received a target dose of 4x106 CAR-T cells as a split dose on day 1 and day 10; the dosing schedule is based on the percentage of bone marrow blasts.[34]
A first interim analysis involved 50 patients enrolled in cohort A of
the study, with a median number of prior lines of treatment, including
42% of patients prior to transplant; at screening, patients had 55% of
BM blasts.[34] 70% of patients achieved a CR; 3% of treated patients had a CRS of grade ≥3.[34] In this study, AUTO1 CAR cell treatment received the commercial designation of abecabtagene autoleucel.[34]Interestingly,
a recent study reported the single-cell transcriptomic analysis of CD19
CAR-T cells of 10 children enrolled in the initial CARPALL study,
studied at the moment of infusion and 1-3 months, 4-6 months, and after
7 months.[35] 87% of patients achieved complete
remission; 46% of responding patients subsequently relapsed, while the
remaining 54% of patients achieved long-lived remissions maintained by
detectable CAR-T cells and concomitant B-cell aplasia.[35] All patients
with long-lived CAR-T cells developed a CD4/CD8 double negative
phenotype with an exhausted-like memory state and a distinct
transcriptomic signature.[35] Interestingly, this
“persistence” signature was also observed in two adult patients with
chronic lymphocytic leukemia with decade-long remission following CD19
CAR-T cell therapy.[35]
CD22-Targeted CAR-T Cell Therapy of B-ALL
CD22
is expressed in the large majority of pediatric and adult B-ALL and,
therefore, represents a suitable target for CAR-T cell therapy of these
leukemias.Initial
studies were based on the use of CD22-targeted/4-1BB cells. The initial
phase I study reported the results on 21 children and adult B-ALL
patients treated with CD22-CAR-T cells. The response to this treatment
was dose-dependent, with 1/6 patients and 10/11 patients responding
with 3x105 and 1x106 CD22-CAR-T cells.[36]
An updated report of this study showed the results obtained in 55 B-ALL
patients, with a CR rate of 70%, a median duration of remission of 6
months, and a mOS of 13.4 months.[37] 24% of patients in CR received allo-HSCT.[37]
63% of patients with CR achieved an MRD-negative status. The rate of
responses and duration were lower in patients with prior CD22-targeted
therapy (inotuzumab).[37] A third of treated patients developed hemophagocytic lymphohistiocytosis about 2 weeks after CAR-T cell infusion.[37]Pan
et al. evaluated the safety and efficacy of CD22-41BB CAR-T cells in 34
pediatric/adult R/R B-ALL patients who, in large majority (91% of
cases, have received prior CD19 CAR-T cell therapy).[34] 70.5% of the enrolled patients achieved a CR, and 11 of these patients were bridged to allo-HSCT.[38] Leukemia-free survival at 12 months for patients who achieved CR was 58%, and for those receiving allo-HSCT was 71.6%.[38] CD22 antigen loss or mutation was not associated with disease relapse.[38]Tan
et al. reported the development of a new CD22-CAR construct with low
immunogenicity and potent activity for treating B-ALL patients who have
failed previous CD19- or CD22-targeted CAR therapies. This construct
was based on the fusion of a full-human anti-CD22 scFv to the
intracellular 4-1BB co-stimulatory and CD3ζ signaling domains to
generate CD22-CARFH80 T cells.[39] These CAR-T cells
were evaluated in 8 patients who were refractory or relapsed after
previous CD19- and CD22-CAR-T cell therapies: 7/8 patients achieved a
response to treatment, and 4 responding patients were bridged to allow
HSCT.[39] The follow-up of these patients was limited to 6 months.[39]
Multitargeting of CD19 and CD22
Targeting
both CD19 and CD22 can be accomplished by four different approaches,
based on: (i) the generation of two cell populations expressing
different CARs and infusion of these cells together (simultaneous
coadministration) or sequentially (sequential coadministration); (ii)
the simultaneous engineering of T cells with two different CAR
constructs (co-transduction), thus generating three different CAR-T
cells, represented by single-expressing and dual-expressing CAR-T
cells; (iii) the development of bicistronic vectors that encode two
different CARs on the same cells; (iv) the encoding in the same
chimeric protein of two different CARs, using a unique expression
vector (bispecific or tandem).[40]Several
studies have explored the safety profile and efficacy of the
coadministration of CD19 and CD22 CAR-T cells to R/R B-ALL patients.
Wang et al. have performed a phase I study involving the sequential
infusion of a cocktail of anti-CD19 and anti-CD22, 2 single-specific,
third-generation CAR (CAR 19/22) T cells in 51 patients R/R with R/R
B-ALL; a CR/CRi was observed in 96% of patients with a PFS of 13.6
months and an OS of 31 months. High-grade CR and neurotoxic events were
observed in 22 and 1 of the cases, respectively.[41]
Liu et al. have investigated in phase I clinical study the therapeutic
efficacy of the combination of CD19 and CD22 CAR-T cell therapy in 27
B-ALL patients who relapsed post-transplant; 27 patients received the
first CD19 CAR-T cells, and 85% achieved a CR; 21 of these patients
received the second CD22 CAR-T cells and were followed by a median of
19.7 months: 14 patients remained in CR, and 7 patients relapsed.[38] EFS and OS at either 12 or 18 months were 67.5% and 88.5%, respectively.[42]A
single-arm, multicenter phase II study involved 195 childhood patients
(≤20 years) with R/R B-ALL treated with a protocol involving
coadministration of CD19 and CD22 CAR-T cell therapy; 99% of these
patients achieved a CR, with MRD negativity.[42] 12-month EFS was 73.5%; relapse occurred in 43 patients (mostly with CD19+/CD22+ or CD19-/CD22+ relapse).[38]
Seventy-eight patients received allo-HSCT and displayed a 12-month EFS
of 85%, while 116 patients were non-transplanted and showed a 12-month
EFS of 69.2%.[42] Favorable outcomes were seen for patients with consistent B-cell aplasia at 6 months.[43] The 12-month EFS was 95% for patients with isolated testicular relapse and 68.6% for patients with CNS relapse.[43]Several
studies reported the development and the clinical use of tandem
CD19/CD22 bispecific CAR-T cells. Preclinical studies have supported
the efficacy of tandem CD22/CD19 CAR-T cells in mediating the killing
of leukemia cells with low CD19 and CD22 antigen density.[44] Dai et al. reported the development of bispecific CD19/CD22 CAR-T cells generated using a tandem CD19/CD22 vector.[41]
In the clinical study of CAR-T cells generated using this vector, all
6/6 R/R B-ALL patients attained CR, with the achievement of MRD
negativity.[45] Three of these patients relapsed
within the first year after CAR-T cell therapy, and one of them with
CD19-negative leukemia cells.[45] Cui et al. reported
the results of an open-label, single-center clinical trial involving
the investigation of the safety and efficacy of tandem CD19/CD22 dual
targets CAR-T cells in 47 R/R B-ALL patients (44% with primary
refractory B-ALL and 57% with high disease burden).[46]
100% of patients responded to treatment, and 85% had MRD-negative
status; grade ≥2 CRS occurred in 17% of patients and neurotoxicity
events only in 1 patient; leukopenia was the most severe common
hematological abnormality.[46] At a follow-up of 21.8
months, the mOS and mLFS were not reached; at 2 years, the OS was
74.5%, and at 1 year, the leukemia-free survival (LFS) was 68%; 72% of
the patients proceeded to bridge allo-HSCT, with 1-year OS of 80.4%; at
1 year, the cumulative incidence of relapse was 23%.[46]Liu
et al. have performed a comparative analysis of the efficacy of
single-target (CD19) or dual-target (tandem or sequential CD19/CD22)
CAR-T cell therapy for R/R B-ALL patients.[47] In
this retrospective analysis, a total of 219 patients, subdivided into
single CD19 CAR-T (147 patients), tandem CD19/CD22 CAR-T (51 patients),
and sequential CD19/CD22 CAR-T (21 patients), all tested at the same
institution, were included.[43] The CR rates in the single-CD19, tandem CD19/CD22 and sequential CD19/CD22 were 83%, 98% and 95%, respectively.[47]
A higher proportion of patients treated with tandem CD19/CD22 CAR-T
(70.5%) was bridged to allo-HSCT compared to those treated with single
CD19 (39%) or sequential CD19/CD22 (28.5%) CAR-T cell therapy.[47] EP300-ZNF384
is originated by a cryptic t(12;22)(p13;q13) chromosome translocation
and is associated at phenotypic level with high CD19 and CD22
expression. This translocation is observed in 4-6% of B-ALL patients;
Zhang et al. reported the successful treatment of two R/R AP300-ZNF384
B-ALL patients with tandem CD19/CD22 CAR-T cell therapy, with bridging
to allo-HSCT.[48]Spiegel
and coworkers have generated a CD19-22.BB,z-CAR comprising a single
cistron encoding the anti-CD19 murine FMC63 scFv and fully human
anti-CD22 m971 scFv, followed by human CD8 hinge and transmembrane
domains, 4-1BB co-stimulation and CD3ζ activation domains.[49]
The study enrolled 17 R/R B-ALL patients, all responding to the
treatment (88% CR and 12% PR); all patients with CR achieved an
MRD-negative status.[49] After a median follow-up of 9.3 months, mOS was 11.8 months, and PFS was 5.8 months.[44]
Ten patients displayed disease progression after CAR-T cell therapy,
and 5 of these patients had low-negative CD19 expression and maintained
CD22 expression on relapsing leukemic cells.[49]Cordoba
et al. developed AUTO3, a CAR-T cell-based treatment with dual
specificity (CD19 and CD22) generated through transduction of
autologous T cells with a bicistronic vector encoding humanized
anti-CD19 and anti-CD22 CARs, both incorporating tumor necrosis factor
receptor co-stimulatory domains.[50] The AMELIA phase
I clinical trial was performed using AUTO-3: at 1 month after AUTO-3
infusion, 86% of patients achieved a CR, with 80% of patients showing
MRD negativity; 9 of the responding patients relapsed, and many of
these patients had low CAR-T cell numbers, thus suggesting that a low
persistence in vivo of these cells could be the predominant cause of treatment failure.[50]Annesley
and coworkers have explored the safety and the efficacy of CAR-T cells
targeting both CD19 and CD22, generated through a co-transduction
approach: autologous T cells were double transduced with lentiviral
vectors encoding for either a CD19-specific or a CD22-specific CAR,
both with 4-1BB co-stimulation.[51] Two types of
CAR-T cell products were explored: SCRI-CAR19x22v1 (leading to
prevalent engraftment of CD19 CAR population, with unsuccessful
eradication of CD19-/CD22+ leukemic cells) and SCRI-CAR19x22v2 /leading
to prevalent engraftment of CD22 CAR-expressing cells).[47] With SCRI-CAR19x22v2, a 91% CR rate was observed, with 100% of MRD negativity in 12 R/R B-ALL patients.[46]
It is of interest to note that the SCRI-CAR19x22v2 product is
predominantly composed of CD22+ and CD19+/CD22+ CAR-T cells, with few
CD19+ cells, while in vivo engraftment is predominated by single CD22 CAR-expressing T cells.[51]Lucchini
et al. developed AUTO1/22, an autologous CAR-T cell product
co-transduced with two different lentiviral vectors encoding a
previously described fast-off rate CD19 CAr and a novel CD22 CAR
designed to recognize targets with low antigen density.[52]
Safety and efficacy of AUTO1/22 in a phase I study in 12 children young
adults with R/R B-ALL; the enrolled patients had a median of 3 prior
lines of therapy; six of these patients had relapsed post-allo-HSCT;
six patients had an extramedullary relapse, and 3 had detectable
CD19-negative disease.[52]
10 of the 12 evaluable patients (83%) achieved MRD-negative complete
remission at 1 month post-infusion; with a follow-up of 8.7 months, 50%
of patients remained alive in MRD-negative CR; the median duration of
response in responding patients was 9.9 months; the OS rate at 6 and 12
months was 75%; Event-free survival was 75% and 60% at 6 and 12 months,
respectively. No patient experienced grade 3 CRS.[52]
Relapse after CAR-T Cell Therapy for B-ALL
The
relapse after single-targeted CD19 CAR-T cell therapy can be subdivided
into three subgroups, differentiated according to CD19 expression on
relapsing leukemic cells and differentiation status: CD19+ relapse and
CD19-/low relapse; a third type of relapse is related to lineage switch
(LS) from a lymphoid to a myeloid phenotype. During the treatment of
R/R B-ALL patients with CD19-directed CAR-T cells, most of the relapses
occurring soon after CAR-T cell therapy were composed of CD19+ leukemic
cells.[53]The
CD19+ relapse usually results from the low potency or short persistence
of CD19 CAR-T cells.[6] In CD19- relapses, which account for about 40-50%
of total relapses, B leukemic cells lose CD19 membrane expression and
escape CAR-T cell-mediated recognition and killing. Molecular studies
have shown that the occurrence of de novo frameshift/missense CD19
mutations, alternative splicing of CD19 mRNA, and hemizygous deletions
spanning the CD19 locus cause impaired CD19 mRNA expression.[54-56] CAR-T
cell therapy is not responsible for a dysregulated CD19 transcription
but favors the emergence of minor CD19- clones preexisting in the
patients and escaping CD19-targeted therapy.[57] This
mechanism was directly supported by single-cell profiling of leukemic
cells of patients with CD19- relapse after CAR-T cell therapy.[57]Pan
et al. have analyzed the outcome of 68 R/R B-ALL children treated with
CD19 CAR-T cells and with a consolidation therapy based either on
allo-HSCT (34 patients) or CD22 CAR-T cells (30 patients): the DFS at 1
year was 79.6%, with 12 relapsing patients with a median time of 6.3
months.[58] 8 of the 12 relapsing patients were
characterized by the presence of TP53 mutations in their pre-therapy
leukemic cells; 7/8 of these patients displayed CD19 negativity on
their relapsed leukemic cells.[58]As
discussed above, for Tisa-Cel-treated patients, high-disease burden and
prior failure in response to blinatumomab therapy are associated with a
reduced response to CAR-T cell therapy.[16] These
observations were confirmed and extended in a wide analysis involving
420 children/young adult B-ALL patients undergoing treatment with
Tisa-Cel and other CD19 CAR-T treatments; 18% of these patients
received prior treatment with blinatumomab.[59] Blinatumomab nonresponders had worse EFS and RFS compared to responders or blinatumomab-naïve patients.[54] A high disease burden was associated with inferior EFS.[59]A
second study based on the analysis of these 420 patients further
extended the analysis of clinical, genetic, and biochemical factors
associated with the relapse of these patients following their treatment
with CAR-T cells. Clinical characteristics associated with worse EFS
included high tumor burden, circulating peripheral blasts, CD19/28ζ CAR
construct type, and poor response to blinatumomab.[60]
Of 420 R/R children/young adult B-ALL patients treated with CD19 CAR-T
cells, 39.5% relapsed; the relapsing patients were 50% CD19+, 41% CD19-
and 7.2% LS relapses.[60] A greater number of prior
complete remissions was associated with CD19+ relapses; high
preinfusion disease burden, prior blinatumomab nonresponse, older age,
and 4-1BB construct were all associated with CD19- relapses; the
presence of KMT2A rearrangements was the only preinfusion risk factor
associated with LS relapses.[60]
The median overall survival following a post-CAR-T relapse was 18.9
months for CD19+ relapses, 9.7 months for CD19- relapses, and 3.7
months for LS relapses.[60]
Primary Resistance to CAR-T Cell Therapy
In
addition to acquired resistance observed in patients initially
responding to CAR-T cell therapy, primary resistance (PR) is observed
in about 10-20% of patients undergoing CD19 CAR-T cell therapy. PR
therapy is characterized by CD19-positive progressive disease. It is
associated with the increased expression of exhaustion markers (LAG3,
TIM3, and PD-1) in the apheresis product used for CAR-T cell
manufacturing or a decreased rate of CAR-T cell expansion in vivo.[61]
Using a genome-wide loss-of-function screening provided evidence that
impaired death receptor signaling in B-ALL leads to rapidly progressive
disease in CD19 CAR-T-treated patients: reduced expression of death
receptor genes was associated with worse overall survival and reduced
T-cell fitness.[62]A
recent study identified a gene expression profile that correlates with
primary resistance to CD19 CAR-T cell therapy in B-ALL samples, related
to the expression of genes typically expressed in hematopoietic
stem/progenitor cells while maintaining a pre-B cell phenotype.[63]
This finding is important because it identified a mechanism of
resistance intrinsic to leukemic cells, preexisting to CAR-T cell
therapy, and may provide a tool to define the eligibility of B-ALL
patients to CAR-T cell therapy.
Expansion and Persistence of CAR-T Cells after in Vivo Infusion
Optimal in vivo expansion
and persistence are two additional important determinants of the
therapeutic efficacy of CAR-T cells in B-ALL patients.
The
kinetics of CAR-T cells after their in vivo infusion shows an initial
expansion after cell infusion, followed by a peak level and then a
decline with persistence at variable levels for years after treatment.
Higher peak CAR-T cell levels and CAR-T cell area under the curve
within the first month of treatment have been associated with response
in most of the studies carried out in B-ALL patients.[64]
In
addition, the studies carried out using Tisa-Cel in pediatric and young
adult patients have clearly supported the role of long-term CAR-T cell
persistence for a durable response.[6-7] In the ELIANA
study, the time to B-cell recovery among responders was 35.3 months,
and the probability of persistent B-cell aplasia (evidencing persistent
functional CAR-T cells) at 12 and 24 months after the infusion, was 71%
and 59%, respectively.[6-7] Furthermore, the duration
of response for allo-HSCY patients with onset of B-cell recovery at
<6 months was clearly shorter compared to those with onset of B-cell
recovery at >6 months.[7]
The Role of Allo-HSCT as a Consolidation Therapy after CAR-T Cell Therapy in B-ALL Patients
Allo-HSCT
represents an important option to consolidate the therapeutic results
obtained using CAR-T cells in B-ALL patients. This consolidation
strategy was adopted in several clinical trials involving CD19 or CD22
CAR-cell therapy in B-ALL patients. The results observed in these
studies on the capacity of allo-HSCT to improve the results of CAR-T
cell therapy, in terms of EFS and OS, are variable. It is important to
note that, to date, no clinical trial has been specifically designed to
evaluate the role of allo-HSCT after CAR-T cell therapy.The
results obtained in the various clinical trials showed variable results
related to the efficacy of allo-HSCT to consolidate the results
achieved by CAR-T cell therapy in terms of EFS and OS. Thus, in the
ELIANA trial carried out in children and young adults treated with
Tisa-Cel, only 15% of the patients who achieved a response underwent
subsequent allo-HSCT with apparent no benefit in EFS related to
transplant.[6-7] In contrast to these findings, Shah
et al., in the National Pediatric trial carried out using the CD19.28ζ
CAR-T, reported consolidative allo-HSCT in 75% of patients achieving a
CR with MRD-negative status, with a mOS of 70.2 months, a 5-yr EFS rate
of 61.9% and a cumulative incidence of relapse of 9.5% at 24 months; 7
patients with MRD-negative CR not undergoing allo-HSCT relapsed with a
median time of relapse of 152 days.[30] Thus, this study concluded that consolidative allo-HSCT is required following CAR-T cell therapy with CD19.28ζ construct.[30]Zheng
et al. reported a retrospective analysis on 52 R/R B-ALL patients
undergoing remission following treatment with CD19 or CD22-targeted
CAR-T cell therapy and processed for allo-HSCT after myeloablative
reduced intensity conditioning; after a median follow-up of 334 days,
1-year OS and EFS were 87.7% and 73%, respectively, with 1-year relapse
rate and transplantation-related mortality of 24.7% and 2.2%,
respectively.[65]The ZUMA-3 trial with Axu-Cel[24,26-27] and the study of Park et al. with CD19.28ζ CAR-T of Park et al.[20]
carried out in adult R/R B-ALL patients failed to show any significant
effect of allo-HSCT in improving EFS and overall survival in patients
who achieved a CR, with a MRD-negative status. However, Hay and
coworkers, in their study with CD19 CAR-T cells with 4.1BB
co-stimulatory domain, carried out on 53 adult R/R B-ALL patients,
showed that patients achieving a CR with MRD negativity and undergoing
allo-HSCT displayed a longer EFS compared to patients with CR and MRD
negativity not undergoing allo-HSCT.[17]Cao
et al. have retrospectively analyzed long-term follow-up data
concerning 97 R/R B-ALL patients who relapsed after a first HSCT and
who have received either CD19- or CD22-targeted CAR-T cell therapy
followed by consolidation with a second allo-HSCT.[66-67]
The second transplant was performed using donors different from the
first transplant. These patients' 4-year OS and OS were 52.6% and
49.8%, respectively.[66-67] These observations
support the view that CAR-T cell therapy followed by consolidation with
a second HSCT for B-ALL patients who have relapsed after first
transplantation may improve long-term survival.
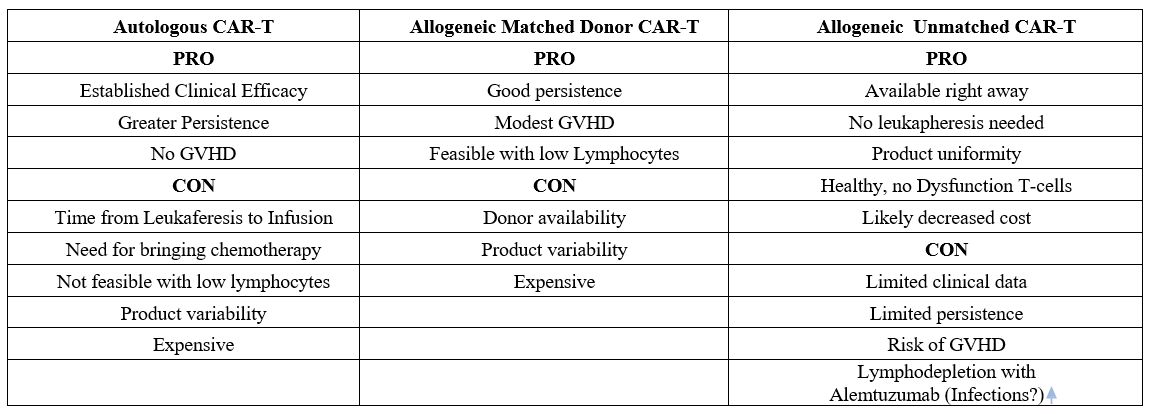
Allogeneic CAR-T Cell therapy
Since
2017 (FDA) and 2018 (EMEA), autologous CAR-T cells have been approved
for commercialization to treat many lymphoid hematological
malignancies, showing impressive clinical efficacy in patients with
relapsed or refractory advanced-stage tumors. However, using the
patient's T cells as starting material (i.e., autologous use) has
important limitations since patients’ T cells may be dysfunctional and
exhausted, influencing CAR-T cell products' potency and variability.
This can be caused by patient age, the number of previous lines of
treatment, the disease itself, and, in solid tumors, local immune
suppression and the effects of prolonged T-cell stimulation. In
addition, autologous CAR-T cell therapies are individualized products,
thus entailing theoretically higher costs and manufacturing time,
usually around 2–3 weeks. Moreover, in patients with refractory
leukemias, there are often large numbers of circulating leukemic cells
that can be extracted along with healthy lymphocytes and thus
contaminate the product. It has been suggested that CAR-transduced
cancer cells present on therapy may be associated with down-regulation
of the target antigen, leading to patient relapse by this newly
generated population. Other
causes of strains could be manufacturing delays, high production costs,
and difficulties in standardizing the preparation process. Furthermore,
the number of T-cells is too low to be harvested in some circumstances.[68]To
take the next step and reach much larger numbers of patients, you
should adopt treatments "off-the-shelf" offering a standardized,
consistent, and cost-effective product to patients.The
use of allogeneic CAR T cells from donors has many potential advantages
over autologous approaches, such as the immediate availability of
cryopreserved batches for patient treatment, possible standardization
of the CAR-T cell product, time for multiple cell modifications,
redosing or combination of CAR T cells directed against different
targets, and decreased cost using an industrialized process.
Furthermore, allogeneic CAR-T cells are the only ones that can be
utilized when the subject is lymphopenic. All autologous and allogeneic
CAR-T cells are genetically modified T cells to express the specific
CAR molecule. Moreover, one of the main strategies to enable the
allogeneic use of CAR-T cells manufactured from healthy donor T cells
involves the addition of extra genetic modifications in the
manufacturing process. The host immune system may rapidly eliminate the
allogeneic T-CAR cells; therefore, the infusion of allogeneic T-CAR
cells must be preceded by a lymphodepletion regimen comprising
fludarabine (F, 90mg/m2) and cyclophosphamide (C,1,500mg/m2)
with or without alemtuzumab (A, 1 mg/kg, or 40mg, or 60mg flat dose),
to improve CAR-T cell engraftment and expansion. However, UCART19
expansion rates in the clinical trials confirm the need for alemtuzumab
to observe UCART19 expansion (along with fludarabine cyclophosphamide).[69]Furthermore,
the allogeneic CAR T cells may cause life-threatening graft-versus-host
disease. Developing next-generation allogeneic CAR T cells to address
these issues is an active area of research. In
recent years, a wide range of different approaches have been studied to
achieve the production of allogeneic CAR-T cell therapies, which could
be classified into two main categories: those involving extra genetic
modifications in addition to CAR transgene introduction and those
relying on the selection of alternative cell sources/subpopulations as
starting material. Different
sources of T cells for optimal allogeneic CAR-T cell therapy and
different technological approaches, mainly based on gene editing, have
been settled to produce allogeneic CAR-T cells with limited potential
for graft-versus-host disease.[70]Novel
strategies, many of which have been reported in the last 5 years,
include the use as cell sources of γδ T cells, Induced pluripotent stem
cells (iPSCs), Umbilical cord blood T (UCB T) cells, memory T cell
subpopulations, Virus-Specific T (VST) cells, and Cytokine-induced
killer cells (CIK) cells. Although genetic modification of T cells is
the most widely used approach, new strategies combining both methods
have emerged. However, further preclinical and clinical research is
needed to establish the most appropriate strategy for producing
allogeneic CAR-T cells, which should minimize the major risks of this
therapy: GvHD and immune rejection. Commercializing this promising
antitumor therapy could extend the availability of CAR-T cells to a
larger number of patients.[70-72]As
previously mentioned, the two main potential problems of the allogeneic
use of T cells are GvHD and immune rejection. The former can be avoided
by eliminating the TCR, usually through the knockout of the constant
domain of one of its chains (α or β), or by replacing some TCR subunits
that impede its antigen recognition function.Regarding
immune rejection, it is avoided by preventing the expression of HLA
class I (HLA-I) molecules by knocking out their common subunit
β2-microglobulin (encoded by the B2M gene), which prevents the
recipient’s T cells from recognizing the therapeutic cells as foreign
through their TCR.In
the context of relapsed and refractory childhood pre-B cell acute
lymphoblastic leukemia (R/R B-ALL), CD19-targeting chimeric antigen
receptor (CAR)-T cells often induce durable remissions, which requires
the persistence of CAR-T cells.[68]The
approach is simpler by utilizing T cells obtained from a compatible
donor as starting material for allogeneic CAR-T cell manufacturing.
However, it depends on donor availability and maintains the main
disadvantages of autologous therapies, such as high cost, lack of
standardization, and manufacturing time. It is not a truly
“off-the-shelf” therapy but has been used experimentally in some
patients.[73-79] Additionally, several studies
investigate using specific cell sources or subpopulations to produce
“off-the-shelf” CAR-T cell therapies. In the following, we describe
these different strategies that, in most cases, are based on selecting
a specific subpopulation that would allow allogeneic use without
causing GvHD.So far, most clinical experience is concerned with using T cells of compatible donors.[76-80]
T-Cells from Compatible Donors
CAR-T
cells from compatible donors demonstrated their efficacy in relapsed
patients after allogeneic stem cell transplantation, increasing
disease-free survival.The
first demonstration that donor allogeneic anti-CD19 CAR-T cells were
able to induce regression of B-cell Lymphoid malignancy in relapse
after allogeneic transplantation was reported by Kochenderfer et al.
(2013), who conducted a clinical trial of allogeneic T cells
genetically modified to express a chimeric antigen receptor (CAR)
targeting the B-cell antigen CD19. T cells for genetic modification
were obtained from each patient's allo-HSCT donor. All patients had a B
cell malignancy that persisted after allo-HSCT and standard donor
lymphocyte infusions (DLIs). Patients did not receive chemotherapy
prior to the CAR T-cell infusions and were not lymphocyte-depleted at
the time of the infusions. The 10 treated patients received a single
infusion of allogeneic anti-CD19-CAR T cells. Three patients had
regressions of their malignancies. One patient with chronic lymphocytic
leukemia (CLL) obtained an ongoing complete remission after treatment
with allogeneic anti-CD19-CAR T cells, another CLL patient had tumor
lysis syndrome as his leukemia dramatically regressed, and a patient
with mantle cell lymphoma obtained an ongoing partial remission—none of
the 10 patients developed graft-versus-host disease (GVHD). Toxicities
included transient hypotension and fever. Cells containing the
anti-CD19-CAR gene were detected in the blood of 8 of 10 patients.
These results showed for the first time that donor-derived allogeneic
anti-CD19-CAR T cells can cause regression of B-cell malignancies
resistant to standard DLIs without causing GVHD.[73]
A similar experience was reported by Cruz et al. (2013) with an
infusion of donor-derived CD19-redirected virus-specimen in patients
who relapsed after allogeneic BMT B-cell malignancies.[74]
Eight patients were treated with allogeneic (donor-derived) CD19.
CAR-VSTs 3 months to 13 years after HSCT. There were no
infusion-related toxicities. VSTs persisted for a median of 8 weeks in
blood and up to 9 weeks at disease sites. Objective antitumor activity
was evident in 2 of 6 patients with relapsed disease during
CD19.CAR-VST persistence, whereas 2 patients who received cells while
in remission remained disease-free.[74]Good
results were also obtained by Brudno et Al. (2016) with the T cells
obtained from each recipient's allo-HSCT donors. Eight of 20 treated
patients obtained remission, which included six complete remissions
(CRs) and two partial remissions. The response rate was highest for
acute lymphoblastic leukemia, with four of five patients obtaining
minimal residual disease-negative CR. None of the patients developed
new-onset acute graft-versus-host disease after CAR T-cell infusion.
Toxicities included fever, tachycardia, and hypotension.[75]Good
results have also been obtained by the sequential infusion of
allogeneic donors followed by autologous and CAR-T cells in a child
suffering from relapsed and refractory B-ALL, severely
lymphoid-depleted.[76]Donor-derived CAR-T infusions after allogeneic transplantation compared with donor lymphocyte infusions. Three Chinese papers [77-79]
demonstrated the superiority of donor-derived anti-CD19 CAR T cells vs
Donor lymphocytes (DLI) for the management of relapsed B-cell acute
lymphoblastic leukemia (B-ALL) after allo-hematopoietic stem cell
transplantation (HSCT) Hua
et Al. (2021) compared B-ALL patients who relapsed after allo-HSCT; 13
were treated with donor-derived anti-CD19 CAR T-cell (study group), and
15 were treated with DLI (DLI group). The rates of MRD-negative
complete remission (61.5%) in the study group were significantly higher
than those in the DLI group (13.3%) (p = 0.02). The complete remission
duration in the study group and DLI group were median of 8.0 months
(range, 3-25 months) and 4.4 months (range, 1-25 months; p = 0.026),
respectively. The overall survival of patients in the study group was
superior to that of the DLI group: 9.5 months (range,3-25 months)
versus 5.5 months (range, 1-25 months; p = 0.030). The study group
identified one patient with grade 1 acute graft-versus-host disease
(aGVHD). At the same time, five (33.3%) patients in the DLI group
developed grades III–IV aGVHD. Three patients (23.07%) developed grade
3 or 4 cytokine release syndrome in the study group. This study
suggested that donor-derived anti-CD19 CAR T-cell therapy is a
promising, safe, and potentially effective treatment for relapsed B-ALL
after allo-HSCT and may be superior to DLI.[77] The
efficacy of anti-CD19-CAR T-cell therapy can be improved by the donor
hemopoietic stem cell infusion (DSI) more than donor lymphocyte
infusion (DLI) therapy, as reported by Li et al. (2023). In total, 22
B-ALL patients who relapsed after allo-HSCT received anti-CD19-CAR
T-cell therapy. Patients who responded to CAR T-cell therapy received
DSI or DLI as maintenance therapy. The two groups' clinical responses,
acute graft versus host disease (aGVHD), expansion of CAR-T-cells, and
adverse events were compared. In our study, 19 patients received
DSI/DLI as maintenance therapy. After DSI/DLI therapy, progression-free
survival and overall survival were higher in the DSI group than in the
DLI group at 365 days. The aGVHD, grades I and II, was observed in four
patients (36.4%) in the DSI group. Only one patient developed grade II
aGVHD in the DLI group. IL-6 and TNF-α levels increased again in nine
of 11 patients after DSI but not in the DLI group. These findings
indicate that for B-ALL patients who relapse after allo-HSCT, DSI is a
feasible maintenance therapy if CR is obtained with CAR-T-cell therapy.[78]A
single-center retrospective study was conducted comparing 12 patients
treated with DLI (control group) and 12 patients treated with
donor-derived CD19 CAR-T cells. The median age of patients was 31. The
event-free survival (EFS) of patients in the experimental group was
longer than that of the control group: 516 days versus 98 days (p =
0.0415). No significant difference in the incidence of infection was
identified between these two groups. Three patients developed GVHD
after CAR-T therapy, including 2 cases of aGVHD (grade 2 and grade 3)
and one severe cGVHD, and were effectively controlled by combinational
therapy with steroids. Among patients of the DLI group, 2 (16.7%) and 7
(58.8%) developed grades I–II and III–IV aGVHD, respectively. Most
patients in the experimental group had only mild cytokine release
syndrome, and post-transplantation relapse was associated with better
EFS. There was no significant difference in EFS between patients
treated with dual-target CAR-T and those with single CD19 CAR-T. In
this study, data supported that donor-derived CAR-T therapy is a safe
and potentially effective treatment for relapsed B-ALL after HSCT and
may be superior to DLI.[79]Another
experience with donor-derived allogeneic CD-19-directed CAR-T cells in
relapsed/refractory B-cell precursor acute lymphoblastic leukemia
(BCP-ALL) has been reported by Del Bufalo et al. from the Bambino Gesù
Hospital in Rome. Donor-derived T cells were transduced with a
second-generation (4.1BB) CD19-directed CAR manufactured in the site
place of care. Thirteen children/young adults (median age 15 years)
received ALLO–CAR-T cells between March 2021 and October 2022. Doses
ranged between 1.0×106 and 3.0×106
CAR-T cells per kg. The toxicity profile was comparable with that of
autologous CAR-T cells, characterized mainly by cytopenia, cytokine
release syndrome (maximum grade 1), and grade 2 immune-effector
cell–associated neurotoxicity syndrome. One case of acute
graft-versus-host disease (GVHD) occurred and was rapidly controlled
with steroids and ruxolitinib. None of the other patients, including 3
treated with ALLO–CAR-T cells from an HLA-haploidentical donor,
experienced GVHD. Two patients received ALLO–CAR-T cells before HSCT
and showed a significant expansion of CAR-T cells without any sign of
GVHD. All patients obtained complete remission (CR) without minimal
residual disease in the bone marrow. With a median follow-up of 12
months (range, 5-21), 8 of 13 patients maintain CR.[80]
T-Cells from Unselected Healthy Donors
UCART19
engineered. Allogeneic CAR-T cells, not HLA compatible with the
recipient, can be utilized only by performing a Genome editing of
allogeneic T cells by the disruption of T cell receptor α chain (TRAC)
to prevent graft-versus-host disease (GVHD) and removal of CD52
(cluster of differentiation 52) by using transcription activator–like
effector nuclease (TALEN) for obtaining a survival advantage in the
presence of alemtuzumab.[81] The mRNA encoding TALENs
is used to knock out the genes encoding the TCR α constant chain and
CD52 to minimize the risk of GVHD by reducing the number of
TCRαβ-positive T cells and to confer resistance to the anti-CD52
monoclonal antibody alemtuzumab.This
method was utilized by the UCART19 Group to treat CD 19 positive ALL.
Qasim et Al. (2017) first treated two infants with relapsed refractory
CD19+ B cell acute lymphoblastic leukemia receiving lymphodepletion
chemotherapy and anti-CD52 serotherapy, followed by a single-dose
infusion of UCART19 cells. Molecular remissions were achieved within 28
days in both infants, and UCART19 cells persisted until conditioning
ahead of successful allogeneic stem cell transplantation. This
bridge-to-transplantation strategy demonstrates the therapeutic
potential of gene-editing technology.[78]Benjamin
et Al. (2020) reported phase 1 trials in pediatric and adult patients
with late-stage relapsed or refractory B-cell acute lymphoblastic
leukemia treated with UCART19. Pediatric
or adult patients were enrolled in two ongoing, multicenter, phase 1
clinical trials to evaluate the safety and antileukemic activity of
UCART19. All patients underwent lymphodepletion with fludarabine and
cyclophosphamide with or without alemtuzumab, then children received
UCART19 at 1·1–2·3 × 10⁶ cells per kg, and adults received UCART19
doses of 6 × 10⁶ cells, 6–8 × 10⁷ cells, or 1·8–2·4 × 10⁸ cells in a
dose-escalation study. Patients not receiving alemtuzumab (n=4) showed
no UCART19 expansion or antileukemic activity. The median duration of
response was 4·1 months, with ten (71%) of 14 responders proceeding to
a subsequent allogeneic stem-cell transplant. Progression-free survival
at 6 months was 27%, and overall survival was 55%.The primary outcome measure was adverse events in the period between the first infusion and data cutoff. Cytokine
release syndrome was the most common adverse event and was observed in
19 patients (91%); three (14%) had grade 3-4 cytokine release syndrome.
Other adverse events were grade 1 or 2 neurotoxicity in eight patients
(38%), grade 1 acute skin graft-versus-host disease in two patients
(10%), and grade 4 prolonged cytopenia in six patients (32%). Two
treatment-related deaths occurred: one caused by neutropenic sepsis in
a patient with concurrent cytokine release syndrome and one from
pulmonary hemorrhage in a patient with persistent cytopenia. 14 (67%)
of 21 patients had a complete response or complete response with
incomplete hematological recovery 28 days after infusionOf
seven children in the PALL study, 1 was lost to follow-up, 2 are alive
and in remission, 4 died, 3 from progressive disease, and 1 from
infection after stem-cell transplant.Of
14 adults in the AALL study, 3 are alive and in remission, 1 relapsed,
11 died, 7 from progressive disease, 2 from non-treatment-related
infection, 1 from treatment-related infection plus cytopenia, 1 from
treatment-related infection plus cytokine release syndrome.These
two studies showed, for the first time, the feasibility of using
allogeneic, genome-edited CAR T cells to treat patients with aggressive
leukemia. UCART19 exhibited in-vivo expansion and antileukemic activity
with a manageable safety profile in heavily pretreated pediatric and
adult patients with relapsed or refractory B-cell acute lymphoblastic
leukemia. The results of this study were considered an encouraging step
forward for the field of allogeneic CAR T cells.[83]This
unicentric experience was repeated in a polycentric study., with the
same modality.84 A phase 1 open-label study was conducted at eight
centers across France, the UK, the USA, and Japan. Adult patients aged
16–70 years with CD19-positive relapsed or refractory B-cell acute
lymphoblastic leukemia who had a morphological relapse or a minimal
residual disease level of at least 1 × 10-³ and had exhausted standard
treatment options were enrolled in the study, which comprised a
dose-escalation phase of up to three UCART19 doses followed by a safety
expansion phase. Patients underwent lymphodepletion, and then 1·8–2·4 ×
10⁸ total CAR T cells were infused intravenously, followed by safety
evaluation and disease response assessments. The primary endpoint was
the incidence and severity of adverse events. Secondary endpoints were
the overall response rate, duration of response, relapse-free survival,
progression-free survival, and overall survival. Between Aug 1, 2016,
and June 30, 2020, 25 patients were enrolled in the study and treated
with UCART19. The median duration of follow-up was 12·8 months (IQR
2·8–24·8). The median age was 37 years (IQR 28–45). 14 (56%) patients
were male and 11 (44%) females. Seventeen (68%) patients were White,
two (8%) were Black, two (8%) were Asian, and four (16%) were from
other racial or ethnic groups. Three patients developed dose-limiting
toxicities (one at each dose level); one had grade 4 cytokine release
syndrome, and two had grade 4 prolonged cytopenia. Grade 3 or higher
cytokine release syndrome was reported in six (24%) patients, and grade
3 or higher neurological toxicity in one (4%) patient. Grade 3 or
higher infections occurred in seven (28%) patients, and grade 4
prolonged cytopenia in four (16%) patients. Two (8%) patients developed
grade 1 acute cutaneous graft-versus-host disease. 14 patients died,
nine from progressive disease and five from infections or other
complications, of which four were considered to be related to UCART19,
lymphodepletion, or both. After a median of follow-up of 12·8 months
(IQR 2·8–24·8), overall response rate was 48% (95% CI 28–69; 12 of 25
patients), duration of response and median relapse-free survival were
7·4 months (95% CI 1·8 to not calculable), progression-free survival
was 2·1 months (95% CI 1·2–2·8), and overall survival was 13·4 months
(95% CI 4·8–23·0).UCART19
had a manageable safety profile and showed evidence of antileukemic
activity in heavily pretreated adult patients with relapsed or
refractory B-cell acute lymphoblastic leukemia. This study shows that
allogeneic off-the-shelf CAR T cells can be used safely to treat
patients with relapsed B-cell acute lymphoblastic leukemia.[84]Dupouys
et Al. (2022) reported twenty-five adult patients with CD19-positive
R/R B-ALL, enrolled from August 2016 to July 2020 in an open-label
nonrandomized phase I/II study conducted in eight clinical centers
across Europe, USA, and Japan. This trial, CALM study, comprised two
phases: a dose escalation investigating three dose levels of UCART19 (6
× 106,6–8 × 107, or 1.8–2.4 × 108 total CAR+ cells) followed by a dose expansion at the recommended dose (6–8 × 107
total CAR+ cells). All patients received a 6-day lymphodepletion
regimen prior to UCART19 infusion (day 0) consisting of fludarabine (F)
30 mg/m2/day i.v. for 3 days (day-7 to day-5) and cyclophosphamide (C) 500 mg/m2/day
i.v. for 3 days (day-4 to day-2), with or without alemtuzumab (A) 1
mg/kg, 40 or 60 mg flat doses (day-7 to day-3). The dose of alemtuzumab
was modified during the trial to balance the infectious complications
related to alemtuzumab use and UCART19 efficacy. An allogeneic stem
cell transplantation (allo-SCT) could be performed at any time
following disease evaluation on day 28 after the UCART19 infusion. In
this trial, UCART19 was administered to 25 adult patients with relapsed
or refractory (R/R) B-cell acute lymphoblastic leukemia (B-ALL). All
patients underwent lymphodepletion with fludarabine and
cyclophosphamide ± alemtuzumab and received one of three ascending
doses of UCART19. Given the allogeneic nature of UCART19, we analyzed
the impact of lymphodepletion, HLA disparities, and host immune system
reconstitution on its kinetics, along with other factors that affect
autologous CAR-T cell clinical pharmacology. Responder patients (12/25)
had higher UCART19 expansion (Cmax) and exposure (AUCT last) than no
responders (13/25), as measured by transgene levels in peripheral
blood. The persistence of CAR+ T cells did not exceed 28 days in 10/25
patients and lasted beyond 42 days in 4/25.No
significant correlation was found between UCART19 kinetics and
administered cell dose, patient and product characteristics, or HLA
disparities. However, the number of prior lines of therapy and the
absence of alemtuzumab negatively impacted UCART19 expansion and
persistence. Alemtuzumab exposure positively affected IL7 and UCART19
kinetics while negatively correlating with host T lymphocyte AUC0-28. UCART19
expansion is a response driver in adult patients with R/R B-ALL. These
results shed light on the factors associated with UCART19 kinetics,
which remain highly affected by the impact of alemtuzumab on IL7 and
host-versus-graft rejection. This study represents the first
description of the clinical pharmacology of a genome-edited allogeneic
anti-CD19 CAR-T cell product, showing the crucial role of an
alemtuzumab-based regimen in sustaining UCART19 expansion and
persistence through increased IL7 availability and decreased host T
lymphocyte population.[85]To
better understand and quantify the impact of the preconditioning
regimen on the engraftment and proliferation of CAR-T cells, Derippe et
Al. built a population-based mechanistic
pharmacokinetic-pharmacodynamic model describing the complex interplay
between lymphodepletion, host immune system, homeostatic cytokines, and
pharmacokinetics of UCART19, an allogeneic product developed against
CD19+ B cells. Data were collected from a phase I clinical trial in
adult relapsed/refractory B-cell acute lymphoblastic leukemia. They
revealed three different UCART19 temporal patterns: (i) expansion and
persistence, (ii) transient expansion with subsequent rapid decline,
and (iii) absence of observed expansion. On the basis of translational
assumptions, the final model was able to capture this variability
through the incorporation of IL-7 kinetics, which is thought to be
increased owing to lymphodepletion, and through an elimination of
UCART19 by host T cells, which is specific to the allogeneic context.
Simulations from the final model recapitulated UCART19 expansion rates
in the clinical trial, confirmed the need for alemtuzumab to observe
UCART19 expansion (along with fludarabine cyclophosphamide), quantified
the importance of allogeneic elimination, and suggested a high impact
of multipotent memory T-cell subpopulations on UCART19 expansion and
persistence. In addition to supporting the role of host cytokines and
lymphocytes in CAR-T cell therapy, such a model could help optimize the
preconditioning regimens in future clinical trials.[86]In
conclusion, UCART19 was shown to proliferate and induce responses in
adult patients with B-ALL following a lymphodepletion regimen,
including fludarabine, cyclophosphamide, and alemtuzumab. Several
factors potentially influencing UCART19 cellular kinetics were
identified, highlighting areas for improvement. Further efforts are
needed to optimize the therapeutic window, allowing appropriate
expansion and persistence of allogeneic CAR-T cells, among which
optimization of the chosen lymphodepletion regimen and strategy of
redosing are key to making allogeneic CAR-T cell therapy a success.CRISPR/Cas9 Technology.
The utility of CRISPR-Cas9 technology has led to a surge in applying
genome editing approaches to combat various genetic disorders and
cancers. Inherited genetic diseases with known gene mutations can be
corrected in these cases. CRISPR/Cas9 technology applications are being
explored in T-cell-based immunotherapies to improve T-cell effector
function and persistence, reduce treatment toxicity, and increase
patient product availability.[87-90]Compared
with TALEN gene editing technology, CRISPR/Cas9 technology has a
simpler design, higher editing efficiency, and wider versatility.
Clinical data from trials of the product generated using CRISPR/Cas9
gene editing technology have demonstrated its safety and feasibility,
including through single-antigen targeting (CD7) in R/R T cell ALL.Hu
et al. developed (2021) CRISPR-edited universal off-the-shelf CD19/CD22
dual-targeted CAR-T cells as a novel therapy for r/r ALL. In their
open-label dose-escalation phase 1 study, universal CD19/CD22-targeting
CAR-T cells (CTA101) with a CRISPR/Cas9-disrupted TRAC region and CD52
gene to avoid host immune-mediated rejection were infused in patients
with r/r ALL. Safety, efficacy, and CTA101 cellular kinetics were
evaluated. Six patients received CTA101 infusions at doses of 1 (3 patients) and 3 (3 patients) × 106
CAR+ T cells/kg body weight. Cytokine release syndrome occurred in all
patients. No dose-limiting toxicity, GvHD, neurotoxicity, or genome
editing-associated adverse events have occurred. The complete remission
(CR) rate was 83.3% on day 28 after the CTA101 infusion. With a median
follow-up of 4.3 months, 3 of the 5 patients who achieved CR or CR with
incomplete hematologic recovery (CR/CRi) remained minimal residual
disease (MRD) negative. The authors concluded that
CRISPR/Cas9-engineered universal CD19/CD22 CAR-T cells exhibited a
manageable safety profile and prominent antileukemia activity.
Universal dual-targeted CAR-T cell therapy may offer an alternative
therapy for patients with r/r ALL.[91] Ottaviano
et Al. deployed next-generation CRISPR-Cas9 editing and linked CAR
expression to multiplexed DNA editing of TRAC and CD52 through the
incorporation of self-duplicating CRISPR guide RNA expression cassettes
within the 3' long terminal repeat of a CAR19 lentiviral vector. Three
cell banks of TT52CAR19 T cells were generated and cryopreserved. A
phase 1, open-label, nonrandomized clinical trial was conducted and
treated six children with relapsed/refractory CD19-positive B cell
acute lymphoblastic leukemia (B-ALL) (NCT04557436). Lymphodepletion
included fludarabine, cyclophosphamide, and alemtuzumab and was
followed by a single infusion of 0.8×106 to 2.0×106
CAR19 T cells per kilogram with no immediate toxicities. Four of six
patients infused with TT52CAR19 T cells exhibited cell expansion,
achieved flow cytometric remission, and then proceeded to receive
allogeneic stem cell transplantation. Two patients required biological
intervention for grade II cytokine release syndrome, one patient
developed transient grade IV neurotoxicity, and one patient developed
skin GVHD, which resolved after transplant conditioning. Other
complications were within expectations, and primary safety objectives
were met.[92]This
study provides a demonstration of the feasibility, safety, and
therapeutic potential of CRISPR-engineered immunotherapy, and none
developed neurotoxicity.
Conclusions
CAR-T
cell therapy has made an important contribution to the therapy of B-ALL
patients, particularly those with R/R disease. Thus, in 2017, Tisa-Cel
was approved for treating pediatric and young adult patients; in 2021,
Brexu-Cel received approval for adult patients with R/R B-ALL.Real-world studies have confirmed the high rates of CR observed using these agents in patients with R/R B-ALL.[93]
Particularly, a real-world report of CAR-T cell treatment in B-ALL
patients from Germany showed that patients with refractory or relapsed
disease also relapsed after allo-HSCT could be rescued with an infusion
of CAR-T cells (Tisa-Cel): patients who relapsed 6 months after HSCT
have a good chance to survive their disease (45% of patients).[94]However,
adverse events such as cytokine release syndrome and immune effector
cell-associated neurotoxicity represent significant challenges to CAR-T
cell therapy. The severity of these adverse events correlates with the
pretreatment tumor burden; this observation supports CAR-T cell therapy
early at low tumor burden and debulking therapies prior to CAR-T cell
infusion to reduce the severity of these adverse events.Furthermore,
a significant proportion of patients rapidly relapse after an initial
response due to early CAR-T cell loss and antigen downregulation and to
enhance CAR-T cell persistence in vivo. The
role of allo-HSCT after CAR-T cell therapy remains an undefined issue;
specific randomized trials are required for the different CAR-T
constructs and for pediatric/young adult and adult B-ALL patients to
assess the role of allo-HSCT in consolidating the therapeutic effects
achieved through CAR-T cell therapy. However, the donor-matched T-CAR
lymphocytes have had a major utilization and should be proposed
whenever the number of patient’s lymphocytes is too low to allow
sufficient harvesting. Their superior efficacy in comparison to simple
lymphocytes has been demonstrated.
References
- June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med 2018; 379: 64-73. https://doi.org/10.1056/NEJMra1706169 PMid:29972754 PMCid:PMC7433347
- Cappell
KM, Kochenderfer JN. Long-term outcomes following CAR T cells therapy:
what we know so far Nat Rev Clin Oncol 2023; 20: 359-371. https://doi.org/10.1038/s41571-023-00754-1 PMid:37055515 PMCid:PMC10100620
- Kantarjian
HM, De Angelo DJ, Stilljes M, et al. Inotuzumab ozogamicin versus
standard therapy for acute lymphoblastic leukemia. N Engl J Med 2016,
375: 740-753. https://doi.org/10.1056/NEJMoa1509277 PMid:27292104 PMCid:PMC5594743
- Kantarjian
HM, Stein A, Gokbugel n, et al. Blinatumumab versus chemotherapy for
advanced acute lymphoblastic leukemia. N Engl J Med 2017; 378: 836-847.
https://doi.org/10.1056/NEJMoa1609783 PMid:28249141 PMCid:PMC5881572
- Pequignot
E, Gill S, Luger SM, Mangau JK, Loren AW, Perl AW, Maude SL, Grupp SA,
et al. Optimizing chimeric antigen receptor T-cell therapy for adults
with acute lymphoblastic leukemia. J Clin Oncol 2019; 28: 417-422.
- Maude
SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and
young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018;
378: 439-448. https://doi.org/10.1056/NEJMoa1709866 PMid:29385370 PMCid:PMC5996391
- Laetsch
TW, Mauda SL, Rives S, Hiramatsu H, Bittencourt H, Beder P, Baruchel A,
Bayer M, De Moorlose B, Qayed M, et al. Three-year update of
tisangenlecleucel in pediatric and young adult patients with
relapsed/refractory acute lymphoblastic leukemia. J Clin Oncol 2022;
41: 1664-1669. https://doi.org/10.1200/JCO.22.00642 PMid:36399695 PMCid:PMC10022844
- Gorashian
S, Jacoby E, De Meerlose B, et al. Tisagenlecleucel therapy for
relapsed or refractory B-cell acute lymphoblastic leukemia in infants
and children younger than 3 years of age at screening: an
international, multicentre, retrospective cohort study. Lancet Haematol
2022; 9: e766-e775. https://doi.org/10.1016/S2352-3026(22)00225-3 PMid:36084658
- Moskop
A, Pommert L, Baggott C, Prabhu S, Placenta HL, Phillips CL, Rossoff J,
Stefanski HE, Talanao JA, Margossian SP, et al. Real-world use of
tisagenlecleucel in infant acute lymphoblastic leukemia. Blood Adv
2022; 6: 4251-4255. https://doi.org/10.1182/bloodadvances.2021006393 PMid:35580324 PMCid:PMC9327536
- Pasquini
MC, Ha ZH, Curran K, Laetsch T, Locke F, Rouce R, Pulsiphaer MA,
Phillips Ch, Kaeting A, Frigault MT, et al. Real-world evidence of
tisagenlecleucel for pediatric acute lymphoblastic leukemia and
non-Hodgkin lymphoma. Blood Adv 2020; 4: 5414-5424. https://doi.org/10.1182/bloodadvances.2020003092 PMid:33147337 PMCid:PMC7656920
- John
S, Pulsipher MA, Moskop A, Hu ZH, Phillips CL, Hall, EM, Margossian SP,
Nikiforow S, Martin PL, Oshrine et al. Real-world outcomes for
pediatric and young adult patients with relapsed and refractory (R/R)
B-cell acute lymphoblastic leukemia (ALL) treated with
Tisagenlecleucel: update from the Center for International Blood Marrow
Transplant Reseach (CIBMTR) Registry. Blood 2021; 138 (supll. 1):
428-429. https://doi.org/10.1182/blood-2021-146393
- Fabrizio
VA, Phillips CL, Lane A, Bagott C, Prabhu S, Egeler ER, Mavroukakis S,
Pacenta H, Rossoff J, Stefanski et al. Tisagenlecleucel outcomes in
relapsed/refractory extramedullary ALL: a Pediatric Real World
Consortium Report. Blood Adv 2022; 6: 601-610. https://doi.org/10.1182/bloodadvances.2021005564 PMid:34794180 PMCid:PMC8791593
- Schultz
LM, Baggott C, Probhu S, Pacenta HL, Phillips CL, Rossoff J, Stefanski
HE, Talano JA, Moskop A, Margossian SP, et al. Disese burden affects
outcomes in pediatric and young adult B-cell lymphoblastic leukemia
after commercial tisagenlecleucel: a pediatric real-world chimeric
antigen receptor consortium report. J Clin Oncol 2021; 40: 945-955. https://doi.org/10.1200/JCO.20.03585 PMid:34882493 PMCid:PMC9384925
- Pulsipher
MA, Han X, Maude SL, Laetsch TW, Qayed M, Rives S, Boyer MW, Hiranatsu
H, Yanik GA, Driscoll T, et al. Next generation sequencing of minimal
residual disease for predicting relapse after tiosagenlecleucel in
children and young adults with acute lymphoblastic leukemia. Blood
Cancer Discov 2022; 3: 66-81. https://doi.org/10.1158/2643-3230.BCD-21-0095 PMid:35019853 PMCid:PMC9924295
- Dekker
L, Calkoen FG, Jiang Y, Blok H, Veldkamp SR, De Konig C, Spoon M,
Admiral R, Hogerbrugge P, et al. Fludarabine exposure predicts outcome
adter CD19 CAR T-cell therapy in children and young adults with acute
leukemia. Blood Adv 2022; 6: 1969-1976. https://doi.org/10.1182/bloodadvances.2021006700 PMid:35134115 PMCid:PMC9006280
- Dourthe
ME, Rabian F, Yakouben K, Chevillon F, Cabannes-Hamy A, Méchinaud F,
Grain A, Gaillou D, Rahal I, Caillat-Zucman S, et al. Determinants of
CD19-positive vs CD19-negative relapse after tisagenlecleucel for
B-cell acute lymphoblastic leukemia. Leukemia 2021; 35: 3383-3393. https://doi.org/10.1038/s41375-021-01281-7 PMid:34002027
- Hay
KA, Gauthier J, Hirayama AV, Voutsinos JM, Vu Q, Li D, Gooley TA,
Cherian S, Chen X, et al. Factors associated with durable EFS in adult
B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell
therapy. Blood 2019; 133: 1652-1663. https://doi.org/10.1182/blood-2018-11-883710 PMid:30728140 PMCid:PMC6460418
- An
F, Wang H, Liu Z, Wu F, Zhang J, Tao Q, Li Y, Shen Y, Ruan Y, Zhang Q,
et al. Influence of patient characteristics of chimeric antigen
receptor T cell therapy in B-cell acute lymphoblastic leukemia. Nat
Commun 2020; 11: 5928. https://doi.org/10.1038/s41467-020-19774-x PMid:33230103 PMCid:PMC7683530
- Hollyman
D, Stefanski J, Przaybylowski M, Bontido S, Borque-Ojeda O, Taylor C,
Yeh R, Capacio V, Olzewska M, Hosey J, et al. Manufacturing validation
of biologically functional T cells targeted to CD19 antigen for
autologous adoptive cell therapy. J Immunotherapy 2009; 32: 169-180. https://doi.org/10.1097/CJI.0b013e318194a6e8 PMid:19238016 PMCid:PMC2683970
- Park
J, Riviere I, Gonen M, Wang X, Sénéchal B, Curran KJ, Sauter C, Wang Y,
Santomasso B, Mead E, et al. Long-term follow-up of CD19 CAR therapy in
acute lymphoblastic leukemia. N Engl J Med 2018; 378: 449-459. https://doi.org/10.1056/NEJMoa1709919 PMid:29385376 PMCid:PMC6637939
- Curran
KJ, Margossian SP, Keman NA,Silverman LB, Williams DA, Shokla N, Kobos
R, Forlenza CJ, Steinherz P, Prockop S, et al. Toxicity and response
after CD19-specific CAR T-cell therapy in pediatric/young
relapsed/refractory B-ALL. Blood 2019; 134: 2361-2368. https://doi.org/10.1182/blood.2019001641 PMid:31650176 PMCid:PMC6933289
- Wudhikam
K, Flynn JR, Rivière I, Gonen M, Wang X, Sonechal B, Curran KJ, Roshal
M, Maslak PG, Geyer MB, et al. Interventions and outcomes of adult
patients with B-ALL progressing after CD19 chimeric antigen receptor
T-cell therapy. Blood 2021; 138: 531-543. https://doi.org/10.1182/blood.2020009515 PMid:33851211 PMCid:PMC8377478
- Shah
BD, Bisho MR, Oluwola OO, Logan AC, Baer MR, Donnellan WB, O'Dwyer KM,
Holmes H, Arellano ML, Ghobadi A, Pagel JM, et al. KTE-X19 anti-CD19
CART-cell therapy in adult relapsed/refractory acute lymphoblastic
leukemia. Blood 2021; 138: 11-22. https://doi.org/10.1182/blood.2020009098 PMid:33827116
- Shah
BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, Leguay T,
Bishop MR, Topp MS, Tzachanis D, et al. KTE-X19 for relapsed or
refractory adult B-cell acute lymphoblastic leukemia: phase 2 results
of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021;
398: 491-502. https://doi.org/10.1016/S0140-6736(21)01222-8 PMid:34097852
- Frey
NV. Approval of brexucabtagene autocel for adults with relapsed and
refractory acute lymphocytic leukemia. Blood 2022; 140: 11-15. https://doi.org/10.1182/blood.2021014892 PMid:35507688
- Shah
BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, Leguay T,
Bishop MR, Topp MS, Tzachanis D, et al. Two-year follow-up of KTE-X19
in patients with relapsed or refractory adult B-cell acute
lymphoblastic leukemia in ZUMA-3 and its contextualization with
SCHOLA-3, an external historical control study. J Hematol Oncol 2022;
15: 170. https://doi.org/10.1186/s13045-22-01379-0 PMid:36494725 PMCid:PMC9734710
- Shah
BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, Leguay T,
Bishop MR, Topp MS, Tzachanis D, et al. Updated outcomes from the
historical control study SCHOLAR-3 contextualizing ZUMA-3 results of
Brexucabtagene autoleucel (KTE-X19) in adult patients with relapsed or
refractory B-cell acute lymphoblastic leukemia (R/R B-ALL). Blood 2022;
140(suppl. 1): 3158-3161. https://doi.org/10.1182/blood-2022-158248
- Wayne
AS, Huyhn V, Hijiya N, Rare RH, Brown P, Krueger J, Kitko CL, Dela Ziga
E, Hermiston ML, Richards MK, et al. Three-year results from phase I of
ZUMA-4: KTE-X19 in pediatric relapsed/refractory acute lymphoblastic
leukemia. Haematologica 2023; 108: 747-760. https://doi.org/10.3324/haematol.2022.280678 PMid:36263840 PMCid:PMC9973494
- Jacoby
E, Bielorai B, Hutt D, Itzhaki O, Adam E, Bar D, Besser MJ, Taren A.
Parameters of long-term response with CD28-based CD19 chimeric antigen
receptor-modified T cells in children and young adults with B-acute
lymphoblastic leukemia. Brit J Haematol 2022; 197: e45-e55. https://doi.org/10.1111/bjh.18105 PMid:35224724
- Shah
NN, Long-term follow-up of CD19-CART-cell therapy in children and young
adults with B-ALL. J Clin Oncol 2021; 39: 1650-1659. https://doi.org/10.1200/JCO.20.02262 PMid:33764809 PMCid:PMC8274806
- Ceolin
V, Brivio E, van Tinteren H, Rheingold SR, Leahy A, Vormoor B, O'Brien
MM, Rubinskin JD, Kalwak K, De Meorlloose B, et al. Outcome of chimeric
antigen receptor T-cell therapy following treatment with inotuzumab
ozogamicin in children with relapsed or refractory acute lymphoblastic
leukemia. Leukemia 2023; 37: 53-60. https://doi.org/10.1038/s41375-022-01740-9 PMid:36310183
- Gorashian
S, Kramer AM, Onuhoa S, Wright G Bartram J, Richardson R, Albon SJ,
Casanovas-Company J, Castro F, Popova B, et al. Enhanced CAR T cell
expansion and prolonged persistence in pediatric patienst with ALL
treated with a low-affinity CD19 CAR. Nat Med 2019; 25: 1408-1414. https://doi.org/10.1038/s41591-019-0549-5 PMid:31477906
- Roddie
C, Dias J, O'Reilly MA, Abbasian M, Cadinanos-Garai A, Vispute K,
Bosshard-Carter L, Mistikakou M, Mehra V, Roddy H, et al. Durable
responses and low toxicity after fast off-rate CD19 chimeric antigen
receptor-T therapy in adults with relapsed or refractory B-cell acute
lymphoblastic leukemia. J Clin Oncol 2021; 31: 3352-3363. https://doi.org/10.1200/JCO.21.00917 PMid:34464155 PMCid:PMC8791810
- Roddie
C, Sandhu KS, Tholouli E, Shaughnessy P, Barba P, Guerreiro MN, Yallop
D, Abbedi M, Chaganti S, Ghobadi A, et al. Safety and efficacy of
obecabtagene autoleucel (obe-cel, AUTO1), a fast-off rate CD19 CAR, in
relapsed/refractory adult B-cell acute lymphoblastic leukemia (r7r
B-ALL): top nline results of the pivotal FELIX study. J Clin Oncol
2023; 41(suppl.16): abst. 7000. https://doi.org/10.1200/JCO.2023.41.16_suppl.7000
- Anderson
ND, Birch J, Accogli T, Criado I, habirova E, Parks C, Wood Y, Young
MD, Porter T, Richardson R, et al. Transcriptional signatures
associated with persisting CD19 CAR-T cells in children with leukemia.
Nat Med 2023; 29: 1770-1709. https://doi.org/10.1038/s41591-023-02415-3 PMid:37407840 PMCid:PMC10353931
- Fry
TT, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S,
Wolters P, Mortin S, Delbrook C, Yates B, et al. CD22-CART cells induce
remissions in CD19-CAR naïve and resistant B-ALL. Nat Med 2018; 24:
20-28. https://doi.org/10.1038/nm.4441 PMid:29155426 PMCid:PMC5774642
- Shah
NN, Highfill SL, Shalabi H. CD4/CD8 T-cell selection affects chimeric
antigen receptor (CAR) T-cell potency and toxicity: updated results
from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol 2020; 38:
1938-1950. https://doi.org/10.1200/JCO.19.03279 PMid:32286905 PMCid:PMC7280047
- Pan
J, Niu Q, Deng B, Liu S, Wu T, Gao Z, Liu Z, Zhang Y, Qu X, Zhang Y, et
al. CD22 CAR T-cell therapy in refractory or relapsed B-acute
lymphoblastic leukemia. Leukemia 2019; 33: 2854-2866. https://doi.org/10.1038/s41375-019-0488-7 PMid:31110217
- Tan
Y, Cai H, Li C, Deng B, Song W, Ling Z, Hu G, Yang Y, Ni P, Meng G, et
al. A novel full-human CD22-CAR T cell therapy with potent activity
against CD22low B-ALL. Blood Cancer J 2021; 11: 71. https://doi.org/10.1038/s41408-021-00465-9 PMid:33839735 PMCid:PMC8036232
- Shah NN, Maatman T, Hari P, Johnson B. Mlti-targeted CAR-T cells for B-cell malignancies. Front Oncol 2019; 9: 146. https://doi.org/10.3389/fonc.2019.00146 PMid:30915277 PMCid:PMC6423158
- Wang
N, Hu X, Cao W, Li C, Xiao Y, Cao Y, Gu C, Zhang S, Chen L, Cheng J,
Wang G, et al. Efficacy and safety of CAR 19/22 T-cell cocktail therapy
in patients with refractory/relapsed B-cell malignancies. Blood 2020;
135: 17-27. https://doi.org/10.1182/blood.2019000017 PMid:31697824
- Liu
S, Deng B, Yin Z, An L, Liu D, Pan J, Yu X, Chen B, Wu T, et al.
Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell
acute lymphoblastic leukemia after allogeneic transplantation. Am J
Hematol 2021; 96: 671-679. https://doi.org/10.1002/ajh.26160 PMid:33725422
- Wang
N, Tang Y, Cai J, Wan X, Hu S, L X, Xie Z, Qiao X, Jiang H, Shao J, et
al. Coadministration of CD19- and CD22-directed chimeric antigen
receptor T-cell therapy in childhood B-cell acute lymphoblastic
leukemia: a single-arm, multicenter, phase II trial. J Clin Oncol 2023;
41: 1670-1683. https://doi.org/10.1200/JCO.22.01214 PMid:36346962
- Hu
P, Xiang Y, Wu D, Zhu Z, Dropulic B, Scneider D. Fully human tandem
CD22-CD19 CAR-T cells with superior sensitivity to low antigen density
derived by optimization of co-stimulation and CAR architecture. Blood
2020; 136 (suppl. 1): 12-13. https://doi.org/10.1182/blood-2020-140836
- Dai
H, Wu Z, Jia H, Tang C, Guo Y, Ti D, Han X, Liu Y, Zhang W, Wang C, et
al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of
adults with relapsed or refractory B cell acute lymphoblastic leukemia.
J Hematol Oncol 2020; 13: 80. https://doi.org/10.1186/s13045-020-00856-8 PMid:32245502 PMCid:PMC7126394
- Cui
W, Zhang X, Dai H, Cui Q, Yin J, Li Z, Yu L, Kang L, Wu D, Tang X.
Tandem CD19/CD22 dual targets CAR T-cells bridging hematopoietic stem
cells transplantation acquires robust remission for relapsed and
refractory B acute lymphoblastic leukemia patients. Blood 2021; 138
(suppl.1): 1753. https://doi.org/10.1182/blood-2021-152565
- Liu
S, Zhang X, Dai H, Cui W, Yin J, Li Z, Yang X, Yang C, Xue S, Qiu H, et
al. Which one is better for refractory/relapsed acute B-cell
lymphoblastic leukemia: single target (CD19) or dual-target (tandem or
sequential CD19/CD22) CAR T-cell therapy? Blood Cancer J 2023; 13: 60. https://doi.org/10.1038/s41408-023-00819-5 PMid:37095120 PMCid:PMC10125987
- Zhang
XY, Dai HP, Zhang L, Liu SN, Dai Y, Wu DP, Tang XW. MRD-negative
remission induced in EP300-ZNF384 positive B-ALL patients by tandem
CD19/CD22 CAR T-cell therapy bridging to allogeneic stem cell
transplantation. Onco Targets Ther 2021; 14: 5197-5204. https://doi.org/10.2147/OTT.S324765 PMid:34744437 PMCid:PMC8565984
- Spiegel
JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, Frank MJ, Shiraz P,
Sahaf B, Craig J, et al. CAR T cells with dual targeting of CD19 and
CD22 in adult patients with recurrent or refractory B cell
malignancies: a phase 1 trial. Nat Med 2021; 27: 1419-1431. https://doi.org/10.1038/s41591-021-01436-0 PMid:34312556 PMCid:PMC8363505
- Cordoba
S, Onuoha S, Thomas S, Soriano Pignataro D, Hough R, Ghorasian S, Vora
A, Bonney D, Veys P, Rao K, et al. CART cells with dual targeting of
CD19 and CD22 in pediatric and young adult patients with relapsed or
refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat
Med 2021; 27: 1797-1805. https://doi.org/10.1038/s41591-021-01497-1 PMid:34642489 PMCid:PMC8516648
- Annesley
A, Summers C, Pulsipher MA, Skiles JL, Li AM, Vatsayan A, Lindgren C,
Mgebroff S, Wilson A, Huang W, et al. SCRI-CAR19x22v2 T cell product
demonstrates bispecific activity in B-ALL. Blood 2021; 138 (suppl.1),
470-472. https://doi.org/10.1182/blood-2021-148881
- Lucchini
G, Gorashian S, Richardson R, Nguyen K, Terris C, Espuela J, Chu J,
Williams L, Ko K, Walding C, et al. Dual Antigen targeting with
co-transduced CD19/CD22 CAR T cells may prevent antigen-negative
relapse after CAR T cell therapy for relapsed/refractory ALL. Eur Bone
Mattow Transpl. 49th Meeting; 2023; OS20-04. https://doi.org/10.1182/blood-2022-164879
- Li
S, Zhang J, Wang M, Fu G, Li Y, Pei L, Xiong Z, Qin D, Zhang R, Tian X,
et al. Treatment of acute lymphoblastic leukemia with the second
generation of CD19 CAR-T containing either CD28 or 4-1BB. Br J Haematol
2018; 181: 360-371. https://doi.org/10.1111/bjh.15195 PMid:29637550
- Orlando
EJ, Han X, Tribouley C, Wood PA, Leary RJ, Riester M, Levine JE, Qayed
M, Grupp SA, Boyer M, et al. Genetic mechanisms of target antigen loss
in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 2018; 24:
1504-1506. https://doi.org/10.1038/s41591-018-0146-z PMid:30275569
- Fischer
J, Paret C, Malki K, Alt F, Wingerter A, Neu MA, Kron B, Russo A,
Lehmann N, Roth L, et al. CD19 isoforms enabling resistance to CART-19
immunotherapy are expressed in B-ALL patients at initial diagnosis. J
Immunother 2017; 40: 187-195. https://doi.org/10.1097/CJI.000000000000169 PMid:28441264 PMCid:PMC5424577
- Asnani
M, Hayer KE, Naqvi AS, Zheng S, Yan SY, Oldridge D, Ibrahim F, Magkakis
M, Gazzara MR, Block KL, et al. Retention of CD19 intron 2 contributes
to CART-19 resistance in leukemias with subclonal frameshift mutations
in CD19. Leukemia 2020; 34: 1202-1207. https://doi.org/10.1038/s41375-019-0580-z PMid:31591467
- Robillaud
T, Potier D, Pankaew S, Nazais M, Loosveld M, Payet-Bornet D.
Single-cell profiling identifies pre-existing CD19-negative subclones
in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat
Commun 2021; 12:865. https://doi.org/10.1038/s41467-021-21168-6 PMid:33558546
- Pan
J, Tan Y, Deng B, Tong C, Hua L, Ling Z, Song W, Xu J, Duan J, Wang Z,
et al. Frequent occurrence of CD19-negative relapse after CD19 CART and
consolidation therapy in 14 TP53-mutated r/r B-ALL children. Leukemia
2020; 34: 3382-3387. https://doi.org/10.1038/s41375-020-0831-z PMid:32346068
- Myers
RM, Taraseviciute A, Steinberg SM, Lamble AJ, Sheppard J, Yates B,
Kovach AE, Wood B, Borowitz MJ, Stetler-Stevenson M, et al.
Blinatumomab nonresponse and high-disease burden are associated with
inferior outcomes after CD19-CAR for B-ALL. J Clin Oncol 2021; 40:
932-944. https://doi.org/10.1200/JCO.21.01405 PMid:34767461 PMCid:PMC8937010
- Lamble
AJ, Myers RM, Travesiciute A, John S, Yates B, Stenberg SM, Sheppard J,
Kovash AE, Wood B, Borowitz MJ, et al. Preinfusion factors impacting
relapse immunophenotype following CD19 CAR T cells. Blood Adv 2023; 7:
575-585. https://doi.org/10.1182/bloodadvances.2022007423 PMid:35482927 PMCid:PMC9979750
- Finney
OC, Brakke H, Rawlings-Rhea R, Hicks R, Doolittle D, Lopez M, Futrell
B, Orentas RJ, Li D, Gardner R, et al. CD19 CART cell product and
disease attributes predict leukemia remission durability. J Clin Invest
2019; 129:2123-2132. https://doi.org/10.1172/JCI125423 PMid:30860496 PMCid:PMC6486329
- Singh
N, Lee YG, Shestova O, Ravikumar P, Hayer KE, Hong SJ, Maggie Lu X,
Pajarillo R, Agarwal S, Kuramitsu S, et al. Impaired death receptor
signaling in leukemia causes antigen-independent resistance by inducing
CART cell dysfunction. Cancer Discov 2020; 10: 552-567. https://doi.org/10.1158/2159-8290.CD-19-0813 PMid:32001516 PMCid:PMC7416790
- Masih
KE, Gardner RA, Chou HC, Abdelmasksouyd A, Song YK, Mariani L,
Gangalapudi V, Gryder BE, Wilson AL, Adebola SO, et al. A stem cell
epigenome is associated with primary nonresponse to CD19 CAR T-cells in
pediatric acute lymphoblastic leukemia. Blood Adv 2023; in press. https://doi.org/10.1182/bloodadvances.2022008977 PMid:36607839 PMCid:PMC10440404
- Cappell
KM, Kochenderfer JN. Long-term outcomes following CAR T cell therapy:
what we know so far. Nat Rev Clin Oncol 2023; 20: 359-371. https://doi.org/10.1038/s41571-023-00754-1 PMid:37055515 PMCid:PMC10100620
- Zheng
Y, Chen H, Song Y, Tan X, Zhao Y, Liu X, Li Z, Yang F, Jiang M, Gao Z,
et al. Chimeric antigen receptor T-cell therapy as a bridge to
hematopoietic stem cell therapy as a bridge to hematopoietic stem cell
transplantation for refractory/relapsed B-cell acute lymphoblastic
leukemia. Brit J Haematol 2020; 189: 146-152. https://doi.org/10.1111/bjh.16339 PMid:31869864
- Cao
X, Zhang J, Zhao Y, Xiong M, Zhou J, Lu Y, Sun R, Wei Z, Liu D, Zhang
X, et al. Analysis benefits of a second allo-HSCT after CAR-T cell
therapy in 97 patients with relapsed/refractory B-cell acute
lymphoblastic leukemia who relapsed after a first transplant. Blood
2022; 140 (suppl. 1): 4802-4803. https://doi.org/10.1182/blood-2022-166993
- Cao
X, Zhang J, Zhao Y, Xiong M, Zhou J, Lu Y, Sun R, Wei Z, Liu D, Zhang
X, et al. Analysis benefits of a second allo-HSCT after CAR-T cell
therapy in patients with relapsed/refractory B-cell acute lymphoblastic
leukemia who relapsed after transplant. Front Immunol 2023 https://doi.org/10.3389/fimmu.2023.1191382 PMid:37469510 PMCid:PMC10352576
- Aparicio
C, Acebal C, González-Vallinas M. Current approaches to develop
"off-the-shelf" chimeric antigen receptor (CAR)-T cells for cancer
treatment: a systematic review. Exp Hematol Oncol. 2023 ;12(1):73. doi:
10.1186/s40164-023-00435-w. https://doi.org/10.1186/s40164-023-00435-w PMid:37605218 PMCid:PMC10440917
- Derippe
T, Fouliard S, Marchiq I, Dupouy S, Almena-Carrasco M, Geronimi J,
Declèves X, Chenel M, Mager DE. Mechanistic Modeling of the Interplay
Between Host Immune System, IL-7 and UCART19 Allogeneic CAR-T Cells in
Adult B-cell Acute Lymphoblastic Leukemia. Cancer Res Commun. 2022;
2(11):1532-1544. doi: 10.1158/2767-9764.CRC-22-0176. https://doi.org/10.1158/2767-9764.CRC-22-0176 PMid:36970053 PMCid:PMC10036133
- Depil
S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic
CAR T cells: development and challenges. Nat Rev Drug Discov. 2020
Mar;19(3):185-199. doi: 10.1038/s41573-019-0051-2. https://doi.org/10.1038/s41573-019-0051-2 PMid:31900462
- Morgan
MA, Büning H, Sauer M, Schambach A. Use of Cell and Genome Modification
Technologies to Generate Improved "Off-the-Shelf" CAR T and CAR NK
Cells. Front Immunol. 2020 Aug 7;11:1965. doi:
10.3389/fimmu.2020.01965. https://doi.org/10.3389/fimmu.2020.01965 PMid:32903482 PMCid:PMC7438733
- Li
W, Zhu X, Xu Y, Chen J, Zhang H, Yang Z, Qi Y, Hong J, Li Y, Wang G,
Shen J and Qian C (2022) Simultaneous editing of TCR, HLA-I/II and
HLA-E resulted in enhanced universal CAR-T resistance to
allo-rejection. Front. Immunol. 13:1052717. doi:
10.3389/fimmu.2022.1052717 https://doi.org/10.3389/fimmu.2022.1052717 PMid:36532006 PMCid:PMC9757162
- Kochenderfer
JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, Hakim FT,
Halverson DC, Fowler DH, Hardy NM, Mato AR, Hickstein DD,
Gea-Banacloche JC, Pavletic SZ, Sportes C, Maric I, Feldman SA, Hansen
BG, Wilder JS, Blacklock-Schuver B, Jena B, Bishop MR, Gress RE,
Rosenberg SA. Donor-derived CD19-targeted T cells cause regression of
malignancy persisting after allogeneic hematopoietic stem cell
transplantation. Blood. 2013 Dec 12;122(25):4129-39. https://doi.org/10.1182/blood-2013-08-519413 PMid:24055823 PMCid:PMC3862276
- Cruz
CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E,
Barrett AJ, Ito S, Shpall EJ, Krance RA, Kamble RT, Carrum G, Hosing
CM, Gee AP, Mei Z, Grilley BJ, Heslop HE, Rooney CM, Brenner MK,
Bollard CM, Dotti G. Infusion of donor-derived CD19-redirected
virus-specific T cells for B-cell malignancies relapsed after
allogeneic stem cell transplant: a phase 1 study. Blood.
2013;122(17):2965-73. https://doi.org/10.1182/blood-2013-06-506741 PMid:24030379 PMCid:PMC3811171
- Brudno
JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH,
Gea-Banacloche JC, Pavletic SZ, Hickstein DD, Lu TL, Feldman SA,
Iwamoto AT, Kurlander R, Maric I, Goy A, Hansen BG, Wilder JS,
Blacklock-Schuver B, Hakim FT, Rosenberg SA, Gress RE, Kochenderfer JN.
Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor
Induce Remissions of B-Cell Malignancies That Progress After Allogeneic
Hematopoietic Stem-Cell Transplantation Without Causing
Graft-Versus-Host Disease. J Clin Oncol. 2016 Apr 1;34(10):1112-21. https://doi.org/10.1200/JCO.2015.64.5929 PMid:26811520 PMCid:PMC4872017
- Zhang
JP, Zhang R, Tsao ST, Liu YC, Chen X, Lu DP, Castillo P, Chang LJ.
Sequential allogeneic and autologous CAR-T-cell therapy to treat an
immune-compromised leukemic patient. Blood Adv. 2018 Jul
24;2(14):1691-1695 https://doi.org/10.1182/bloodadvances.2018017004 PMid:30026294 PMCid:PMC6058233
- Hua
J, Zhang J, Zhang X, Wu X, Zhou L, Bao X, Han Y, Miao M, Li C, Fu C,
Chen S, Tang X, Wu D, Qiu H. Donor-derived anti-CD19 CAR T cells
compared with donor lymphocyte infusion for recurrent B-ALL after
allogeneic hematopoietic stem cell transplantation. Bone Marrow
Transplant. 2021 May;56(5):1056-1064. https://doi.org/10.1038/s41409-020-01140-6 PMid:33235353
- Li
Q, Lyu C, Liu M, Wang J, Mou N, Jiang E, Zhang R, Deng Q. Donor
Hematopoietic Stem Cell/Lymphocyte Maintenance Treatment After CAR
T-Cell Therapy in Patients With B-Cell Acute Lymphoblastic Leukemia
Relapse Following Stem Cell Transplant. Cell Transplant. 2023
Jan-Dec;32:9636897231158155. doi: 10.1177/09636897231158155. https://doi.org/10.1177/09636897231158155 PMid:36879459 PMCid:PMC9996720
- Liang
Z, Xu H, Zhou X, Yang J, Tu S, He Y, Zhou L, Li Y. Donor-derived CAR-T
therapy improves the survival of relapsed B-ALL after allogeneic
transplantation compared with donor lymphocyte infusion. Hum Cell. 2023
Sep;36(5):1716-1728. https://doi.org/10.1007/s13577-023-00934-2 PMid:37418233
- Del
Bufalo F, Becilli M, Rosignoli C, De Angelis B, Algeri M, Hanssens L,
Gunetti M, Iacovelli S, Li Pira G, Girolami E, Leone G, Lazzaro S,
Bertaina V, Sinibaldi M, Di Cecca S, Iaffaldano L, Künkele A, Boccieri
E, Del Baldo G, Pagliara D, Merli P, Carta R, Quintarelli C, Locatelli
F. Allogeneic, donor-derived, second-generation, CD19-directed CAR-T
cells for the treatment of pediatric relapsed/refractory BCP-ALL.
Blood. 2023 Jul 13;142(2):146-157 https://doi.org/10.1182/blood.2023020023 PMid:37172203
- Poirot
L, Philip B, Schiffer-Mannioui C, et al. Multiplex genome edited T-cell
manufacturing platform for "off-the-shelf" adoptive T-cell
immunotherapies. Cancer Res 2015; 75: 3853-64. https://doi.org/10.1158/0008-5472.CAN-14-3321 PMid:26183927
- Qasim
W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, Butler K,
Rivat C, Wright G, Somana K, Ghorashian S, Pinner D, Ahsan G, Gilmour
K, Lucchini G, Inglott S, Mifsud W, Chiesa R, Peggs KS, Chan L,
Farzeneh F, Thrasher AJ, Vora A, Pule M, Veys P. Molecular remission of
infant B-ALL after infusion of universal TALEN gene-edited CAR T cells.
Sci Transl Med. 2017 Jan 25;9(374):eaaj2013 https://doi.org/10.1126/scitranslmed.aaj2013 PMid:28123068
- Benjamin
R, Graham C, Yallop D, et al. ; UCART19 Group. Genome-edited,
donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in
pediatric and adult B-cell acute lymphoblastic leukemia: results of two
phase 1 studies. Lancet. 2020 Dec 12;396(10266):1885-1894. doi:
10.1016/S0140-6736(20)32334-5. https://doi.org/10.1016/S0140-6736(20)32334-5 PMid:33308471
- Benjamin
R, Jain N, Maus MV, Boissel N, Graham C, Jozwik A, Yallop D, Konopleva
M, Frigault MJ, Teshima T, Kato K, Boucaud F, Balandraud S,
Gianella-Borradori A, Binlich F, Marchiq I, Dupouy S, Almena-Carrasco
M, Pannaux M, Fouliard S, Brissot E, Mohty M; CALM Study Group.
UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen
receptor T-cell therapy for adults with relapsed or refractory B-cell
acute lymphoblastic leukaemia (CALM): a phase 1, dose-escalation trial.
Lancet Haematol. 2022 Nov;9(11):e833-e843. doi:
10.1016/S2352-3026(22)00245-9.ar;19(3):185-199. doi:
10.1038/s41573-019-0051-2.) https://doi.org/10.1016/S2352-3026(22)00245-9 PMid:36228643
- Dupouy
S, Marchiq I, Derippe T, Almena-Carrasco M, Jozwik A, Fouliard S, Adimy
Y, Geronimi J, Graham C, Jain N, Maus MV, Mohty M, Boissel N, Teshima
T, Kato K, Benjamin R, Balandraud S. Clinical Pharmacology and
Determinants of Response to UCART19, an Allogeneic Anti-CD19 CAR-T Cell
Product, in Adult B-cell Acute Lymphoblastic Leukemia. Cancer Res
Commun. 2022 Nov 30;2(11):1520-1531. doi:
10.1158/2767-9764.CRC-22-0175. https://doi.org/10.1158/2767-9764.CRC-22-0175 PMid:36970059 PMCid:PMC10035397
- Derippe
T, Fouliard S, Marchiq I, Dupouy S, Almena-Carrasco M, Geronimi J,
Declèves X, Chenel M, Mager DE. Mechanistic Modeling of the Interplay
Between Host Immune System, IL-7 and UCART19 Allogeneic CAR-T Cells in
Adult B-cell Acute Lymphoblastic Leukemia. Cancer Res Commun. 2022 Nov
30;2(11):1532-1544. doi: 10.1158/2767-9764.CRC-22-0176. https://doi.org/10.1158/2767-9764.CRC-22-0176 PMid:36970053 PMCid:PMC10036133
- Xu
L, Wang J, Liu Y, Xie L, Su B, Mou D, et al. CRISPR-edited stem cells
in a patient with HIV and acute lymphocytic leukemia. N Engl J Med
2019;381:1240-7. https://doi.org/10.1056/NEJMoa1817426 PMid:31509667
- Lu
Y, Xue J, Deng T, Zhou X, Yu K, Deng L, Huang M, Yi X, Liang M, Wang Y,
et al. Safety and feasibility of CRISPR-edited T cells in patients with
refractory non-small-cell lung cancer. Nat Med. 2020;26:732-40 https://doi.org/10.1038/s41591-020-0840-5 PMid:32341578
- Stadtmauer
EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, Mangan PA,
Kulikovskaya I, Gupta M, Chen F, et al. CRISPR-engineered T cells in
patients with refractory cancer. Science. 2020;367:eaba7365 https://doi.org/10.1126/science.aba7365 PMid:32029687
- Dimitri
A, Herbst F, Fraietta JA. Engineering the next-generation of CAR
T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 2022 Mar
18;21(1):78. doi: 10.1186/s12943-022-01559-z. https://doi.org/10.1186/s12943-022-01559-z PMid:35303871 PMCid:PMC8932053
- Hu
Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, Wei G, Han L, Wang H, Yu S,
Chen Y, Wang Y, He X, Zhang X, Gao M, Yang J, Li X, Ren J, Huang H.
CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell
Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia.
Clin Cancer Res. 2021 May 15;27(10):2764-2772. https://doi.org/10.1158/1078-0432.CCR-20-3863 PMid:33627493
- Ottaviano
G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, Preece R, Chu J,
Kubat A, Adams S, Veys P, Vora A, Rao K, Qasim W; TT52 CRISPR-CAR
group. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T
cells for treatment of children with refractory B cell leukemia. Sci
Transl Med. 2022 Oct 26;14(668):eabq3010.
- Roloff
G, Faramand R, Aldoss I. Outcomes following brexucabtagene autoleucel
administered as an FDA-approved therapy for adults with
relapsed/refractory B-ALL. J Clin Oncol 2023; 41(suppl 16): 7001. https://doi.org/10.1200/JCO.2023.41.16_suppl.7001
- Bader
P, Rossig C, Hutter M, Ayuk FA, Baldus CD, Bucklein VL, Bonig H, Cario
G, Einsele H, Holtich U, Koenecke C, et al. CD19 CAR T cells are an
effective therapy for posttransplant relapse in patients with B-lineage
ALL: real-world data from Germany. Blood Adv 2023; 7: 2436-2448. https://doi.org/10.1182/bloodadvances.2022008981 PMid:36607834 PMCid:PMC10242498