Amiya
Nayak, Pratyusha Gudapati, Swapnil Tripathi, Jasmita Dass, Mukul
Aggarwal and Pradeep Kumar.
All
India Institute of Medical Sciences, New Delhi. India.
Correspondence to: Dr. Pradeep Kumar. All India Institute of Medical Sciences, New Delhi. India. E-mail:
doctorpkgmu@gmail.com
Published: March 01, 2024
Received: November 26, 2023
Accepted: February 08, 2024
Mediterr J Hematol Infect Dis 2024, 16(1): e2024033 DOI
10.4084/MJHID.2024.033
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
To the editor
Autoimmune
lymphoproliferative syndrome (ALPS) is a rare hereditary disease due to
a defect in immune regulation defined by disrupted lymphocyte
homeostasis. This condition is characterized by the accumulation of
abnormally active lymphocytes in lymphoid organs due to an alteration
of the extrinsic apoptotic pathway mediated by the protein receptor FAS
which results in the autoimmune lymphoproliferative syndrome and more
recently in defects affecting the intrinsic apoptotic pathway mediated
by RAS proteins.[1,2]
This apoptotic defect induces the persistence of autoreactive cells,
with an increase (>1.5 % of total Lymphocytes or 2.5% of CD3+
lymphocytes in the setting of normal or elevated lymphocyte counts) in
TCRαβ+CD4-CD8- double negative T (DNT) lymphocytes. These cells are a
hallmark of ALPS and other ALPS-like
disorders. The abnormal accumulation of DNT lymphocytes produces a
myriad of clinical manifestations including lymphadenopathy,
splenomegaly, autoimmune cytopenias and propensity for lymphoid
malignancies. The most common defect responsible for ALPS is germline
heterozygous mutation in FAS, which shows an autosomal dominant pattern
of inheritance. Somatic FAS mutations turn out to be the 2nd most
common cause. Other pathogenic genes include FASL, CASP10, CASP8 and
NRAS.[3,4]
Germline KRAS pathway mutations have been described in
association with cardio facio-cutaneous or Noonan syndrome.[5] Some rare
cases of ALPS-like disease caused by somatic KRAS mutation have been
published.[6,7]
Monogenic lupus is a kind of SLE that commonly
manifests in childhood, usually before the age of five, with severe
disease symptoms. This type of lupus is caused by a genetic variation
in a specific gene. KRAS mutation is an uncommon cause of the
aforementioned phenomenon.[8] We describe a case of probable ALPS with
systemic lupus erythematosus in a child, who was found to have a KRAS
G13C mutation.
Case
A one-and-a-half-year-old boy of Indian origin was brought
by his parents with a 1-year history of fever on and off and gradually
progressive abdominal distension for 10 months. Fever was mild to moderate
grade and intermittent in nature. Parents have consulted multiple centers for
the same and the child has been prescribed multiple antibiotics including
anti-tubercular therapy during the last 1 year. He had also received 1 aliquot
of packed red blood cells at 6 months of age and multiple random donor platelets
in the last 2 months. Family history was insignificant. General physical
examination revealed pallor and multiple petechial spots over both the lower
limbs. On per abdomen examination, the spleen was palpable 7 cm below the left
costal margin and the liver was palpable 5 cm below the right costal margin.
Complete blood count showed hemoglobin of 3.4 mmol/L (5.5 g/dl), WBC counts of
17×10⁹/L, and platelet count of 3×10⁹/L.
Peripheral smear showed spherocytes and numerous nucleated RBCs. Serum
LDH level was 441 mg/dL and direct Coomb’s test was significantly positive
(4+). A provisional diagnosis of Evan’s syndrome was made, and he was started
on steroids. Because of a prolonged history starting from infancy and
organomegaly, the possibility of autoimmune disorders including ALPS was
considered. Peripheral blood immunophenotyping determined 4.8% double negative αβ+
T cells out of total CD3+ cells (Figure 1). The autoimmune panel was positive
for antinuclear antibody (>1:40) and high anti-dsDNA antibody (ELISA) (200
IU/ml). 2D echo didn’t show any evidence of valvular defect but showed mild
pericardial effusion. Based on SLICC criteria, he was diagnosed with SLE.
Clinical exome sequencing was sent. Even after one week of steroid therapy,
there was inadequate response, for which IVIG (2 g/kg) was given over 2 days.
There’s gradual improvement in hemoglobin and platelet count and he became
transfusion independent. During the fever workup, his blood culture grew
Escherichia coli and it was treated with sensitive antibiotics. Clinical exome
sequencing by next-generation sequencing detected a pathogenic variant of KRAS
(Gly13Cys) at exon 2. The patient has been following up with us and doing well
with low-dose steroids.
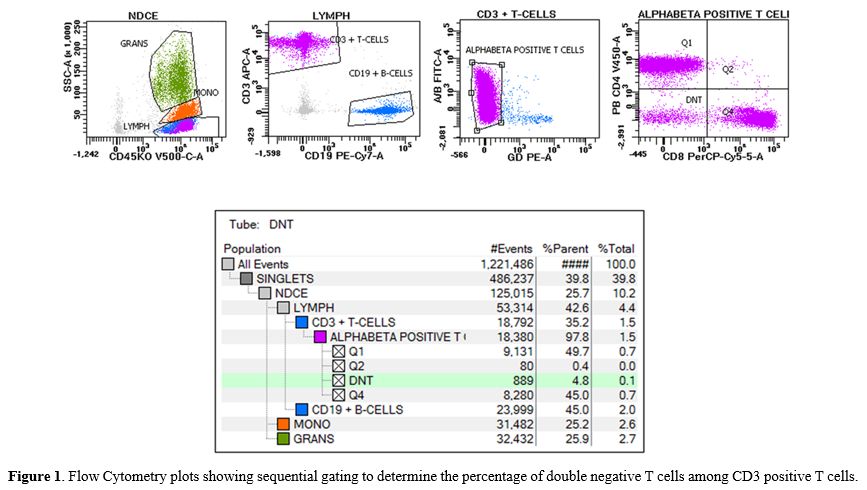 |
- Figure
1. Flow Cytometry plots showing sequential gating to determine the
percentage of double negative T cells among CD3 positive T cells.
|
Discussion
Our
patient showed severe autoimmunity in the form of immunological
cytopenia and hepatosplenomegaly, which was identical to ALPS in
certain ways. The patient also fulfilled SLE categorization criteria,
including the presence of anti-ds-DNA, which is thought to be
reasonably specific for SLE. Considering childhood onset of disease,
simultaneous occurrence of two immune dysregulated disorders, and a
genetically proven pathogenic mutation, a monogenic cause is the most
likely explanation for the described phenotype.
There are around
16 case reports in the literature describing RALD (RAS Associated
Leukoproliferative Disorder) secondary to KRAS mutation.[6,7]
This entity has partial similarity to ALPS but usually doesn’t match
the requisite double negative T cell criterion. For the same reason,
NRAS-associated ALPS, which was formerly classified as Type IV ALPS, is
now considered under this entity. However, our patient did have
increased TCRαβ+-DNT cells. Hence, the diagnosis of 'probable ALPS' is more apt here.[2-4]
Monogenic
lupus is a group of unique genetic defects that cause similar clinical
symptoms and culminate in autoantibody production. It is characterized
by childhood onset SLE with predominant renal, neurological, and
hematological manifestations. It is reasonable to believe that in
childhood-onset disease, genetic factors may be more relevant than
hormonal and environmental influences.[8,9] With the
development of sequencing techniques in recent years, multiple
pathogenic genes have been identified including genes involved in the
complement pathways, genes responsible for ALPS, interferonopathies,
and many more.[10]
Mutations in KRAS at amino
acid position G13 have been linked to cancer, RALD, and Noonan
syndrome. Noonan syndrome is characterized by distinctive facies,
cardiovascular disease, and various skeletal anomalies - none of which
our present possessed. Position G13 is found within the KRAS protein's
p-loop and participates in GTP-hydrolysis. When Glycine is replaced
with another amino acid (e.g Cysteine as in our case), it reduces the
enzymatic activity of KRAS GTPase. This causes growth
factor-independent activation of downstream pathways, which helps in
increasing cellular development and suppressing T-cell death.[11]
The
literature search revealed around 9 previously reported cases of RALD
related to p.G13C KRAS mutations. Among these, 5 patients did have
evidence of pericardial effusion, similar to our case.[7] One patient also had associated SLE and massive lymphadenopathy with sinus histiocytosis.[11] However, enlarged lymph nodes were absent in our patient.
As
autoimmunity is the primary mechanism behind the clinical spectrum,
immunosuppressive therapy remains the cornerstone of treatment. Various
combinations of steroids, IVIG, Rituximab, and calcineurin inhibitors
have been tried in the past with good results.[3,4]
Recently, progress has been achieved in the development of drugs that
can target KRAS mutations seen in cancer. We believe that further
clinical trials of these medications will be highly effective for
patients with KRAS-mediated immune defects.[10]
To
the best of our knowledge, this is the first case in India with SLE
with probable ALPS caused by a KRAS mutation. This case highlights the
necessity of constantly evaluating a monogenic origin for autoimmunity,
especially when disease signs begin early in childhood and do not
follow a conventional clinical course.
References
- Speckmann C, Borkhardt A, Gaspar HB et al (2017)
Genetic disorders of immune regulation. In: Primary immunodeficiency
diseases. Springer-Verlag Berlin Heidelberg 2017:295-338 https://doi.org/10.1007/978-3-662-52909-6_5
- Fleisher TA, Oliveira JB. Monogenic defects in lymphocyte apoptosis. Curr Opin Allergy Clin Immunol. 2012 Dec;12(6):609-15. https://doi.org/10.1097/ACI.0b013e3283588da0 PMid:22918222
- Oliveira,
Joao B et al. Revised diagnostic criteria and classification for the
autoimmune lymphoproliferative syndrome (ALPS): report from the 2009
NIH International Workshop. Blood vol. 116,14 (2010): e35-40. https://doi.org/10.1182/blood-2010-04-280347 PMid:20538792 PMCid:PMC2953894
- Oliveira
Mendonça L, Matucci-Cerinic C, Terranova P, Casabona F, Bovis F, Caorsi
R, Fioredda F, Palmisani E, Grossi A, Guardo D, Bustaffa M, Volpi S,
Ceccherini I, Ravelli A, Dufour C, Miano M, Gattorno M. The challenge
of early diagnosis of autoimmune lymphoproliferative syndrome in
children with suspected autoinflammatory/autoimmune disorders.
Rheumatology (Oxford). 2022 Feb 2;61(2):696-704. https://doi.org/10.1093/rheumatology/keab361 PMid:33909886
- Fabozzi
F., Mastronuzzi A. Genetic predisposition to hematologic malignancies
in childhood and adolescence. Mediterr J Hematol Infect Dis 2023,
15(1): e2023032 https://doi.org/10.4084/MJHID.2023.032 PMid:37180200 PMCid:PMC10171214
- Neven,
Quentin et al. Clinical Spectrum of Ras-Associated Autoimmune
Leukoproliferative Disorder (RALD). Journal of Clinical Immunology
vol. 41,1 (2021): 51-58. https://doi.org/10.1007/s10875-020-00883-7 PMid:33011939
- Masatoshi
Takagi, Kunihiro Shinoda, Jinhua Piao, Noriko Mitsuiki, Mari Takagi,
Kazuyuki Matsuda, Hideki Muramatsu, Sayoko Doisaki, Masayuki Nagasawa,
Tomohiro Morio, Yoshihito Kasahara, Kenichi Koike, Seiji Kojima, Akira
Takao, Shuki Mizutani; Autoimmune lymphoproliferative syndrome-like
disease with somatic KRAS mutation. Blood 2011; 117 (10): 2887-2890 https://doi.org/10.1182/blood-2010-08-301515 PMid:21063026
- Alperin JM, Ortiz-Fernández L and Sawalha AH (2018) Monogenic Lupus: A Developing Paradigm of Disease. Front. Immunol. 9:2496. https://doi.org/10.3389/fimmu.2018.02496 PMid:30459768 PMCid:PMC6232876
- Webb
R, Kelly JA, Somers EC, Hughes T, Kaufman KM, Sanchez E, et al. Early
disease onset is predicted by a higher genetic risk for lupus and is
associated with a more severe phenotype in lupus patients. Ann Rheum
Dis. (2011) 70:151-6. https://doi.org/10.1136/ard.2010.141697 PMid:20881011 PMCid:PMC3034281
- E.
Niemela, L. Lu, T.A. Fleisher, J. Davis, I. Caminha, M. Natter, L.A.
Beer, K.C. Dowdell, S. Pittaluga, M. Raffeld, V.K. Rao, J.B. Oliveira,
Somatic KRAS mutations associated with a human nonmalignant syndrome of
autoimmunity and abnormal leukocyte homeostasis, Blood 117 (2011)
2883-2886. https://doi.org/10.1182/blood-2010-07-295501 PMid:21079152 PMCid:PMC3062298
- Ragotte
RJ, Dhanrajani A, Pleydell-Pearce J, Del Bel KL, TarailoGraovac M, van
Karnebeek C, et al. The importance of considering monogenic causes of
autoimmunity: a somatic mutation in KRAS causing pediatric
Rosai-Dorfman syndrome and systemic lupus erythematosus. Clin Immunol.
2017;175:143-6 https://doi.org/10.1016/j.clim.2016.12.006 PMid:28043923
- Huang,
Lamei et al. KRAS mutation: from undruggable to druggable in cancer.
Signal Transduction and Targeted Therapy vol. 6,1 386. 15 Nov. 2021. https://doi.org/10.1038/s41392-021-00780-4 PMid:34776511 PMCid:PMC8591115