Mireille Yayo-Aye1,2, Adia Eusèbe Adjambri1, Boidy Kouakou3, Rebecca N’guessan-Blao1, Louis Missa Adjé2, Tairatou Kamagaté1, Vincent Yapo1 and Duni Sawadogo1,2.
1 Department of Hematology, Faculty of Pharmacy, Felix Houphouet Boigny University, Abidjan, Côte d’Ivoire.
2 Hematology Unit, Central Laboratory, Yopougon University Hospital, Abidjan, Côte d’Ivoire.
3 Department of Clinical Hematology, Yopougon University Hospital, Abidjan, Côte d’Ivoire.
Correspondence to:
Dr. Mireille Yayo-Ayé (Pharm D), Department of Hematology, Faculty of
Pharmacy, Felix Houphouet Boigny University, Cocody, P.O. Box 2308,
Abidjan 08, Côte d’Ivoire. Tel: +225 07 08 06 54 01. Fax: +225 22 41 05
79. E-mail:
yayoaye@yahoo.fr
Published: March 01, 2024
Received: December 27, 2023
Accepted: February 12, 2024
Mediterr J Hematol Infect Dis 2024, 16(1): e2024026 DOI
10.4084/MJHID.2024.026
This is an Open Access article distributed
under the terms of the Creative Commons Attribution License
(https://creativecommons.org/licenses/by-nc/4.0),
which permits unrestricted use, distribution, and reproduction in any
medium, provided the original work is properly cited.
|
To the editor
Sickle
cell disease results from a point mutation in the sixth codon of the
β-globin gene, leading to the synthesis of mutated hemoglobin known as
hemoglobin S (Hb S), which can polymerize under deoxygenated
conditions. Hb S is responsible for hemolysis and vaso-occlusive events
in individuals with sickle cell disease.[1] It stands
as the world's leading genetic disease, posing a significant public
health challenge in Africa. Indeed, prevalence rates of Hb S in at
least 40 African countries range between 2% and 30%, which explains the
high level of mortality and morbidity due to sickle cell disease.[2]
Currently, the only curative treatments are bone marrow transplantation
and gene therapy, which unfortunately cannot be carried out in Côte
d'Ivoire due to the high cost and insufficient technical
infrastructure. Formerly used in certain hematologic malignancies,
hydroxyurea (HU) remains an effective alternative in the treatment of
sickle cell disease.[3] Biological monitoring of HU is necessary to ensure a better quality of life for children with sickle cell disease.
It
was a prospective observational study that took place from November
2017 to April 2019. Children of both sexes, aged 5 to 15 years,
experiencing at least 3 vaso-occlusive crises (VOC) per year were
included in the study after obtaining informed and written consent from
their parents. Each patient received a daily dose of 15mg/kg of
hydroxyurea for 12 months in the form of 500mg capsules. For each
patient, a venous blood sample was taken for hematological and
biochemical tests. In our trial involving children with SSFA2 (Hb SS)
and SFA2 (Sβ 0 thalassemia) sickle cell disease, subjects received a
dose of 15mg/kg/day of hydroxyurea for 12 months due to challenges in
handling the maximum tolerated dose in developing countries. In
contrast, American authors often use doses ranging from 35 to 40
mg/kg/day.[4] A total of 45 children aged 5 to 10 years were selected. The mean age was 9 years.
The SSFA2 hemoglobin phenotype is predominant in our study (Table 1), in accordance with its historical prevalence among major forms of sickle cell disease.[5]
The administration of hydroxyurea led to a modification of clinical and
biological parameters. With hydroxyurea, we observed a reduction or
absence of vaso-occlusive crises, hospitalizations, and transfusions.
After 6 months of hydroxyurea treatment, our study found rates of
84.4%, 100%, and 97.8%, respectively, for the absence of VOC,
hospitalizations, and transfusions (Table 2).
 |
Table 1. Distribution of subjects by sex, age, and hemoglobin phenotype. |
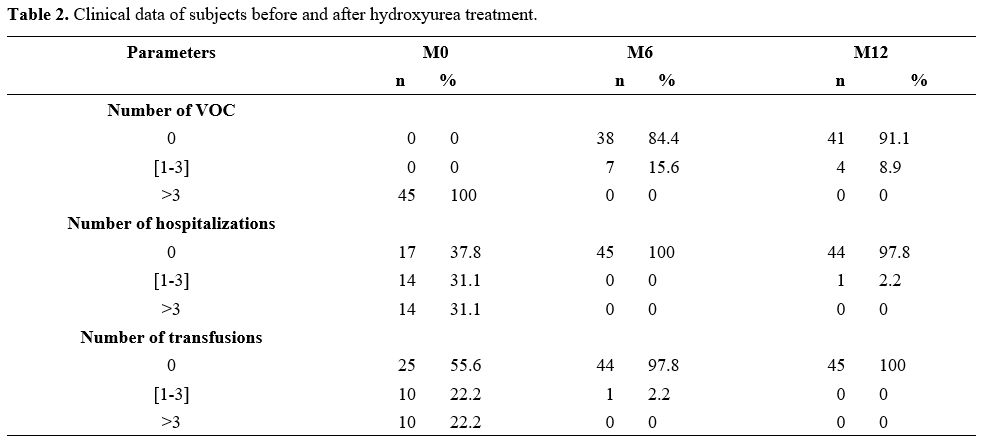 |
Table 2. Clinical data of subjects before and after hydroxyurea treatment.
|
These rates of
reduction in sickle-cell-related events were statistically significant
in our study and consistent with findings from various authors.[6]
These clinical improvements were even more pronounced after 12 months
of treatment overall. The reduction in the frequency of vaso-occlusive
crises and hospitalizations can have a positive impact on the quality
of life of these patients. On a biological level (Table 3),
we observed a reduction in white blood cell (WBC) and platelet counts.
These reductions were statistically significant at 6 months. It is
worth noting that a high WBC count is an unfavorable prognostic factor
for patients.[7] The increase of these parameters,
hematocrit, mean corpuscular hemoglobin concentration, and hemoglobin
level, is beneficial as it reduces the frequency of blood transfusions
by raising the threshold at which a transfusion should be considered in
these patients.[8] The changes in hemogram parameters in our study are similar to those reported by other authors.[6]
In our study, the fetal hemoglobin (Hb F) level doubled from 6 months
and remained stable until 12 months of hydroxyurea treatment. This
induction of Hb F production has been observed by other authors.[9]
This increase in Hb F reduces complications related to this disease.
Hydroxyurea induces the production of Hb F and increases the volume of
red blood cells, thus reducing the risk of Hb S polymerization.[10]
This induction of Hb F leads to a decrease in Hb S. In our series, the
mean Hb S level decreased from 87.5% to 75.7% after 6 months of
treatment, a result beneficial for the patients. Creary et al.[9] also observed this decrease in Hb S with hydroxyurea.
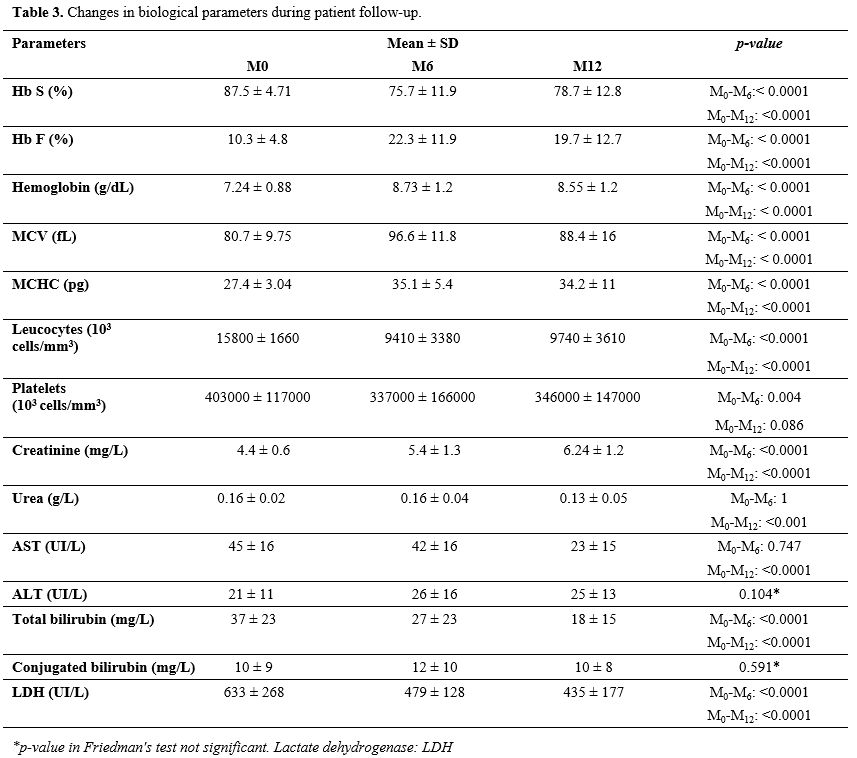 |
Table 3. Changes in biological parameters during patient follow-up. |
During
this clinical trial, we monitored renal and hepatic functions as well
as some biochemical parameters. We did not observe any disruption in
these functions. The concentrations of creatinine, urea, alanine
aminotransferase, aspartate aminotransferase, bilirubin, and Lactate
dehydrogenase did not increase after 6 months and 12 months of
treatment. Although hydroxyurea is mainly eliminated by renal
excretion, instances of kidney toxicity are exceedingly rare, as is
hepatic toxicity.[11] At the beginning of the study
(M0), a slight increase in bilirubin was noted, reflecting the
sub-icterus commonly seen in sickle cell disease. The trend towards a
reduction in bilirubin from M0 to M12 suggests the absence of
hemolysis, emphasizing the protective effect of hydroxyurea on red
blood cells from pathological lysis.[12] Hydroxyurea
treatment is deemed safe for patients and has a positive impact on the
quality of life for children with sickle cell disease by reducing the
frequency of hospitalizations, vaso-occlusive crises, and transfusions.
References
- Elion J, Laurance S, Lapouméroulie C. Revue
générale physiopathologie de la drépanocytose spécial drépanocytose.
Med Trop 2010;70 (5): 454‑458. https://www.jle.com/fr/MedSanteTrop/2010/70.5-6/454-458%20Physiopathologie%20de%20la%20dr%C3%A9panocytose%20(Elion).pdf PMID: 21520646.
- Modell,
Bernadette, et Matthew Darlison. Global epidemiology of haemoglobin
disorders and derived service indicators. Bull World Health Organ
2008;86 (6): 480‑487. https://doi.org/10.2471/BLT.06.036673 PMid:18568278 PMCid:PMC2647473
- Cannas
G, Poutrel S and Thomas X. Hydroxycarbamine: from an old drug used in
malignant hemopathies to a current standard in sickle cell disease.
Mediterr J Hematol Infect Dis 2017, 9(1): e2017015. https://doi.org/10.4084/mjhid.2017.015 PMid:28293403 PMCid:PMC5333733
- Jayabose
S, Tugal O, Sandoval C, Patel P, Puder D, Lin T, Visintainer P.
Clinical and hematologic effects of hydroxyurea in children with sickle
cell anemia. J Pediatr 1996;129:559-565. https://doi.org/10.1016/S0022-3476(96)70121-X PMid:8859263
- Sangare
A, Koffi KG, Sanogo I, Toure AH, Allangba O, Tolo A, Coulibaly FH,
N'dhatz, Elenga JP. Essai thérapeutique de la buprenorphine (Temgesic)
dans le traitement des crises douloureuses drépanocytaires. Méd Afr
Noire 1998;45(2) :138-143. http://www.santetropicale.com/Resume/24512.pdf
- Tshilolo
L, Tomlinson G, Williams TN, Santos B, Olupot-Olupot P, Lane A, Aygun
B, Stuber SE, Latham TS, McGann PT. Hydroxyurea for Children with
Sickle Cell Anemia in SubSaharan Africa. N Engl J Med
2019;380(2):121-131. https://doi.org/10.1056/NEJMoa1813598 PMid:30501550 PMCid:PMC6454575
- Miller
ST, Sleeper LA, Pegelow CH, Enos LE, Wang WC, Weiner SJ, Wethers DL,
Smith J, Kinney TR. Prediction of adverse outcomes in children with
sickle cell disease. N Engl J Med 2000; 342(2):83-89. https://doi.org/10.1056/NEJM200001133420203 PMid:10631276
- Wang
WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, Rana S,
Thornburg CD, Rogers ZR, Kalpatthi RV, Barredo JC, R Clark Brown RC,
Sarnaik SA, Howard TH, Wynn LW, Kutlar A, Armstrong FD, Files BA,
Goldsmith JC, Waclawiw MA, Huang X, and Thompson BW for the BABY HUG
Investigators. Hydroxycarbamide in very young children with sickle-cell
anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet
2011; 377:1663-1672. https://doi.org/10.1016/S0140-6736(11)60355-3 PMid:21571150
- Creary
SE, Beeman C, Stanek J, King K, McGann PT, O'Brien SH, Liem RI, Holl J,
Badawy SM. Impact of hydroxyurea dose and adherence on hematologic
outcomes for children with sickle cell anemia. Pediatr Blood Cancer
2022; 69(6): e29607. https://doi.org/10.1002/pbc.29607 PMid:35373884 PMCid:PMC9038671
- Carden MA, and Little J. Emerging disease-modifying therapies for sickle cell disease. Haematologica. 2019;104(9):1710-1719. https://doi.org/10.3324/haematol.2018.207357 PMid:31413089 PMCid:PMC6717563
- Patrick T McGann, and Russell E Ware. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf 2015;14(11): 1749-1758. https://doi.org/10.1517/14740338.2015.1088827 PMid:26366626 PMCid:PMC5868345
- Lal
A, Patterson L, Goldrich A, and Marsh A. Point-of-Care End-Tidal Carbon
Monoxide Reflects Severity of Hemolysis in Sickle Cell Anemia. Pediatr
Blood Cancer 2015 ; 62(5): 912-914. https://doi.org/10.1002/pbc.25447 PMid:25683629 PMCid:PMC4376621