A 12-year-old male patient from southern China presented with transfusion-dependent hemolytic anemia. His family history was unremarkable, and his parents were non-consanguineous. He first developed symptoms of anemia at 3 months old. Laboratory tests showed microcytic hypochromic anemia and no HbH bands were detected during the Hb analysis. A—-SEA/αCSα with βCD17/βN genotype was detected in the thalassemia gene. The patient was thus diagnosed with HbH disease with β-thalassemia. He subsequently received regular transfusions almost every month and iron-removal therapy.
The patient was evaluated for hematopoietic stem cell transplant in our hospital. During the examination in our hospital, clinical examination revealed anemia, and the liver and spleen were 4 cm and 5 cm below the costal margin, respectively. Routine blood tests and serum biochemical tests revealed Hb 8.2 g/dL, red blood cell count 3.39 × 1012/L, reticulocyte percentage 1.28%, lactate dehydrogenase 213 U/L, total bilirubin 31.6 μmol/L, indirect bilirubin 24.5 μmol/L, and erythropoietin 44.51 mIU/mL. Hb analysis did not detect HbH bands.
Based on his severe clinical phenotype and HbH incompatibility, we further screened for other possible causes of exacerbated anemia. Laboratory tests for iron deficiency anemia, autoimmune hemolytic anemia, glucose-6-phosphate dehydrogenase deficiency, megaloblastic anemia, pure red cell aplasia, paroxysmal nocturnal hemoglobinuria, and hepatopathy were negative. Acid hemolysis was positive. Bone marrow aspiration showed markedly active erythroid hyperplasia and evidence of dysplastic erythropoiesis, including forms of binucleation, nuclear budding, and karyorrhexis (Figure 1). The patient and his parents underwent next-generation sequencing to detect any other underlying hemolytic anemia-related variants. In addition to thalassemia gene mutations, a compound heterozygous variant of the SEC23B gene, c.74C > A (p.Pro25His), inherited from the father, and c.1508G > A (p.Arg503Gln) from his mother were confirmed by Sanger sequencing. The patient was thus diagnosed with CDA II and HbH disease with β-thalassemia. A hematopoietic stem cell transplant was planned after a bone marrow transplant consultation. The laboratory data are summarized in Table 1.
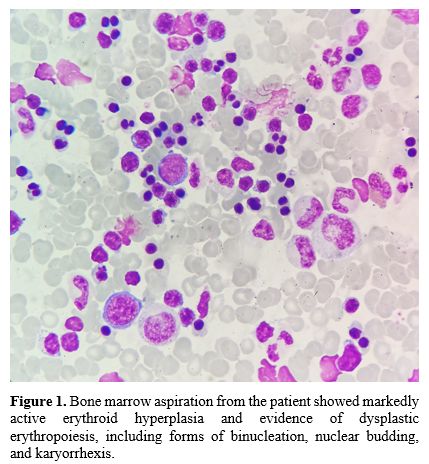 |
Figure 1. Bone marrow aspiration from the patient showed markedly active erythroid hyperplasia and evidence of dysplastic erythropoiesis, including forms of binucleation, nuclear budding, and karyorrhexis. |
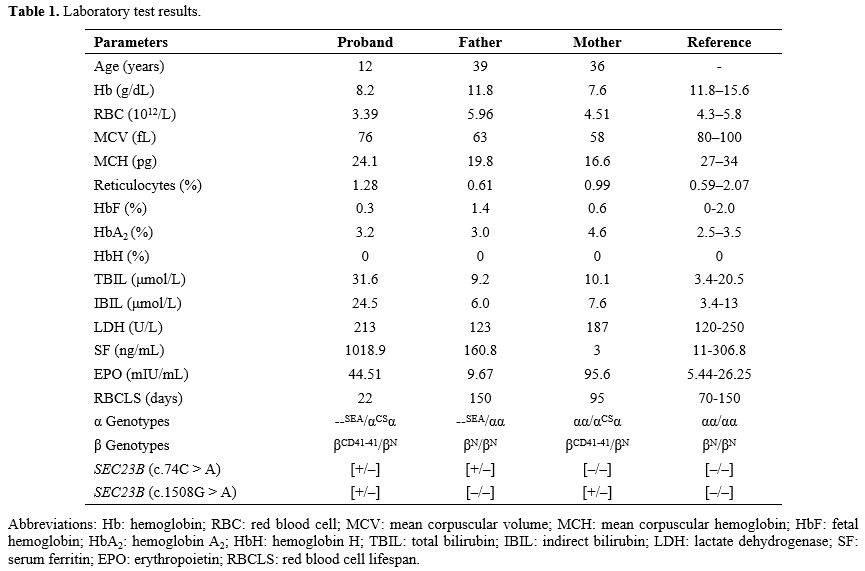 |
Table 1. Laboratory test results. |
HbH disease is the most common form of α-thalassemia syndrome, resulting from compound heterozygosity of α-thalassemia due to a loss of two linked α-globin genes and either a single α-gene deletion or a non-deletional mutation on the other alleles.[3,4] Instability and oxidization ability of the β-chain tetramer produce intracellular precipitates that affect the integrity of the red cell membrane in early erythroid cells, leading to ineffective erythropoiesis and erythroid cell death, which in turn eventually cause acute hemolytic anemia, marked microcytosis, and hypochromia. When combined with β-thalassemia, the symptoms of HbH can be reduced, and the detection of HbH bands may decrease.[5] In the current case, no HbH band was detected by multiple tests due to the patient’s comorbid β-thalassemia, which may have reduced the clinical phenotype of HbH Constant Spring disease. Notably, however, the patient's clinical symptoms were not relieved, and he showed transfusion dependence. In addition, the reticulocyte count was relatively low in multiple blood tests, suggesting the possibility of other congenital hemolytic diseases.
CDA is characterized by ineffective erythropoiesis and dyserythropoiesis in the bone marrow, and CDA II is the most common form. Clinical presentation of patients with CDA II is highly heterogeneous, ranging from symptomless to transfusion-dependent anemia.[6,7] The phenotypic presentation of Hb H disease and CDA II is similar and often presents with anemia, pallor, jaundice, splenomegaly, etc. Amwal et al. reported a patient with non-deletion HbH who had 17% binucleated normoblasts on bone marrow images.[8] Morphology alone is thus not sufficient to diagnose CDA II, and genetic results are needed to verify a diagnosis. In the current proband, two missense mutations in the SEC23B gene (c.74C > A and c.1508G > A) were found by genetic analysis. The bone marrow showed vigorous hematopoiesis and increased erythropoietin levels, consistent with ineffective hematopoiesis. In addition, the proband had a positive acid hemolysis test, suggesting a diagnosis of CDA II. To date, more than 60 different pathogenic mutations have been identified worldwide, and there appears to be a correlation between type and phenotype.[6,7] Previous studies reported two patients with the c.74C > A mutation, both of whom presented with mild anemia with Hb levels of 10.8 g/dL and 9.5 g/dL, respectively.[9,10] In addition, Lolascon et al.[11] reported a patient with a c.1508G > A heterozygous mutation, with slight clinical manifestations and an Hb level of 13.2 g/dL. CDA is also reported to be inherited with hemolytic anemias, modifying the clinical phenotype.[12] The main clinical manifestation in the proband was transfusion dependence, and he started monthly blood transfusions at 3 months old, gradually increasing to twice a month after the age of 10. Symptoms may thus worsen in patients with co-inherited HbH disease and CDA II.
In summary, we report the case of a patient in whom CDA II was initially overlooked due to comorbid HbH disease. This highlights the need to pay attention to subtle differences and overlaps in the clinical phenotypes and laboratory findings in patients with thalassemia and CDAs before making a final diagnosis.