Sickle cell disease (SCD) is one of the most frequently reported inherited diseases worldwide, affecting approximately 300,000 newborns yearly.[3] Saudi Arabia is reported to have a high prevalence of sickle cell trait, ranging from 2% to 27%, with up to 1.4% having SCD in some areas.[6-10] Allogeneic hematopoietic stem cell transplantation (HSCT) remains the only curative option for SCD, and HLA-identical, sibling donor transplant is the standard of care with excellent overall survivals.[2,11-14] However, the procedure is not without risks that include severe chronic graft-versus-host-disease (GVHD), graft rejection, onset of transplant-related complications, and mortality.[2,11]
The Pediatric Stem Cell Transplantation unit at King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia (KFSH&RC-R) is the major tertiary care referral center for children with SCD indicated for HSCT with a suitable donor. In this study, we present a pediatric cohort of patients (age at transplant ≤ 14 years) who underwent HSCT for SCD from a fully matched-related donor (MRD).
Material and Methods
We report a single-center, retrospective review of pediatric patients (aged ≤ 14 years) who underwent HSCT from MRD for SCD from January 2006 to December 2020. Written informed consent was taken from the patient at the time of transplantation that explained to the patient/family the risks and benefits involved with the procedure; in addition, it was explained to the family that data is being collected anonymously for all patients undergoing transplant that will be used for HSCT outcomes research. The study was approved by the institutional review board as a retrospective chart review study with a waiver of written/verbal consent (IRB Approval No. 2221219).Study data was collected from the institutional medical charts using the case report form, which was divided into three sections: 1) Demographic data, transplant indication, donor-type, conditioning regimen, graft-versus-host disease (GVHD) prophylaxis, and cell dose (CD34 x 106/kg and TNC x 108), 2) Transplant-related outcomes data included engraftment data (neutrophil and platelet recovery) and chimerism data, 3) Transplant-related complications data included graft-versus-host-disease (GVHD), veno-occlusive disease (VOD), posterior reversible encephalopathy syndrome (PRES) and infectious toxicity.
Study endpoints and definitions. The primary endpoint of the study was 10-year overall survival (OS), measured from the date of stem cell infusion to the last contact, and death due to any cause was considered as an event. In contrast, events for event-free survival (EFS) were graft failure and death. Secondary endpoints of the study were stem cell dose, neutrophil recovery defined as the first of three consecutive days with a neutrophil count of ≥0.5×109/L, platelet recovery defined as the first of three consecutive days with platelets count of >20×109/L, sustained without transfusion for a minimum of seven-days, chimerism assessment was done using short tandem repeats. Full donor chimerism was defined as donor content of ≥95%, and mixed donor chimerism was defined as donor content of <95% of both myeloid and lymphoid cells.[15] Graft rejection was considered in symptomatic patients who showed evidence of recurrence of disease with changes in Hb level and Hb electrophoresis results, which were further confirmed by a reduction of donor cells (lymphoid /myeloid cells). Post-transplant infectious and non-infectious complications were also measured in this study.
Pre-Transplant evaluation and clearance. All patients had a pre-transplantation evaluation, including hemoglobin electrophoresis, molecular studies, and MRI evaluations. Prior to each patient's HSCT, a multidisciplinary team meeting was conducted with a validated medical decision.
Transplant conditioning regimen. All patients were conditioned using myeloablative regimen that included Busulfan (BU) IV dose 16mg/kg divided over 4 days Day -5, - 2 and cyclophosphamide (CY) dose 200 mg /kg over 4 days Day -10, -7), after 2007 subsequent patients 88% of the patients had additional ATLG- GRAFALON (10 mg/kg/day×4 days Day -5, -2). GVHD prophylaxis consisted of a short MTX dose given on days 1, 3, and 6. Cyclosporin A (CSA) was started on day -3 and continued for 6- 12 months post-HSCT. Levetiracetam (Keppra) was administered in all patients as prophylaxis to mitigate the risk of seizures in all patients for 6-12 months. Graft monitoring was done by molecular PCR.
Statistical methods. Quality assurance measures were applied to ensure data completeness and accuracy. Baseline clinical characteristics and demographic data were described using frequencies and percentages for categorical values, while continuous data were described as non-parametric tests. Descriptive statistics were used to report the incidence of infectious and non-infectious toxicities. Kaplan-Meier survival analysis was used to estimate the 5-year OS and EFS. The Breslow (Generalized Wilcoxon) test was utilized to test the significance of differences between the survival times between groups, and the p-value of <0.05 was considered statistically significant in this study. All data was analyzed using the IBM SPSS Statistics for Windows, version 20.0 (IBM Corporation, Armonk, N.Y., USA).
Results
In total, 129 children underwent allogeneic MRD-HSCT between January 2006 and December 2020. There were 62 males (48%) and 67 females (52%). The median age at transplantation was 9.1 years (range: 1.5 to 13.9 years). Disease phenotype, as determined by Hemoglobin electrophoresis, showed Hemoglobin SS in 123 (95%) patients and Hemoglobin Sβ in 6 (5%); confirmatory testing by molecular PCR was achieved in 109 (89%) patients. The primary transplant indication was cerebral vasculopathy in 57% (74), followed by vascular occlusive disease (VOC) in 36% (47), recurrent acute coronary syndrome (ACS) in 4% (5), and osteonecrosis/avascular necrosis (AVN) in 2% (3), respectively (Table 1). Variable compliance (60%) to hydroxyurea (HU) was observed in the cohort prior to transplantation. Regular monthly blood transfusion with iron chelation therapy was observed in 52% (66) of the patients with cerebral vasculopathy.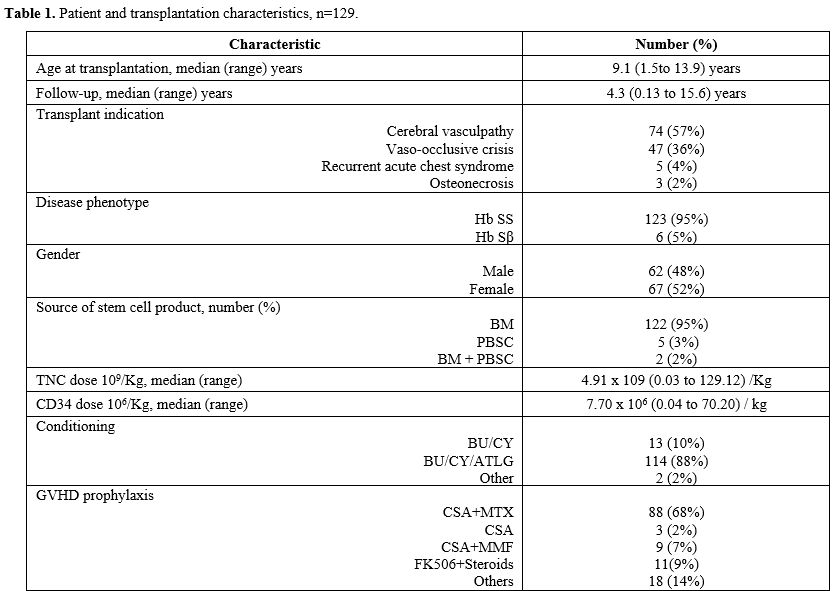 |
|
All donors were HLA-identical siblings in 82% (106) cases, followed by parents in 18% (23) cases. The sickle cell trait was observed at 67% (86), and the remaining demonstrated normal hemoglobin electrophoresis patterns. Bone marrow (BM) was the main source of stem cells in 95% (122), followed by peripheral blood stem cells (PBSC) in 4% (5), and BM + PBSC in 1% (2). Mean TNC and CD34 doses of 4.91 x 10^9/kg and 7.7 x 10^6/kg were observed, respectively (Table 1).
All patients were engrafted, with a median time to neutrophil recovery of 14 days (range, 9 to 29 days) and platelet recovery of 24 days (range, 14 to 100 days). Initial chimerism assessment on Day 100 revealed full chimerism in 70% (91) of patients and mixed chimerism in 28% (36); primary graft failure was observed in 2% (2) of patients. Among the patients who were alive at the last follow-up, 70% (85) of patients continued to maintain full chimerism, and mixed chimerism in 27% (33) and 2% (2) had secondary graft failure. Amongst patients with full to mixed chimerism, a minimum of 33% donor cell content was observed to maintain stable hemoglobin electrophoresis with no evidence of disease. Two patients with primary graft failure were symptomatic and on HU. One patient with secondary graft failure did not require any support, and another patient is responding to HU.
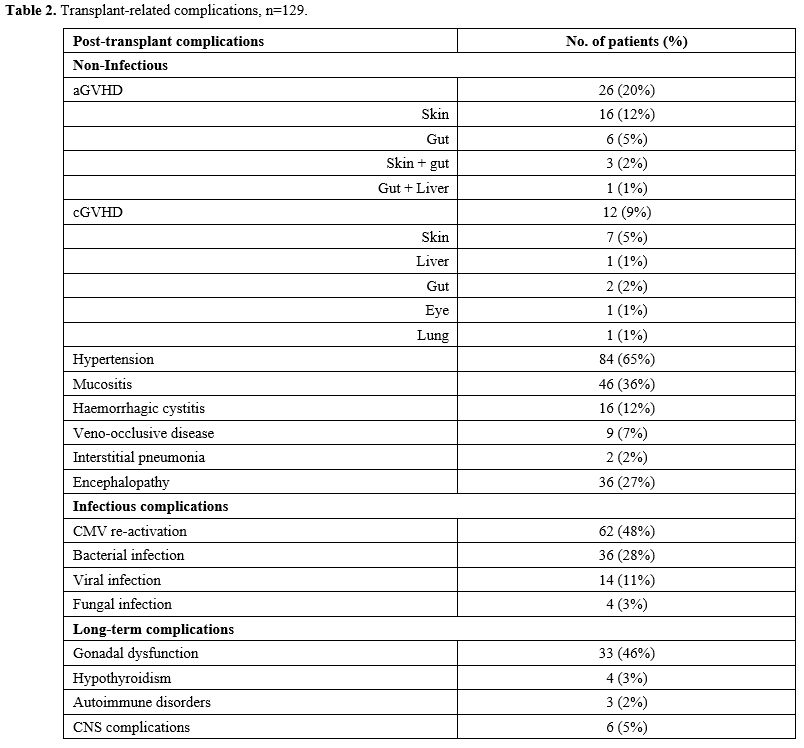
The cumulative incidence of acute GVHD was 20% (26). Grade I-II, aGVHD was seen in 17% (22) and grade III-IV in 3% (4) patients. Skin was the major organ involved in 12% (16) of patients, the gut in 5% (6), and multiple organs in 3% (4). Chronic (c) GVHD was observed in 9% (12) patients, and 4 out of 12 patients developed extensive cGVHD with multi-organ involvement (Table 2).
 |
|
Age at transplant and donor gender disparity were not found to be associated with the incidence of aGVHD (P=0.693 and P=0.547, respectively), although female donor to male recipient was associated with a higher incidence of aGVHD (8 of 30, 27%). However, the difference was not statistically significant (P = 0.310). Similarly, age at transplant and donor gender disparity were not associated with a higher incidence of cGVHD (P=0.273 and P=0.313, respectively). However, having a mother as a donor was associated with an increased risk for severe aGVHD and cGVHD, which was statistically significant (P=0.001 and P=0.025, respectively).
Early non-infectious complications included hypertension in 65% (84), mucositis in 36% (46), encephalopathy in 27% (seizures, 16 (12 %); headache 15 (11%); PRES, 7 (5%), hemorrhagic cystitis in 12% (16), VOD in 7% (9), interstitial pneumonia 2% (2). Infectious complications included CMV re-activation in 48% (62), bacterial infections in 28% (36), viral infections in 11% (14), and fungal infections in 3% (4) (Table 2). Our center adopted the practice of exchange transfusion prior to transplant in 2019 (May), and this was not associated with reducing the risk of PRES (P=0.307).
At a median follow-up of 4.36 (range: 0.13 to 15.5) years, 122 (95%) of patients were alive, and 7 (5%) died. Five-year OS and EFS of the cohort were 94.3% and 91.2%, respectively. Overall and event-free survival was significantly better in patients receiving BU/CY/ATLG when compared to BU/CY (OS: 97.4%±1.5%, 3 of 114 events vs. 76.2%±12.1%, 3 of 13 events, P=0.003 and EFS: 94.7%±2.1%, 6 of 114 events vs. 76.2%±12.1%, 3 of 13 events, P=0.019), respectively (Figure 1 and 2).
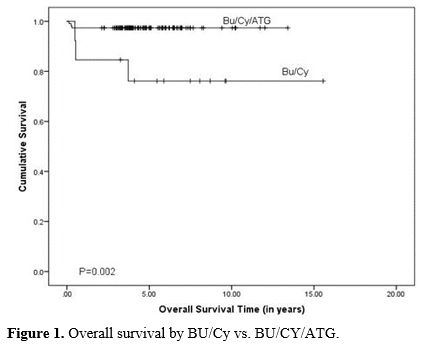 |
Figure 1. Overall survival by BU/Cy vs. BU/CY/ATG. |
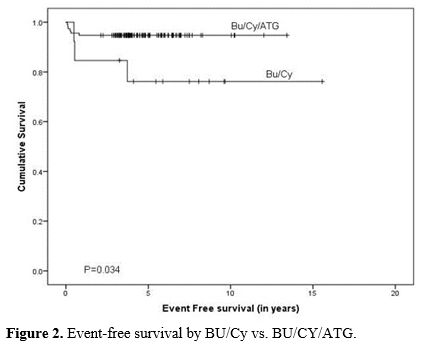 |
Figure 2. Event-free survival by BU/Cy vs. BU/CY/ATG. |
A statistically significant difference in OS was not observed for recipient gender (female: 94.0%±2.9% vs. male: 94.5%±3.2%, P=0.664), age at HSCT (< 8 years: 97.3%±2.7% and >= 8 years: 91.8%±3.2%, P=0.075) and donor sickle cell trait (SCT) status (SCT: 93.7±2.8% and normal Hgb electrophoresis: 95.3±3.2%, P=0.823). Similarly, EFS statistically significant difference was not observed for gender (female: 91.0%±3.5% vs. male: 91.9%±3.5%, P=0.827), age at HSCT (< 8 years: 92.9%±3.4% vs. >=8 years: 90.4%±3.4%, P=0.544) and donor sickle cell trait (SCT) status (SCT: 95.2±2.3% and normal Hgb electrophoresis: 100%, P=0.151%). Similarly, no significance in OS and EFS was observed when compared by disease phenotype and male patients to the female donor group (P=>0.05).
Among the seven patients who died, the cause of death was severe GVHD in two patients, refractory septic shock in two patients, pulmonary hemorrhage, and multi-organ failure in one patient each. One patient developed chronic lung disease with fibrosis four years after transplant and died.
Long-term complications. At the last follow-up, 55% (Female, 44 and Male, 27) of patients were above the age of 13 and were reviewed for long-term complications. A total of 33 (46%) patients showed long-term complications: 13 patients with ovarian failure on hormonal therapy and 8 patients with short stature needing growth hormone therapy. It was noted that patients who developed short stature and gonadal failure were transplanted at a median age of 9 (range: 8 to 13) years. Four patients were having hypothyroidism on replacement therapy. In terms of Central Nervous System (CNS) complications, four patients continued to have seizures, and two- patients had psychomotor disabilities. A post-HSCT comparison was made between patients who received exchange transfusion and those who did not, and a statistically significant difference was not observed. Three patients had autoimmune disorders (Autoimmune hemolytic anemia (AIHA), neutropenia, and arthropathy) and pulmonary restrictive disease in one patient. None of the patients developed a malignancy (Table 2).
Discussion
We report a cohort of 129 pediatric patients with severe SCD who had HSCT, representing the largest single-center experience from Saudi Arabia. Results from this study confirm and extend findings from similar published studies[12,16-18] with 10-year overall and event-free survival rates of 94.3% and 91.2%, respectively. HSCT is considered the standard of care treatment option in symptomatic children with SCD[13,19,20] when a fully HLA-matched related donor is available.[16,21,22] Earlier intervention has been shown to have an advantage in terms of overall and event-free survival outcomes.[16] At a median age at transplant of 9.1 years, an earlier age was associated with a survival advantage, similar to the outcomes reported by Vermylen et al.[16] We found cerebral vasculopathy (53%) was the major indication for transplantation, followed by recurrent vaso-occlusive crises (40%) when compared to the reported literature.[12,16-18]A multicenter study reported outcomes from a retrospective review of patients post-HSCT for SCD, showing excellent outcomes by adding anti-thymocyte globulins (ATG) to busulfan and cyclophosphamide in terms of GVHD and graft rejection.[12] Moreover, a large multicenter study that included 1000 SCD recipients of HLA match sibling donors mostly received myeloablative conditioning in 87% of the patients with a 5-year OS and EFS of 92.9% and 91.4%, respectively.[2]
Transplantation conditioning in our cohort of patients consisted of myeloablative conditioning with busulfan (BU) and cyclophosphamide (CY), with or without anti-thymocyte globulin (ATG). Our findings confirm the favorable survival outcomes with the addition of ATLG to BU/CY conditioning.[12]
Our results showed that all patients achieved engraftment, with a median time to neutrophil engraftment of 14 days and platelet recovery of 24 days. Chimerism data was routinely collected after HSCT in all patients, and sustained donor cell content was demonstrated at the last follow-up. Graft rejection was observed in 3% of the patients and was the main cause of transplant failure in our cohort. The most common post-HSCT complications observed were hypertension (65%), hemorrhagic cystitis (12%), seizures (12%), veno-occlusive disease (7%), encephalopathy (27%), and infectious complications such as CMV re-activation in 48%, bacterial infection in 28%, and other viral infections in 11% and fungal infections in 3%. These findings were comparable to those of other studies.[12,16-18] However, CNS sequelae, especially PRES, were less common in our patients, with a 5% incidence, although it was reported to be higher (10% to 32%) in a similar study.[23] Noteworthy, we have observed an increased risk of severe aGVHD and cGVHD when donors were the mothers.
Long-term complications were reviewed in a subset of patients aged 13 years or older. Among those patients, 46% experienced long-term complications, including ovarian failure requiring hormonal therapy, which is comparable to a published report by Dedeken et al.[24] Other complications included short stature, seizures and psychomotor disability, autoimmune disorders, and pulmonary restrictive disease. None of the patients developed a secondary malignancy post-HSCT.
Furthermore, it was noted that patients who developed short stature and gonadal failure had their transplantation at an older age of >9 years, and studies had shown that the gonads were significantly affected after myeloablative conditioning in patients who had HSCT and post-pubertal BU-based conditioning.[21,25,26] Nevertheless, our results show that pre-pubertal patients can be affected, and an earlier age of transplantation is preferable. Our findings advocate the utilization of gonadal cryopreservation to preserve fertility in eligible candidates.[27,28]
The study's findings are consistent with previous reports on the efficacy of HSCT in treating severe SCD from fully matched related donors. However, the study also highlights the importance of considering the potential long-term complications associated with HSCT, especially in patients who undergo the procedure at a younger age, which was considered the strongest predictor of EFS in fully matched HLA-identical sibling donors with more favorable outcomes.[4,5,29,30]
Reduced toxicity, myeloablative, and reduced intensity conditioning regimen studies have been reported with favorable outcomes in patients with SCD trying to avoid BU and CY complications. However, the increased frequency of mixed chimerism and the potential for rejection require further studies and a longer follow-up.[12,31-33] Therefore, prospective trials for the development of less toxic conditioning regimens and supportive care are warranted.
Conclusions
HSCT for children with sickle cell disease (SCD) from fully matched siblings offers the best outcome using myeloablative conditioning. The use of BU/CY/ATLG has proven to be a successful and effective conditioning regimen for patients with sickle cell disease. The outcome of the addition of ATLG to BU/CY was superior in terms of survival, rejection, and GVHD. This is in line with reports of studies from EBMT. However, significant toxicities were observed secondary to myeloablative regimens, in particular long-term complications, which demands further exploring the use of less toxic regimens. HSCT for pre-school-age patients is highly recommended to achieve the best outcome and reduce long-term complications.References
- Inusa BPD, Hsu LL, Kohli N, Patel A, Ominu-Evbota
K, Anie KA, Atoyebi W. Sickle Cell Disease-Genetics, Pathophysiology,
Clinical Presentation and Treatment. Int J Neonatal Screen.
2019;5(2):20. https://doi.org/10.3390/ijns5020020 PMid:33072979 PMCid:PMC7510211
- Gluckman
E, Cappelli B, Bernaudin F, Labopin M, Volt F, Carreras J, Pinto Simoes
B, Ferster A, Dupont S, de la Fuente J, Dalle JH, Zecca M, Walters MC,
Krishnamurti L, Bhatia M, Leung K, Yanik G, Kurtzberg J, Dhedin N,
Kuentz M, Michel G, Apperley J, Lutz P, Neven B, Bertrand Y, Vannier
JP, Ayas M, Cavazzana M, Matthes-Martin S, Rocha V, Elayoubi H, Kenzey
C, Bader P, Locatelli F, Ruggeri A, Eapen M, Eurocord tPWPotESfB,
Marrow T, the Center for International B, Marrow Transplant R. Sickle
cell disease: an international survey of results of HLA-identical
sibling hematopoietic stem cell transplantation. Blood.
2017;129(11):1548-56. https://doi.org/10.1182/blood-2016-10-745711 PMid:27965196 PMCid:PMC5356458
- Brandow AM, Liem RI. Advances in the diagnosis and treatment of sickle cell disease. J Hematol Oncol. 2022;15(1):20. https://doi.org/10.1186/s13045-022-01237-z PMid:35241123 PMCid:PMC8895633
- Cappelli
B, Volt F, Tozatto-Maio K, Scigliuolo GM, Ferster A, Dupont S, Simoes
BP, Al-Seraihy A, Aljurf MD, Almohareb F, Belendez C, Matthes S, Dhedin
N, Pondarre C, Dalle JH, Bertrand Y, Vannier JP, Kuentz M, Lutz P,
Michel G, Rafii H, Neven B, Zecca M, Bader P, Cavazzana M, Labopin M,
Locatelli F, Magnani A, Ruggeri A, Rocha V, Bernaudin F, de La Fuente
J, Corbacioglu S, Gluckman E, Eurocord tCT, Immunobiology Working P,
the Paediatric Diseases Working Party of the E. Risk factors and
outcomes according to age at transplantation with an HLA-identical
sibling for sickle cell disease. Haematologica. 2019;104(12):e543-e6. https://doi.org/10.3324/haematol.2019.216788 PMid:31018975 PMCid:PMC6959194
- Brazauskas
R, Scigliuolo GM, Wang HL, Cappelli B, Ruggeri A, Fitzhugh CD, Hankins
JS, Kanter J, Meerpohl JJ, Panepinto JA, Rondelli D, Shenoy S, Walters
MC, Wagner JE, Tisdale JF, Gluckman E, Eapen M. Risk score to predict
event-free survival after hematopoietic cell transplant for sickle cell
disease. Blood. 2020;136(5):623-6. https://doi.org/10.1182/blood.2020005687 PMid:32518950 PMCid:PMC7393258
- Jastaniah W. Epidemiology of sickle cell disease in Saudi Arabia. Ann Saudi Med. 2011;31(3):289-93. https://doi.org/10.5144/0256-4947.2011.289 PMid:21623060 PMCid:PMC3119971
- Lehmann H, Maranjian G, Mourant AE. Distribution of sickle-cell hemoglobin in Saudi Arabia. Nature. 1963;198:492-3. https://doi.org/10.1038/198492b0 PMid:13929353
- Al-Qurashi
MM, El-Mouzan MI, Al-Herbish AS, Al-Salloum AA, Al-Omar AA. The
prevalence of sickle cell disease in Saudi children and adolescents. A
community-based survey. Saudi Med J. 2008;29(10):1480-3. https://doi.org/10.4103/0256-4947.55163 PMid:19700891 PMCid:PMC2860399
- Alhamdan
NA, Almazrou YY, Alswaidi FM, Choudhry AJ. Premarital screening for
thalassemia and sickle cell disease in Saudi Arabia. Genet Med.
2007;9(6):372-7. https://doi.org/10.1097/GIM.0b013e318065a9e8 PMid:17575503
- el-Hazmi MA. Clinical and haematological diversity of sickle cell disease in Saudi children. J Trop Pediatr. 1992;38(3):106-12. https://doi.org/10.1093/tropej/38.3.106 PMid:1380566
- Angelucci
E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini
MD, Dalle JH, Di Bartolomeo P, de Heredia CD, Dickerhoff R, Giardini C,
Gluckman E, Hussein AA, Kamani N, Minkov M, Locatelli F, Rocha V,
Sedlacek P, Smiers F, Thuret I, Yaniv I, Cavazzana M, Peters C, Error
EI, Parties EPW. Hematopoietic stem cell transplantation in thalassemia
major and sickle cell disease: indications and management
recommendations from an international expert panel. Haematologica.
2014;99(5):811-20. https://doi.org/10.3324/haematol.2013.099747 PMid:24790059 PMCid:PMC4008115
- Bernaudin
F, Socie G, Kuentz M, Chevret S, Duval M, Bertrand Y, Vannier JP,
Yakouben K, Thuret I, Bordigoni P, Fischer A, Lutz P, Stephan JL,
Dhedin N, Plouvier E, Margueritte G, Bories D, Verlhac S, Esperou H,
Coic L, Vernant JP, Gluckman E, Sfgm TC. Long-term results of related
myeloablative stem-cell transplantation to cure sickle cell disease.
Blood. 2007;110(7):2749-56. https://doi.org/10.1182/blood-2007-03-079665 PMid:17606762
- Kanter
J, Liem RI, Bernaudin F, Bolanos-Meade J, Fitzhugh CD, Hankins JS,
Murad MH, Panepinto JA, Rondelli D, Shenoy S, Wagner J, Walters MC,
Woolford T, Meerpohl JJ, Tisdale J. American Society of Hematology 2021
guidelines for sickle cell disease: stem cell transplantation. Blood
Adv. 2021;5(18):3668-89. https://doi.org/10.1182/bloodadvances.2021004394C PMid:34581773 PMCid:PMC8945587
- Vallee
T, Schmid I, Gloning L, Bacova M, Ahrens J, Feuchtinger T, Klein C,
Gaertner VD, Albert MH. Excellent outcome of stem cell transplantation
for sickle cell disease. Ann Hematol. 2023;102(11):3217-27. https://doi.org/10.1007/s00277-023-05447-4 PMid:37726493 PMCid:PMC10567813
- Miura
S, Ueda K, Minakawa K, Nollet KE, Ikeda K. Prospects and Potential for
Chimerism Analysis after Allogeneic Hematopoietic Stem Cell
Transplantation. Cells. 2024;13(11). https://doi.org/10.3390/cells13110993 PMid:38891125 PMCid:PMC11172215
- Vermylen
C, Cornu G, Ferster A, Brichard B, Ninane J, Ferrant A, Zenebergh A,
Maes P, Dhooge C, Benoit Y, Beguin Y, Dresse MF, Sariban E.
Haematopoietic stem cell transplantation for sickle cell anaemia: the
first 50 patients transplanted in Belgium. Bone Marrow Transplant.
1998;22(1):1-6. https://doi.org/10.1038/sj.bmt.1701291 PMid:9678788
- Walters
MC, Sullivan KM, Bernaudin F, Souillet G, Vannier JP, Johnson FL,
Lenarsky C, Powars D, Bunin N, Ohene-Frempong K, et al. Neurologic
complications after allogeneic marrow transplantation for sickle cell
anemia. Blood. 1995;85(4):879-84. https://doi.org/10.1182/blood.V85.4.879.bloodjournal854879 PMid:7849310
- Panepinto
JA, Walters MC, Carreras J, Marsh J, Bredeson CN, Gale RP, Hale GA,
Horan J, Hows JM, Klein JP, Pasquini R, Roberts I, Sullivan K, Eapen M,
Ferster A, Non-Malignant Marrow Disorders Working Committee CfIB,
Marrow Transplant R. Matched-related donor transplantation for sickle
cell disease: report from the Center for International Blood and
Transplant Research. Br J Haematol. 2007;137(5):479-85. https://doi.org/10.1111/j.1365-2141.2007.06592.x PMid:17459050
- de
la Fuente J, Gluckman E, Makani J, Telfer P, Faulkner L, Corbacioglu S,
Paediatric Diseases Working Party of the European Society for B, Marrow
T. The role of haematopoietic stem cell transplantation for sickle cell
disease in the era of targeted disease-modifying therapies and gene
editing. Lancet Haematol. 2020;7(12):e902-e11. https://doi.org/10.1016/S2352-3026(20)30283-0 PMid:33242447
- Leonard
A, Tisdale JF. Stem cell transplantation in sickle cell disease:
therapeutic potentia l and challenges faced. Expert Rev Hematol.
2018;11(7):547-65. https://doi.org/10.1080/17474086.2018.1486703 PMid:29883237 PMCid:PMC8459571
- Walters
MC, Storb R, Patience M, Leisenring W, Taylor T, Sanders JE, Buchanan
GE, Rogers ZR, Dinndorf P, Davies SC, Roberts IA, Dickerhoff R, Yeager
AM, Hsu L, Kurtzberg J, Ohene-Frempong K, Bunin N, Bernaudin F, Wong
WY, Scott JP, Margolis D, Vichinsky E, Wall DA, Wayne AS, Pegelow C,
Redding-Lallinger R, Wiley J, Klemperer M, Mentzer WC, Smith FO,
Sullivan KM. Impact of bone marrow transplantation for symptomatic
sickle cell disease: an interim report. Multicenter investigation of
bone marrow transplantation for sickle cell disease. Blood.
2000;95(6):1918-24.
- Walters
MC, Patience M, Leisenring W, Eckman JR, Buchanan GR, Rogers ZR,
Olivieri NE, Vichinsky E, Davies SC, Mentzer WC, Powars D, Scott JP,
Bernaudin F, Ohene-Frempong K, Darbyshire PJ, Wayne A, Roberts IA,
Dinndorf P, Brandalise S, Sanders JE, Matthews DC, Appelbaum FR, Storb
R, Sullivan KM. Barriers to bone marrow transplantation for sickle cell
anemia. Biol Blood Marrow Transplant. 1996;2(2):100-4.
- Gaziev
J, Marziali S, Paciaroni K, Isgro A, Di Giuliano F, Rossi G, Marziali
M, De Angelis G, Alfieri C, Ribersani M, Andreani M, Palmieri MG,
Placidi F, Romigi A, Izzi F, Floris R, Mercuri NB. Posterior Reversible
Encephalopathy Syndrome after Hematopoietic Cell Transplantation in
Children with Hemoglobinopathies. Biol Blood Marrow Transplant.
2017;23(9):1531-40. https://doi.org/10.1016/j.bbmt.2017.05.033 PMid:28602890
- Dedeken
L, Le PQ, Azzi N, Brachet C, Heijmans C, Huybrechts S, Devalck C, Rozen
L, Ngalula M, Ferster A. Haematopoietic stem cell transplantation for
severe sickle cell disease in childhood: a single centre experience of
50 patients. Br J Haematol. 2014;165(3):402-8. https://doi.org/10.1111/bjh.12737 PMid:24433465
- Dallas
MH, Triplett B, Shook DR, Hartford C, Srinivasan A, Laver J, Ware R,
Leung W. Long-term outcome and evaluation of organ function in
pediatric patients undergoing haploidentical and matched related
hematopoietic cell transplantation for sickle cell disease. Biol Blood
Marrow Transplant. 2013;19(5):820-30. https://doi.org/10.1016/j.bbmt.2013.02.010 PMid:23416852 PMCid:PMC4712646
- Phelan
R, Mann E, Napurski C, DeFor TE, Petryk A, Miller WP, Wagner JE,
Verneris MR, Smith AR. Ovarian function after hematopoietic cell
transplantation: a descriptive study following the use of GnRH agonists
for myeloablative conditioning and observation only for
reduced-intensity conditioning. Bone Marrow Transplant.
2016;51(10):1369-75. https://doi.org/10.1038/bmt.2016.150 PMid:27272448 PMCid:PMC5299542
- Grundy
R, Gosden RG, Hewitt M, Larcher V, Leiper A, Spoudeas HA, Walker D,
Wallace WH. Fertility preservation for children treated for cancer (1):
scientific advances and research dilemmas. Arch Dis Child.
2001;84(4):355-9. https://doi.org/10.1136/adc.84.4.355 PMid:11259242 PMCid:PMC1718722
- Mathiesen
S, Andres-Jensen L, Nielsen MM, Sorensen K, Ifversen M, Jahnukainen K,
Juul A, Muller K. Male Gonadal Function After Pediatric Hematopoietic
Stem Cell Transplantation: A Systematic Review. Transplant Cell Ther.
2022;28(8):503 e1- e15. https://doi.org/10.1016/j.jtct.2022.05.036 PMid:35644480
- Krishnamurti
L, Neuberg DS, Sullivan KM, Kamani NR, Abraham A, Campigotto F, Zhang
W, Dahdoul T, De Castro L, Parikh S, Bakshi N, Haight A, Hassell KL,
Loving R, Rosenthal J, Smith SL, Smith W, Spearman M, Stevenson K, Wu
CJ, Wiedl C, Waller EK, Walters MC. Bone marrow transplantation for
adolescents and young adults with sickle cell disease: Results of a
prospective multicenter pilot study. Am J Hematol. 2019;94(4):446-54. https://doi.org/10.1002/ajh.25401 PMid:30637784 PMCid:PMC6542639
- Shenoy
S, Eapen M, Panepinto JA, Logan BR, Wu J, Abraham A, Brochstein J,
Chaudhury S, Godder K, Haight AE, Kasow KA, Leung K, Andreansky M,
Bhatia M, Dalal J, Haines H, Jaroscak J, Lazarus HM, Levine JE,
Krishnamurti L, Margolis D, Megason GC, Yu LC, Pulsipher MA, Gersten I,
DiFronzo N, Horowitz MM, Walters MC, Kamani N. A trial of unrelated
donor marrow transplantation for children with severe sickle cell
disease. Blood. 2016;128(21):2561-7. https://doi.org/10.1182/blood-2016-05-715870 PMid:27625358 PMCid:PMC5123194
- Bhatia
M, Jin Z, Baker C, Geyer MB, Radhakrishnan K, Morris E, Satwani P,
George D, Garvin J, Del Toro G, Zuckerman W, Lee MT, Licursi M, Hawks
R, Smilow E, Baxter-Lowe LA, Schwartz J, Cairo MS. Reduced toxicity,
myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT
from sibling donors in children with sickle cell disease. Bone Marrow
Transplant. 2014;49(7):913-20. https://doi.org/10.1038/bmt.2014.84 PMid:24797180
- Strocchio
L, Zecca M, Comoli P, Mina T, Giorgiani G, Giraldi E, Vinti L, Merli P,
Regazzi M, Locatelli F. Treosulfan-based conditioning regimen for
allogeneic haematopoietic stem cell transplantation in children with
sickle cell disease. Br J Haematol. 2015;169(5):726-36. https://doi.org/10.1111/bjh.13352 PMid:25818248
- Marzollo A., Calore E., Tumino M., Pillon M., GazzolaM.V., Destro R., Colombatti R., Marson P., Tison T., Colpo A., Mainardi C., Gabelli M., Boaro M.P., Rossin S., Strano A., Quaglia N., Menzato F., Basso G., Sainati L., Messina C.Treosulfan-based conditioning regimen in sibling and alternative donor hematopoietic stem cell transplantation for children with sickle cell disease.Mediterr J Hematol Infect Dis 2017, 9(1): e2017014 https://doi.org/10.4084/mjhid.2017.014 PMid:28293402 PMCid:PMC5333731