Thalidomide is an antiangiogenic, anti-inflammatory, and immunomodulatory drug that has shown promise in increasing fetal hemoglobin levels and reducing transfusion requirements in patients with β-thalassemia.[3] Furthermore, thalidomide has been shown to significantly reduce spleen length and increase platelet levels, indicating its potential utility in managing thrombocytopenia in β-thalassemia patients with hypersplenism.[4] However, there are no reported studies on the role of thalidomide in managing thrombocytopenia secondary to hypersplenism in children with β-thalassemia. Herein, we report three cases of transfusion-dependent β-thalassemia (TDT) patients with thrombocytopenia secondary to hypersplenism who experienced a reduction in spleen length and an increase in platelet levels after treatment with thalidomide.
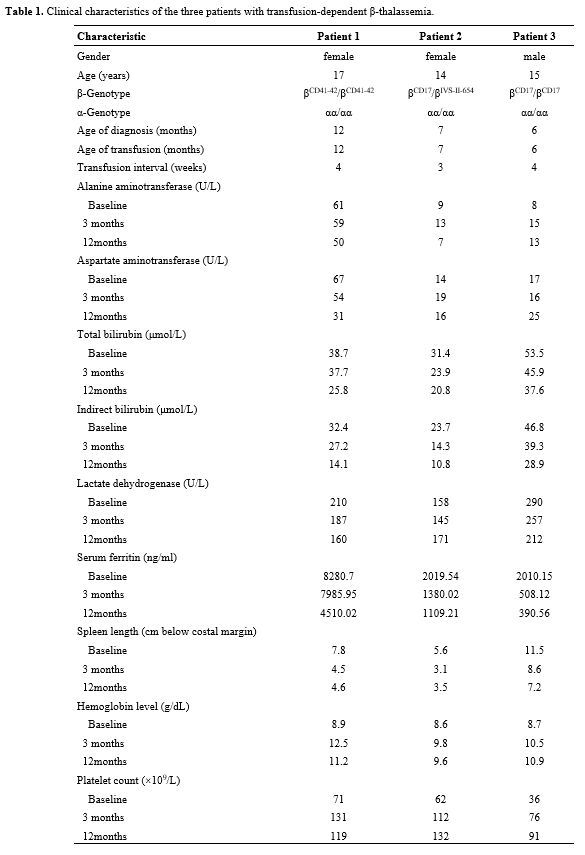
The clinical characteristics of the three patients with TDT are outlined in Table 1. Thalidomide was administered daily at an initial dose of 100 mg/day. Patient 1 showed a significant increase in hemoglobin concentration after one month of treatment and was gradually weaned off transfusions. By three months, hemoglobin increased from 8.9 g/dL to 12.5 g/dL. Spleen length was reduced from 7.8 cm to 4.5 cm, and platelet count normalized, rising from 71×109/L to 131×109/L. The thalidomide dose was then reduced from 100 mg/day to 50 mg/day, and treatment was continued. Beyond hematologic improvements, liver function also showed positive changes. After 12 months, serum ferritin levels declined from 8280.7 ng/mL to 4510.02 ng/mL, indicating reduced iron overload and potential long-term benefits of treatment.
 |
|
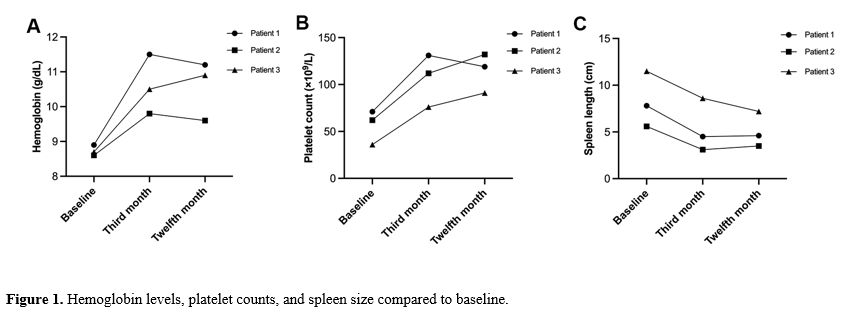
Similarly, patients 2 and 3 experienced significant increases in hemoglobin levels and were weaned off blood transfusions after two months of treatment. At three months, patient 2’s hemoglobin increased from 8.6 g/dL to 9.8 g/dL, while patient 3’s hemoglobin rose from 8.7 g/dL to 10.5 g/dL. Spleen length in patient 2 decreased from 5.6 cm to 3.1 cm, and platelet count increased from 62×109/L to 112×109/L. In patient 3, spleen length shrank from 11.5 cm to 8.6 cm, and platelet count increased from 36×109/L to 76×109/L. Following these improvements, the thalidomide doses for patients 2 and 3 were reduced to 75 mg/day and 50 mg/day, respectively, for maintenance therapy. Both patients also showed low levels of hemolysis markers, including bilirubin and lactate dehydrogenase, alongside significant reductions in iron overload. By 12 months, all three patients exhibited sustained improvements in hemoglobin levels, platelet counts, and spleen size compared to baseline (Figure 1).
 |
|
Massive blood transfusions, along with iron chelation therapy, remain the primary treatment for thalassemia. While early and standardized blood transfusion therapy may help prevent splenomegaly in some patients, hypersplenism can still progress. Hypersplenism due to thalassemia is not uncommon, and in these cases, splenectomy may improve the pancytopenia associated with splenic enlargement. However, the potential side effects of splenectomy, including thromboembolism and postoperative infections, should not be overlooked. The patients in this study experienced a significant reduction in spleen length and a marked increase in platelet count following thalidomide treatment for thrombocytopenia secondary to hypersplenism. No serious adverse events were observed during the study, and thalidomide was well tolerated. Based on these findings, we suggest that thalidomide may have potential as a new therapeutic option for treating thrombocytopenia associated with hypersplenism in patients with β-thalassemia.
The results of our study indicate that thalidomide improved platelet counts in patients with secondary hypersplenism, which was accompanied by a progressive decrease in spleen length throughout the treatment. We propose that this platelet improvement resulted from a reduction in hypersplenism, leading to less platelet sequestration in the spleen. The spleen typically stores around 30% of the body's platelets, and increased sequestration due to splenomegaly is well-recognized as a cause of thrombocytopenia.[5] In patients with β-thalassemia, extramedullary hematopoiesis increases to compensate for anemia, leading to greater production and clearance of abnormal red blood cells (RBCs), contributing to hypersplenism and spleen enlargement.[6] Thalidomide has been shown to increase RBC production and maturation in patients with TDT, while simultaneously reducing erythropoietin levels and reticulocytes.[7] This improved erythropoiesis likely results in more effective RBC production. Beyond its effects on erythropoiesis, thalidomide may also help manage iron overload, improving organ function in these patients, which may indirectly support platelet production.[7-9] Furthermore, thalidomide has been shown to significantly inhibit the secretion of IFN-γ and IL-17 in immune thrombocytopenia while also preventing antiplatelet antibody-mediated platelet destruction by reducing macrophages, which are central to platelet phagocytosis.[10] In our previous studies, we observed a significant reduction in endothelial activation and stress index in patients with TDT following thalidomide treatment.[8] Thus, thalidomide’s effects on hypersplenism and thrombocytopenia in patients with β-thalassemia may also be attributed to its anti-inflammatory, immunomodulatory, and antiangiogenic properties.
In conclusion, thalidomide represents a promising therapeutic option for patients with thrombocytopenia secondary to hypersplenism in β-thalassemia. Our case series indicates that thalidomide effectively improves both splenomegaly and thrombocytopenia associated with hypersplenism in children with TDT. However, further studies are needed to confirm these findings and establish optimal treatment regimens and long-term safety.
Acknowledgments
We would like to thank the participating families for their continuous support and assistance in this study.Ethics Statement
The study protocol was approved by the Medical Ethics Committee of the First People’s Hospital of Zigong. The participating families provided written informed consent.References
- Kattamis A, Kwiatkowski
JL, Aydinok Y. Thalassaemia. Lancet. 2022;399:2310-24. https://doi.org/10.1016/S0140-6736(22)00536-0
PMid:35691301
- Pines
M, Sheth S. Clinical Classification, Screening, and Diagnosis in
Beta-Thalassemia and Hemoglobin E/Beta-Thalassemia. Hematol Oncol Clin
North Am. 2023;37:313-25. https://doi.org/10.1016/j.hoc.2022.12.003
PMid:36907605
- Chen
JM, Zhu WJ, Liu J, Wang GZ, Chen XQ, Tan Y, Xu WW, Qu LW, Li JY, Yang
HJ, Huang L, Cai N, Wang WD, Huang K, Xu JQ, Li GH, He S, Luo TY, Huang
Y, Liu SH, Wu WQ, Lu QY, Zhou MG, Chen SY, Li RL, Hu ML, Huang Y, Wei
JH, Li JM, Chen SJ, Zhou GB. Safety and efficacy of thalidomide in
patients with transfusion-dependent beta-thalassemia: a randomized
clinical trial. Signal Transduct Target Ther. 2021;6:405. https://doi.org/10.1038/s41392-021-00811-0
PMid:34795208 PMCid:PMC8602273
- Chen
Y, Cai N, Lai Y, Xu W, Li J, Huang L, Huang Y, Hu M, Yang H, Chen J.
Thalidomide for the Treatment of Thrombocytopenia and Hypersplenism in
Patients With Cirrhosis or Thalassemia. Front Pharmacol. 2020;11:1137. https://doi.org/10.3389/fphar.2020.01137
PMid:32792958 PMCid:PMC7394185
- Smock
KJ, Perkins SL. Thrombocytopenia: an update. Int J Lab Hematol.
2014;36:269-78. https://doi.org/10.1111/ijlh.12214
PMid:24750673
- Kolnagou
A, Michaelides Y, Kontoghiorghe CN, Kontoghiorghes GJ. The importance
of spleen, spleen iron, and splenectomy for determining total body iron
load, ferrikinetics, and iron toxicity in thalassemia major patients.
Toxicol Mech Methods. 2013;23:34-41. https://doi.org/10.3109/15376516.2012.735278
PMid:23039902
- Yang
K, Liu X, Peng W, Hua F, Li L, Chen K, Zhang J, Luo S, Li W, Ding Y,
Chen J, Xiao J. Effects of Thalidomide on Erythropoiesis and Iron
Homeostasis in Transfusion-Dependent beta-Thalassemia. Mediterr J
Hematol Infect Dis. 2024;16:e2024001. https://doi.org/10.4084/MJHID.2024.001
PMid:38223482 PMCid:PMC10786143
- Chen
J, Kong W, Xiao J, Liu X, Yang K. Effects of Thalidomide on Endothelial
Activation and Stress Index in Children with beta-Thalassemia Major.
Mediterr J Hematol Infect Dis. 2024;16:e2024076. https://doi.org/10.4084/MJHID.2024.076
PMid:39534709 PMCid:PMC11556424
- Che
J, Luo T, Huang L, Lu Q, Yan D, Meng Y, Xie J, Chen W, Chen J, Long L.
Magnetic Resonance Imaging Quantification of the Liver Iron Burden and
Volume Changes Following Treatment With Thalidomide in Patients With
Transfusion-Dependent ss-Thalassemia. Front Pharmacol. 2022;13:810668. https://doi.org/10.3389/fphar.2022.810668
PMid:35250561 PMCid:PMC8894715
- Xu M, Wang X, Xu X, Wei G, Lu W, Luo Q, Li X, Liu Y, Ju W, Li Z, Xu K, Zeng L, Qiao J. Thalidomide prevents antibody-mediated immune thrombocytopenia in mice. Thromb Res. 2019;183:69-75. https://doi.org/10.1016/j.thromres.2019.09.035 PMid:31670229