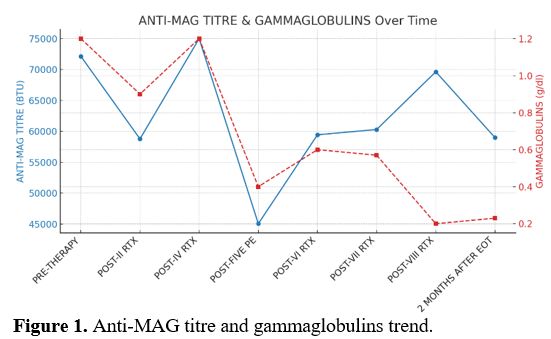
Here we describe a case of a 61 years old male with monoclonal gammopathy IgM kappa and sensorimotor polyneuropathy (PN), unresponsive to high-dose corticosteroids and high-dose intravenous immunoglobulins (IVIGs) in February 2023. Blood count, renal function, calcium levels, kappa/lambda free light chain ratio, and cerebrospinal fluid physical-chemical examination showed no abnormalities. Bence-Jones proteinuria was absent. A total body CT scan with contrast medium revealed hepatomegaly (LD right lobe 18 cm) with steatosis and no other abnormalities. A high titer (72142 BTU) of anti-myelin-associated glycoprotein antibodies (anti-MAG) and a positivity for anti-sulfatide IgM antibodies were reported. Bone marrow needle aspiration and biopsy showed a small lymphoplasmacytic clone (< 5%). Therefore, a diagnosis of monoclonal gammopathy of neurological significance (MGNS) was made. Then, a cycle of four weekly doses of Rituximab (RTX) (375 mg/m² IV) was administered, with initial symptomatic and biochemical response (58773 BTU) followed by a flare-up (75000 BTU) confirmed by worsening of electroneuromyographic evaluation. Subsequently, in May 2023, 5 sessions of plasmapheresis (PE) were performed (45058 BTU). Hence, 4 monthly RTX (375 mg/m² IV), each preceded by 2 PE sessions, were performed with a clinical improvement but no biochemical (59000 BTU) or electromyographic response (Figure 1). After 18 months of follow-up, the patient maintained clinical improvement and biochemical stability.
 |
|
PE was performed using the Fresenius continuous filtration system, exchanging one blood volume per session with human serum albumin in saline.
MGNS is defined as a PN caused by a monoclonal gammopathy without a diagnosis of malignancy (e.g., Waldenström Macroglobulinemia), which means it is a diagnosis of exclusion. They are classified on the basis of the type of monoclonal protein (IgM vs IgG/IgA) and the type of neurological damage (demyelinating, axonal, mixed).[4] Generally, PN is sensory rather than motor, symmetrical, length-dependent, and of slow progression in the context of IgM paraproteinemia, which can be indistinguishable from chronic idiopathic demyelinating polyneuropathy (CIPD). A nerve biopsy should be performed to assess the causality link between MG and the PN; however, it is not routinely performed as it is associated with permanent sensory or motor deficits and pain in the area distal to the biopsy.
The most common form is the IgM-related MGNS, in which anti-MAG, anti-ganglioside, and/or anti-sulphate-3-glucuronyl paragloboside antibodies can be found. It's characterized by demyelinating PN. Rarely, it can cause a syndrome characterized by chronic ataxic neuropathy, ophthalmoplegia, IgM paraprotein, cold agglutinins, and disialosyl antibodies (CANOMAD).[5]
There is no consensus or guidelines on the treatment of MGNS. IVIGs and corticosteroids proved of no or little help, as did alkylating agents and nucleoside analogs.[4-6] It is long known that PE and RTX are useful in the management of MG-associated PN. PE is useful for managing acute symptoms, reducing rapidly the titre of paraprotein, and RTX can lead to a functional benefit of variable duration in 30-50% of patients.[4-7]
A recurrent somatic point mutation of the myeloid differentiation factor 88 (MYD88) gene, leading to an amino acid change from leucine to proline (L265P), is present in 50% of cases of MGNS.[8] This mutation is present in > 90% of cases of Waldenström Macroglobulinemia (WM), too, underlying the presence of biological similarities between the two entities.[4] As in WM, Bruton kinase inhibitors (BTKi) could be a potential therapeutic strategy. In small case series, promising results with ibrutinib and acalabrutinib were reported. In particular, a recent prospective single-arm trial enrolling 7 patients showed that the combination of acalabrutinib and rituximab could be promising (86% hematologic responses, 57% neurological improvement).[9] For these reasons, in the absence of standardized treatment regimens, it is recommended to follow WM guidelines.[4-5,10-12]
MGNS represents a novel entity and an unmet clinical need without a codified therapy. The available therapies have not been particularly effective, as shown in our experience. BTKi could be promising drugs in this setting, as in WM.
References
- Chen LY, Drayson M,
Bunce C, Ramasamy K. Monoclonal
gammopathy of increasing significance: time to screen? Haematologica.
2023 Jun 1;108(6):1476-1486. doi: 10.3324/haematol.2022.281802. https://doi.org/10.3324/haematol.2022.281802
PMid:36373250 PMCid:PMC10233333
- Latov
N, Braun PE, Gross RB, Sherman WH, Penn AS, Chess L. Plasma cell
dyscrasia and peripheral neuropathy: identification of the myelin
antigens that react with human paraproteins. Proc Natl Acad Sci U S A.
1981 Nov;78(11):7139-42. doi: 10.1073/pnas.78.11.7139. https://doi.org/10.1073/pnas.78.11.7139
PMid:6273914 PMCid:PMC349211
- Sabatelli
M, Laurenti L, Luigetti M. Peripheral Nervous System Involvement in
Lymphoproliferative Disorders. Mediterr J Hematol Infect Dis. 2018 Sep
1;10(1):e2018057. doi: 10.4084/MJHID.2018.057. https://doi.org/10.4084/mjhid.2018.057
PMid:30210750 PMCid:PMC6131106
- Cibeira
MT, Rodríguez-Lobato LG, Alejaldre A, Fernández de Larrea C.
Neurological manifestations of MGUS. Hematology Am Soc Hematol Educ
Program. 2024 Dec 6;2024(1):499-504. doi:
10.1182/hematology.2024000665. https://doi.org/10.1182/hematology.2024000665
PMid:39644073 PMCid:PMC11665721
- Castillo
JJ, Callander NS, Baljevic M, Sborov DW, Kumar S. The evaluation and
management of monoclonal gammopathy of renal significance and
monoclonal gammopathy of neurological significance. Am J Hematol. 2021
Jul 1;96(7):846-853. doi: 10.1002/ajh.26155 https://doi.org/10.1002/ajh.26155
PMid:33709474 PMCid:PMC8252623
- Nobile-Orazio
E, Bianco M, Nozza A. Advances in the Treatment of Paraproteinemic
Neuropathy. Curr Treat Options Neurol. 2017 Oct 16;19(12):43. doi:
10.1007/s11940-017-0479-9. PMID: 29034435.
https://link.springer.com/article/10.1007/s11940-017-0479-9 https://doi.org/10.1007/s11940-017-0479-9
PMid:29034435
- Dyck
PJ, Low PA, Windebank AJ, Jaradeh SS, Gosselin S, Bourque P, Smith BE,
Kratz KM, Karnes JL, Evans BA, et al. Plasma exchange in polyneuropathy
associated with monoclonal gammopathy of undetermined significance. N
Engl J Med. 1991 Nov 21;325(21):1482-6. doi:
10.1056/NEJM199111213252105. https://doi.org/10.1056/NEJM199111213252105
PMid:1658648
- Vos
JM, Notermans NC, D'Sa S, Lunn MP, van der Pol WL, Kraan W, Reilly MM,
Chalker J, Gupta R, Kersten MJ, Pals ST, Minnema MC. High prevalence of
the MYD88 L265P mutation in IgM anti-MAG paraprotein-associated
peripheral neuropathy. J Neurol Neurosurg Psychiatry. 2018
Sep;89(9):1007-1009. doi: 10.1136/jnnp-2017-316689 https://doi.org/10.1136/jnnp-2017-316689
PMid:29018161
- Shayna
R Sarosiek, Andrew R. Branagan, Christopher Doughty, Catherine A.
Flynn, Megan Little, Katherine Stockman, Timothy P. White, Kirsten
Meid, Steven P Treon, Jorge J. Castillo, Prospective Study of
Acalabrutinib with Rituximab in Patients with Symptomatic Anti-MAG
Mediated IgM Peripheral Neuropathy, Blood, Volume 142, Supplement 1,
2023, Page 213, https://doi.org/10.1182/blood-2023-185113
- Bibas
M, Sarosiek S, Castillo JJ. Waldenström Macroglobulinemia - A
State-of-the-Art Review: Part 1: Epidemiology, Pathogenesis,
Clinicopathologic Characteristics, Differential Diagnosis, Risk
Stratification, and Clinical Problems. Mediterr J Hematol Infect Dis.
2024 Jul 1;16(1):e2024061. doi: 10.4084/MJHID.2024.061. https://doi.org/10.4084/MJHID.2024.061
PMid:38984103 PMCid:PMC11232678
- Kastritis
E, Leblond V, Dimopoulos MA, Kimby E, Staber P, Kersten MJ, Tedeschi A,
Buske C. Waldenström's macroglobulinaemia: ESMO Clinical Practice
Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019 May
1;30(5):860-862. doi: 10.1093/annonc/mdy466. Erratum for: Ann Oncol.
2018 Oct 1;29(Suppl 4):iv41-iv50. doi: 10.1093/annonc/mdy146. https://doi.org/10.1093/annonc/mdy146
PMid:29982402
- Heyman BM, Opat SS, Wahlin BE, Dimopoulos MC, Castillo JJ, Tedeschi A, Tam CS, Buske C, Owen RG, Leblond V, Trotman J, Barnes G, Chan WY, Schneider J, Allewelt H, Cohen A, Matous JV. Peripheral neuropathy in the phase 3 ASPEN study of Bruton tyrosine kinase inhibitors for Waldenström macroglobulinemia. Blood Adv. 2025 Feb 25;9(4):722-728. doi: 10.1182/bloodadvances.2024014115. https://doi.org/10.1182/bloodadvances.2024014115 PMid:39626287 PMCid:PMC11869863