Non-deletional Hemoglobin H (Hb H) disease arises from the coexistence of a⁰-thalassemia with a point mutation or a small insertion/deletion affecting either HBA1 or HBA2 on the other chromosome 16. In Thailand, the most common non-deletional mutations are Hemoglobin Constant Spring (Hb CS) and Hemoglobin Pakse (Hb Pakse). Both involve alterations in the stop codon of the a2-globin gene, resulting in the addition of 31 extra amino acids at the C-terminus of the α-globin chain.[4] Specifically, Hb CS results from a TAA→CAA mutation, while Hb Pakse arises from a TAA→TAT mutation. Another non-deletional mutation, Hemoglobin Pak Num Po (Hb PNP) (HBA1:c.396_397insT), is an α⁺-α-α-thalassemia mutation characterized by the insertion of a thymine (+T) nucleotide at codons 131/132 of the α1-globin gene.[5]
The clinical spectrum of Hb H disease varies widely, ranging from non-transfusion-dependent thalassemia (NTDT) to transfusion-dependent thalassemia (TDT), and, in rare cases, to hydrops fetalis (“Hb H hydrops”). Clinical severity may also be influenced by the co-inheritance of β-globin gene mutations, resulting in Hb H/β-thalassemia. Notably, non-deletional Hb H disease is generally associated with more severe clinical manifestations than deletional variants.[1,6,7]
Although several studies have reported the genetic diversity and clinical manifestations of Hb H disease in Thailand, data from the northeastern region remain limited. This study aims to describe the clinical spectrum, hematological findings, transfusion requirements, and genetic heterogeneity of Hb H disease in northeastern Thailand.
Methods
A retrospective cross-sectional study was conducted by reviewing medical records of patients aged 0–18 years with Hemoglobin H (Hb H) disease who received follow-up care at the Pediatric Hemato-Oncology Clinic, Srinagarind Hospital, Faculty of Medicine, Khon Kaen University, the sole tertiary referral center in northeastern Thailand. The study period spanned January 2010 to November 2019. Patients with incomplete hematological data or unavailable molecular DNA analysis were excluded. DNA analysis for a-thalassemia using multiplex PCR was not performed in all cases due to parental financial constraints.Data collected included a-thalassemia genotypes, clinical characteristics, hematological parameters, and transfusion history. Clinical data comprised sex, age at diagnosis, presenting symptoms, neonatal jaundice, anthropometric measurements, facial changes, and hepatosplenomegaly. Disease-related complications reviewed included gallstones (detected by routine ultrasonography in patients >10 years), growth failure, and delayed puberty. Growth failure was defined as a height velocity below the expected range for age and sex, or a decrease of >2 percentile lines on standardized Thai growth charts.[8] Delayed puberty was defined as the absence of secondary sexual characteristics after 13 years in girls and 13.5 years in boys.
Transfusion-related data included age at first transfusion, history of transfusions, and transfusion frequency. Patients were categorized as: (1) never transfused; (2) occasional transfusions (<6 times/year) for transient severe anemia triggered by physiological stress; and (3) regular transfusions (8–12 times/year) for clinical indications such as growth failure, skeletal deformities, or progressive splenomegaly.[10] Transfusion-related complications, including autoimmune hemolytic anemia and alloimmunization, were recorded.
Iron overload was assessed using serum ferritin and, when available, MRI with T2* analysis for liver iron concentration. Iron overload was defined as serum ferritin >800 ng/mL for non-transfusion-dependent thalassemia (NTDT) and >1,000 ng/mL for transfusion-dependent thalassemia (TDT). Serum ferritin was used as an indirect marker of total body iron burden.
Hematological parameters included hemoglobin (Hb), hematocrit (Hct), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), red cell distribution width (RDW), reticulocyte count, and red blood cell (RBC) count, recorded during steady-state conditions (absence of recent transfusion or acute illness).[11]
Genotypic analysis for common a-thalassemia mutations was performed using multiplex-gap PCR to detect a⁰-thalassemia deletions — Southeast Asian (--SEA) and Thai (--THAI) — and ⁺a-thalassemia deletions (-a³·⁷ and -a⁴·²). Allele-specific PCR was used to identify non-deletional mutations, including Hb Constant Spring (Hb CS), Hb Pakse, and Hb Pak Num Po.[12,13]
Descriptive statistics summarized the data as mean ± SD, median with interquartile range, or percentage. Normality of continuous variables was assessed using the Shapiro–Wilk test. Group comparisons used the Chi-square or Fisher’s exact test for categorical variables and the Kruskal–Wallis test for non-normally distributed continuous variables. A p-value <0.05 was considered significant. Analyses were performed using SPSS version 17.0.1 and Microsoft Excel.
Results
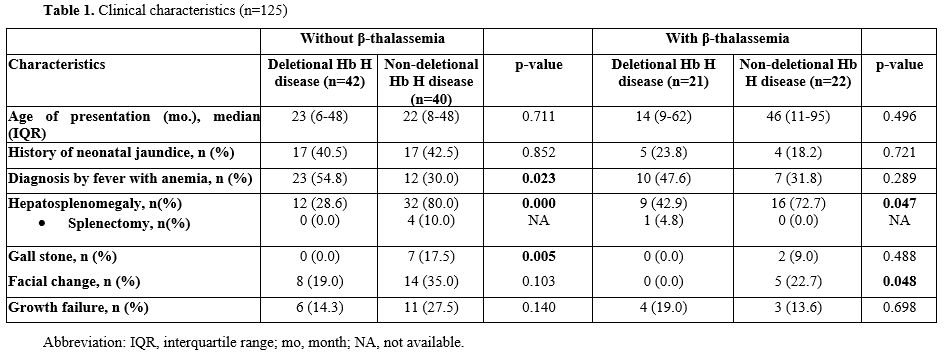
Genotype Distribution. A total of 208 patients with alpha-thalassemia were followed at the Pediatric Hemato-Oncology Clinic, Srinagarind Hospital. Genotyping was successfully performed in 125 patients (60.0%), including 68 males (54.4%) and 57 females (45.6%). Among them, 42 (33.6%) had deletional Hb H disease, 40 (32.0%) had non-deletional Hb H, 21 (16.8%) had deletional Hb H with β-thalassemia, and 22 (17.6%) had non-deletional Hb H with β-thalassemia. The most common a⁰-thalassemia mutation was the Southeast Asian (--SEA) deletion (97.6%), followed by the Thai (--THAI) deletion (2.4%). Among ⁺a-thalassemia mutations, -a³.⁷ deletion predominated (88.9%), followed by -a⁴.² deletion (11.1%). Non-deletional alpha-globin mutations were mainly Hb Constant Spring (Hb CS, 83.9%), Hb Pakse (11.2%), and Hb Pak Nam Po (4.8%). Co-inherited β-globin mutations included Hb E (95.3%), Hb Hope (2.3%), and β-codon 17 combined with Hb E (2.3%).Clinical Characteristics. Clinical features are summarized in Table 1.
 |
|
No significant differences were observed in median age at presentation or history of neonatal jaundice among the four groups. Anemia following febrile illness was more frequent in deletional Hb H disease than non-deletional Hb H, both without and with β-thalassemia [54.8% vs. 30.0% (p=0.023), 47.6% vs. 31.8% (p=0.289)]. Hepatosplenomegaly was more common in non-deletional Hb H patients. Splenectomy was performed in 4 (10.0%) non-deletional Hb H patients without β-thalassemia due to hypersplenism and 1 (4.8%) deletional Hb H patient with β-thalassemia for immune thrombocytopenia; no thrombotic complications occurred. Gallstones were seen only in non-deletional Hb H patients [7 (17.5%) without β-thalassemia, p=0.005; 2 (9.0%) with β-thalassemia, p=0.488]. Facial bone changes were more frequent in non-deletional Hb H disease, whereas growth failure was similar across groups. Hydrops fetalis and leg ulcers were not observed.
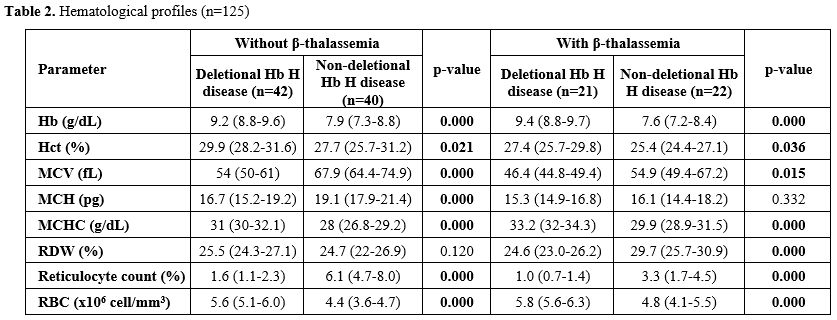
Hematological Profiles (Table 2). Non-deletional Hb H disease exhibited lower hemoglobin (Hb), hematocrit (Hct), mean corpuscular hemoglobin concentration (MCHC), and red blood cell count, but higher mean corpuscular volume (MCV) and reticulocyte counts compared to deletional Hb H disease, regardless of β-thalassemia.
 |
|
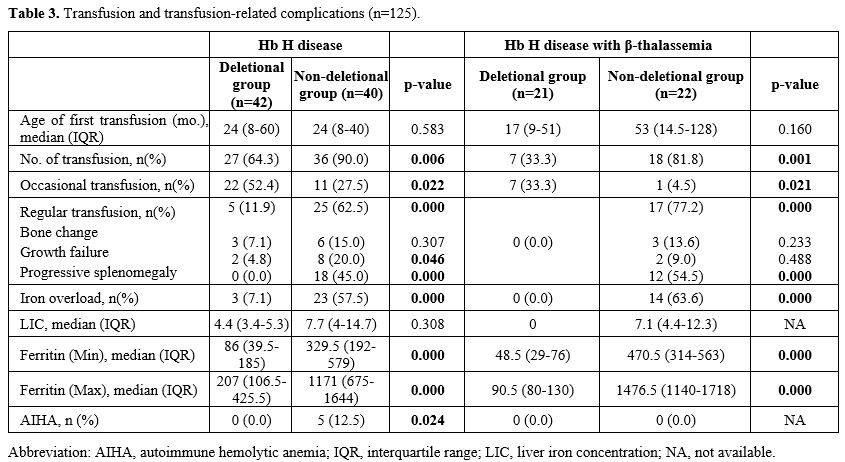
Transfusion Requirements (Table 3). Although the median age at first transfusion was similar, more patients in the non-deletional Hb H group required transfusions. Deletional Hb H patients received occasional transfusions [7 (33%) vs. 1 (4.5%), p=0.021; 22 (52.4%) vs. 11 (27.5%), p=0.022], whereas non-deletional Hb H patients more often required regular transfusions for bone changes, growth failure, or splenomegaly.
Iron Overload and AIHA (Table 3). Non-deletional Hb H patients had higher liver iron concentration and serum ferritin levels, which correlated with transfusion frequency. No iron overload was observed in deletional Hb H with β-thalassemia. Autoimmune hemolytic anemia occurred in 5 (12.5%) non-deletional Hb H patients without β-thalassemia.
 |
|
Discussion
Hemoglobin H (Hb H) disease poses a significant healthcare challenge in Southeast Asia, particularly in Thailand. Hockham et al.[14] projected that by 2020, the burden of Hb H disease would be highest in the northeast region, encompassing both deletional and non-deletional forms. The increasing prevalence of Hb H disease can be attributed to the lack of comprehensive national prenatal and postnatal screening programs, leading to underdiagnosis and suboptimal disease management that exacerbate the regional healthcare burden.This study provides valuable insights into the clinical spectrum and genotypic heterogeneity of alpha-thalassemia in pediatric patients from Northeast Thailand. Consistent with findings from other regions, the most prevalent α⁰-thalassemia mutation was the Southeast Asian (--SEA) deletion (97.6%), whereas the 3.7 kb deletion (-α³·⁷) represented the most common α⁺-α-thalassemia mutation (88.9%).[3,15,16] Non-deletional Hb H disease was mainly caused by Hb Constant Spring (Hb CS) (83.9%), consistent with its reported prevalence of 1–8% in the general Thai population.[17]
Most β-thalassemia trait co-inheritance involved Hb E (95.3%), consistent with its high frequency in Northeast Thailand, approaching 40–50% in some minority populations.[18] The broad range of clinical manifestations observed in HbH disease highlights its genetic and phenotypic complexity. Non-deletional Hb H disease tended to present with more severe clinical features than deletional forms, likely due to the instability of α-globin chains produced by mutated genes, leading to greater red cell destruction, splenomegaly, and increased transfusion requirements.[3,11,15,16] Interestingly, co-inheritance with β-thalassemia appeared to mitigate disease severity, producing milder manifestations.[1,16] However, Songdej et al.[19] reported that Hb H disease co-inherited with Hb E could exhibit more severe anemia, though their findings were limited by incomplete molecular testing.
Patients with deletional Hb H disease generally presented with mild anemia, modest hepatosplenomegaly, and minimal skeletal changes. Growth disturbance remains a significant feature in untreated thalassemia, particularly during puberty. The benefits of regular transfusions for growth and bone development were first described in 1965.[20] In this study, growth disturbance occurred in approximately 20% of patients, with no significant difference between deletional and non-deletional groups. Febrile episodes often preceded the first diagnosis in deletional Hb H disease, suggesting that many patients maintain a near-normal life until infection or physical stress triggers hemolysis requiring transfusion.
A small subset of patients exhibited growth failure, hepatosplenomegaly, or facial bone deformities, necessitating periodic transfusions. Gallstones were more common in non-deletional Hb H disease, particularly among those without β-thalassemia co-inheritance, likely due to increased hemolysis and elevated reticulocyte counts. Conversely, gallstone prevalence was lower in deletional HbH patients and was not strongly influenced by β-globin gene mutations. Although Gilbert syndrome (UGT1A1 polymorphisms) has been associated with gallstone risk,[4] this alone does not explain the observed variation. Splenectomy was rarely performed — mainly for hypersplenism in non-deletional Hb H without β-thalassemia or for immune thrombocytopenia with intracerebral hemorrhage in deletional Hb H with β-thalassemia — with no post-surgical thrombotic complications.
Three patients with Hb H disease due to the Pak Nam Po mutation exhibited more severe manifestations than those with Hb Constant Spring (CS) or Hb Pakse (PS) mutations, consistent with Singha et al.[7] Two were identical twins co-inheriting heterozygous Hb E, while the third was a girl. Symptoms appeared early — two at 6 months and one at 2 months — with marked hepatosplenomegaly, poor growth, and regular red cell transfusion requirements every four weeks. All three developed severe iron overload comparable to transfusion-dependent thalassemia (TDT) patients.
Iron overload in Hb H disease may result from both increased intestinal absorption and transfusion therapy. In this study, iron overload was more frequent among non-deletional HbH patients, consistent with their higher transfusion requirements. These findings underscore the necessity of regular iron monitoring and timely chelation therapy to reduce complications associated with iron overload, particularly in patients with severe non-deletional genotypes such as Pak Nam Po.
This study has several limitations. Its retrospective design limited the depth of data collection and analysis. Molecular testing was not available for all patients, possibly leading to underestimation of true case numbers and genotypic diversity. The incidence of gallstones may also be underestimated, as abdominal ultrasonography was performed only in patients aged 10 years or older. Despite these constraints, the study provides a comprehensive overview of the clinical and genetic spectrum of Hb H disease in northeastern Thai children. The findings highlight the variability of disease expression and the importance of genetic diagnosis, long-term follow-up, and early implementation of screening and management programs to reduce morbidity and improve quality of life in affected individuals.
Conclusions
Non-deletional Hb H disease, with or without β-thalassemia, causes more severe symptoms, often requiring regular transfusions and leading to iron overload similar to TDT. Deletional Hb H disease usually needs only occasional transfusions during growth. Close, regular follow-up is essential for monitoring disease progression and managing complications in both groups.Data Availability Statement
All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.Ethics approval and consent to participate
This study protocol was reviewed and approved by the Khon Kaen University ethics committee. Written informed consent to participate in this study was obtained from participants and from parents/legal guardians for all participants aged under 18. Ethics approval number (HE 681617).References
- Vichinsky E. Complexity of alpha thalassemia:
growing health problem with new approaches to screening, diagnosis, and
therapy. Ann NY Acad Sci 2010;1202:180-7. https://doi.org/10.1111/j.1749-6632.2010.05572.x PMid:20712791
- Fucharoen
S, Winichagoon P, Sirithanaratkul N, Chowthaworn J, Pootrakul P: α- and
β-thalassemia in Thailand. Ann NY Acad Sci 1998;850:412-4. https://doi.org/10.1111/j.1749-6632.1998.tb10507.x PMid:9668570
- Laosombat
V, Viprakasit V, Chotsampancharoen T, Wongchanchailert M, Khodchawan S,
Chinchang W, et al. Clinical features and molecular analysis in Thai
patients with HbH disease. Ann Hematol 2009;88:1185-92. https://doi.org/10.1007/s00277-009-0743-5 PMid:19390853
- Lal
A, Goldrich ML, Haines DA, Azimi M, Singer ST, Vichinsky EP.
Heterogeneity of Hemoglobin H Disease in Childhood. N Engl J Med
2011;364:710-8. https://doi.org/10.1056/NEJMoa1010174 PMid:21345100
- Singha
K, Wiangnon S, Fucharoen G, Jetsrisuparb A, Komwilaisak P, Fucharoen
S.Severe thalassemia syndrome caused by Hemoglobin Pak Num Po AEBart's
disease: A hematological, molecular, and diagnostic aspects. Int J Lab
Hematol. 2020;42(4):e173-e176 https://doi.org/10.1111/ijlh.13224
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica 2013;98(6):833-44. https://doi.org/10.3324/haematol.2012.066845 PMid:23729725 PMCid:PMC3669437
- Chui DHK. Alpha-thalassemia and population health in Southeast Asia. Annals of Human Biology 2005;32:123-130. https://doi.org/10.1080/03014460500075084 PMid:16096207
- National
Growth References for children aged 5-19 years, 2020, Bureau of
Nutrition, Department of Health: Ministry of Public Health.
- Kliegman RM, ST Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM. Nelson Textbook of pediatrics. 21th ed. Elsevier; 2019. https://evolve.elsevier.com/cs/product/9780323846257
- Taher
A, Musallam K, Karimi M, El-Beshlawy A, Belhoul K, Daar S, et al.
Overview on practices in thalassemia intermedia management aiming for
lowering complication rates across a region of endemicity: the OPTIMAL
CARE study. Blood 2010;115(10):1886-92. https://doi.org/10.1182/blood-2009-09-243154 PMid:20032507
- Taher
A, Musallam K, Cappellini MD. Guidelines for the management of non
transfusion dependent thalassemia (NTDT). 3rd ed. Nicoria: Thalassemia
International Federation publishing; 2023.
- Yamsri
S, Sanchaisuriya K, Fucharoen G, et al. Prevention of severe
thalassemia in northeast Thailand: 16 years of experience at a single
university center. Prenat Diagn. 2010;30:540-546 https://doi.org/10.1002/pd.2514 PMid:20509153
- Sae-ung
N, Fucharoen G, Sanchaisuriya K, et al. α0-thalassemia and related
disorders in northeast Thailand: a molecular and hematological
characterization. Acta Haematol. 2007;117:78-82 https://doi.org/10.1159/000096857 PMid:17106191
- Hockham
C, Ekwattanakit S, Bhatt S, Penman BS, Gupta S, Viprakasit V, et al.
Estimating the burden of alpha-thalassemia in Thailand using a
comprehensive prevalence database for Southeast Asia. e-Life research
communication 2019;8:e40580. https://doi.org/10.7554/eLife.40580 PMid:31120421 PMCid:PMC6533055
- Trivaree
C, Boonyawat B, Monsereenusorn C, Rujkiyanont P, Photia A. Clinical and
molecular genetic features of Hb H and AEBart's disease in central Thai
children. Appl Clin Genet 2018;11:23-30. https://doi.org/10.2147/TACG.S161152 PMid:29662324 PMCid:PMC5892615
- Charoenkwan
P, Taweephon R, Sae-Tung R, Thanarattanakorn P, Sanguansermsri T.
Molecular and clinical features of Hb H disease in northern Thailand.
Hemoglobin 2005;29(2):133-140. doi:10.1081/HEM-200058583 https://doi.org/10.1081/HEM-200058583 PMid:15921165
- Laig
M, Pape M, Hundrieser J, Flatz G, Sanguansermsri T, Das BM, et al. The
distribution of the Hb Constant Spring gene in Southeast Asian
populations.Hum Genet.1990;84: 188-190. https://doi.org/10.1007/BF00208939 PMid:2298455
- Tritipsombut
J, Sanchaisuriya K, Phollarp P, Bouakhasith D, Sanchaisuriya P,
Fucharoen G, et al. Micromapping of thalassemia and hemoglobinopathies
in different regions of northeast Thailand and Vientiane, Lao PDR.
Hemoglobin 2012;36(1):47-56. https://doi.org/10.3109/03630269.2011.637149 PMid:22122810
- Duantida
Songdej D, Teawtrakul N, Laoaroon N, Komvilaisak P, Sripornsawan P,
Surapolchai P, et al. Impact of HbE mutation on the clinical severity
of HbH disease: A multicentre study from Thailand. Br J
Haematol. 2025;206(2):703-712. https://doi.org/10.1111/bjh.19869 PMid:39478290
- Saxena A. Growth Retardation in Thalassemia Major Patients. Int J Hum Genet 2003;3(4):237-46. https://doi.org/10.1080/09723757.2003.11885858