Gaucher disease (GD) and Acid Sphingomyelinase
Deficiency (ASMD) are autosomal recessive lysosomal storage disorders (LSDs)
caused by biallelic pathogenic variants in GBA1 and SMPD1,
respectively. The resulting enzymatic defects lead to progressive accumulation
of undegraded sphingolipids within macrophages and parenchymal cells, producing
chronic, multisystemic, and often irreversible organ damage.[1,2] Diagnostic
delays remain common worldwide and contribute to significant morbidity,
impaired quality of life, and increased healthcare burden.[3] Clinical overlap
between GD and ASMD, particularly splenomegaly, hepatomegaly, cytopenias, bone
involvement, and constitutional symptoms, further complicates early
recognition. Dried blood spot (DBS) enzymatic assays provide a practical,
first-line tool for screening for these conditions and allow simultaneous
measurement of glucocerebrosidase (GCase) and acid sphingomyelinase (ASM)
activities.[4,5] When enzymatic results are borderline or discordant with
clinical suspicion, molecular testing is required to confirm diagnosis,
identify carriers, and facilitate cascade testing within families.[5] Multiple
biomarkers, including ferritin, chitotriosidase, and CCL18, are frequently used
to support diagnosis and monitor disease activity; more recently, glucosylsphingosine
(Lyso-GB1) has emerged as the most specific and sensitive marker for GD, with
strong correlation to disease burden and therapeutic response.[6-8] Analogous
biomarkers for ASMD include lysosphingomyelin (Lyso-SM) and its derivative
Lyso-SM-509.[9]
Within this context, we conducted the Ichnos
Project, a multicenter observational initiative designed to evaluate whether a
structured, cross-departmental diagnostic approach could enhance early
detection of GD and ASMD in Sardinia, a genetically and geographically
distinctive region. The project involved major hospitals across the island and
incorporated standardized referral criteria and a unified diagnostic algorithm
(Figure 1).
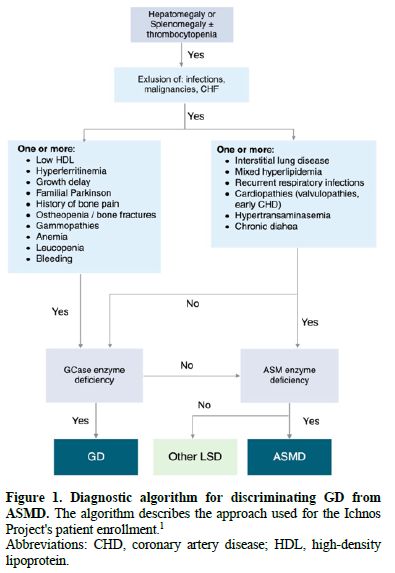
Abbreviations: CHD, coronary artery disease; HDL, high-density lipoprotein.
Between April 2022 and May 2023, 196 individuals presenting with
clinical findings suggestive of LSDs, including unexplained splenomegaly or
hepatomegaly, cytopenias, bone pain, hyperferritinemia, or multisystemic
features, were screened across six major hospitals in Sardinia. Participating
departments included Hematology, Oncology, Internal Medicine, Pediatrics,
Rheumatology, Gastroenterology, Orthopedics, Transfusion Medicine, Pathology,
Hepatology, Neuropsychiatry, and Rare Disease Units. DBS assays measured GCase
and ASM activity. Pathological values were defined as ≤2.5 nmol/h/mL for GCase
activity and ≤1.7 µmol/h/L for ASM. Individuals with decreased enzyme activity
or highly suggestive clinical features underwent molecular analysis for GBA1
or SMPD1. Quantification of Lyso-GB1 or Lyso-SM509 was performed based
on one or more of the following criteria: (i) reduced GCase or ASM enzymatic
activity below the normal range; (ii) detection of at least one pathogenic or
likely pathogenic genetic variant; (iii) familial relationship with a
genetically confirmed case; or (iv) the presence of clinical manifestations
highly suggestive of GD or ASMD, particularly skeletal involvement. Written
informed consent was obtained from all participants (or their legal guardians
for minors) in accordance with institutional and national ethical regulations
and the principles of the Declaration of Helsinki. Statistical analysis was
performed with R Core Team (2021). R: A Language and Environment for
Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria.
Continuous variables were summarized as median and interquartile range (IQR)
due to non-normal distribution, while categorical variables were expressed as
absolute frequencies and percentages.
The median age of enrolled patients was 50.5
years (IQR: 15.7-66), with 41.8% male sex (Table 1). Most referrals originated
from hematology units (45%), followed by Internal Medicine (24%) and Pediatrics
(20%) (Figure 2). As shown in Figure 3, hematologic abnormalities,
hepatosplenic involvement, and skeletal manifestations represented the most
frequent reasons for referral. At enrollment, anemia and thrombocytopenia were
reported in 38 (19.4%) and 29 (14.8%) of screened subjects, respectively.
Moreover, splenomegaly was present in 23.5% of individuals and hepatomegaly in
19.4%. Bone involvement affected 27.6% of patients, while hyperferritinemia was
reported in 31.6%.
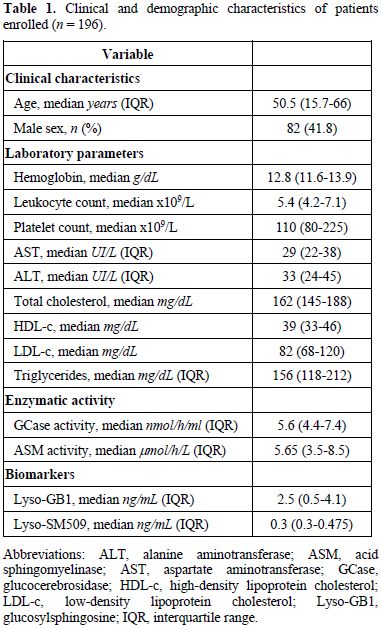
Table 1. Clinical and demographic characteristics of patients enrolled (n = 196)
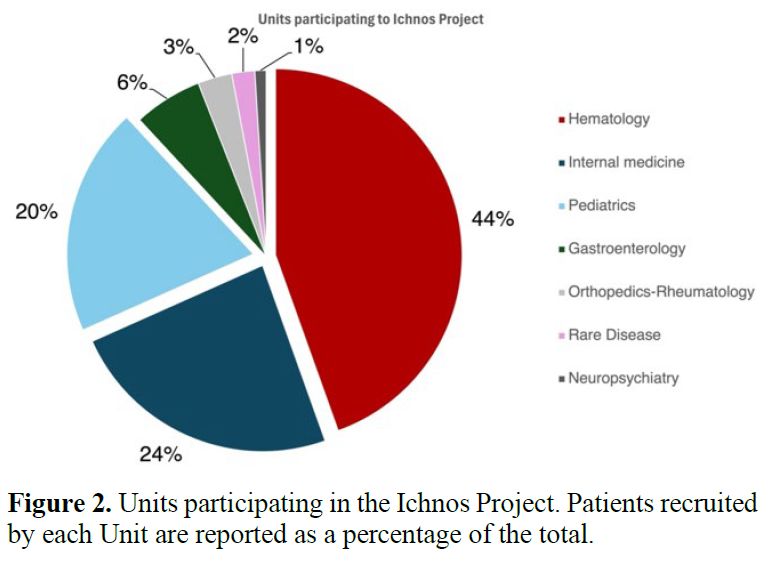 | Figure 2. Units participating in the Ichnos Project. Patients recruited by each Unit are reported as a percentage of the total. |
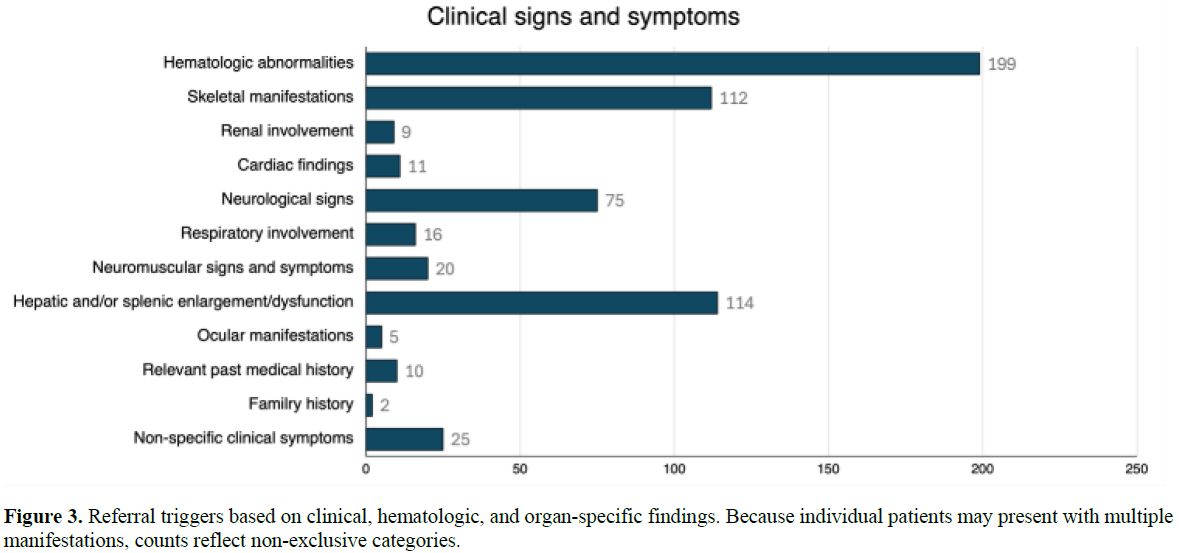 |
Figure 3. Referral triggers based on clinical, hematologic, and organ-specific findings. Because individual patients may present with multiple manifestations, counts reflect non-exclusive categories. |
In the analyzed cohort, the median GCase
activity was 5.6 nmol/h/mL (IQR: 4.4–7.4). Overall, 10 out of 196 patients
(10/196, 5.1%; 95% CI, 2.5–9.3%) presented reduced GCase activity. ASM activity
was measured in 168 out of the 196 enrolled patients. The median ASM activity
for the analyzed cohort was 5.65 µmol/h/L (IQR: 3.5–8.5). The overall
prevalence of pathological ASM activity was 1.2% (2/168; 95% CI, 0.1-4.2%),
with two patients exhibiting pathological values of 1.6 and 1.4 µmol/h/L,
respectively. Among 57 patients evaluated, the median Lyso-GB1 value was 2.5
ng/mL (IQR: 0.5-4.1), with only one patient exhibiting a markedly elevated
Lyso-GB1 concentration of 495 ng/mL. Median Lyso-SM509 values were 0.3 ng/mL
(IQR: 0.3-0.475).
Molecular genetic testing was performed in 57
of the 196 enrolled patients (29.1%). Among these, GBA1 gene analysis
was primarily conducted in 34 individuals who exhibited GCase activity ≤ 3.6
nmol/h/mL. Genetic analysis was also conducted in relatives of patients with
double mutation and in 10 patients who presented particularly suspicious
clinical signs (such as skeletal involvement, splenomegaly, or a family history
of Parkinson’s disease), or unsuitability of the DBS sample for enzymatic
testing. Overall, 2 of 57 patients tested (3.5%) carried biallelic pathogenic
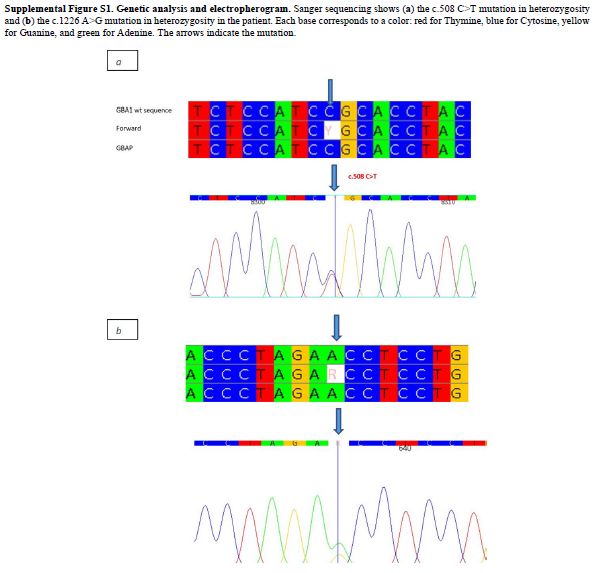
or likely pathogenic GBA1 variants. Patient KE142 harbored two
pathogenic variants:NM_000157.4(GBA1):c.508C>T (p.Arg170Cys), located
in the exon 5; and the NM_000157.4(GBA1):c.1226A>G (p.Asn409Ser)
located in the exon 9 (Supplemental Figure S1). Patient KB874 also carried two
heterozygous variants in exon 10 of GBA1: NM_000157.4(GBA1):c.1483G>C
(p.Ala495Pro) and NM_000157.4(GBA1):c.1497G>C (p.Val499Val). The
GCase enzymatic activity of 3.2 nmol/h/mL, and Lyso-GB1 concentration of 6.7
ng/mL were within reference limits, supporting the interpretation that these
variants are present in a monoallelic configuration. Segregation analysis in
his first-degree relatives (sister and son) confirmed the absence of these
variants, excluding compound heterozygosity. Four additional patients were
heterozygous carriers of GBA1 variants. Two were first-degree relatives
(father and sister) of patient KE142, both carrying the NM_000157.4(GBA1):c.1226A>G,
p.(Asn409Ser) variant. Another unrelated patient carried the same
heterozygous variant. The fourth patient harbored NM_000157.4(GBA1):c.349G>A,
p.(Val117Met), located in exon 4. Overall, one confirmed case of GD was
identified among the 196 screened individuals, corresponding to a diagnostic
rate of 10% (1/10) among subjects with low enzymatic activity and an overall
prevalence of 0.51% (95% CI, 0.01–2.83%). Finally, SMPD1 analysis was
performed in 10 patients with ASM activity ≤ 2.5 µmol/h/L, including four who
simultaneously exhibited GCase activity ≤ 3.6 nmol/h/mL. No pathogenic variants
were identified in the SMPD1 coding region in any of these cases.
This multicenter study represents the first
coordinated effort to implement an integrated diagnostic strategy for GD and
ASMD in Sardinia, a geographically isolated region with a relatively
homogeneous genetic background. The initiative successfully involved multiple
clinical departments, demonstrating the importance of education and
collaboration among physicians. Integrating biochemical screening into clinical
practice can reduce unnecessary investigations, guide appropriate genetic
testing, and shorten the “diagnostic odyssey” experienced by many patients with
GD.[10]
The proportion of patients in our cohort with
organomegaly, hyperferritinemia, or metabolic abnormalities aligns with
previously reported “red flag” profiles for GD in non-specific clinical
settings.[11,12] Our findings also support the central role of DBS enzymatic
assays integrated with a second-tier biomarker. This combined approach enabled
simultaneous assessment of GCase and ASM activity in all enrolled patients,
reduced pre-analytical limitations, and facilitated rapid, structured triage toward
confirmatory biomarker testing and targeted sequencing. In our cohort, 5.1% of
participants exhibited subnormal GCase activity, markedly lower than the 17.3%
reported by Motta et al.[12] in a phenotype-enriched population selected for
splenomegaly and/or thrombocytopenia. Likewise, the overall GD detection rate
of 0.51% observed in the Ichnos Project is substantially lower than the 3.3%
prevalence reported by Motta et al..[12] This discrepancy is expected and most
likely reflects the broader, multispecialty referral pattern of our study,
which inherently lowers the pre-test probability of GD and reduces the overall
diagnostic yield.
We acknowledge the limitations of this study.
First, the relatively small sample size and the identification of only a single
confirmed case do not allow for generalizability of the findings. Second,
enzyme activity was measured on DBS, which, despite its practicality, may
introduce pre-analytical variability and borderline results in leukopenic
patients. Third, not all patients with reduced enzymatic activity underwent
complete genetic confirmation, potentially leading to an underestimation of
disease frequency. However, the Ichnos Project provides an instructive example
of the logistical and epidemiological barriers to screening rare disorders in
small populations. It also demonstrates that even identifying a single affected
individual carries substantial public health significance when the disease is
underdiagnosed and treatable. Future efforts should prioritize continued education
of frontline clinicians, refinement of biomarker thresholds to improve pre-test
probability, and the establishment of centralized diagnostic hubs that can
efficiently integrate biochemical, genetic, and clinical data. This
network-based model could serve as a scalable framework for early detection of
other rare metabolic diseases within a similar healthcare system.
Acknowledgments
The study was conducted thanks to the following specialists who contributed to the study: Alagna Giuliano, Campus Simona, Congia Mauro, Corpino Mara, Corrias Maria Gabriella, Cossu Fausto, Dedoni Maurizio, Di Francesco Alessandra, Dellacà Paola, Furcas Maria,Marras Tatiana, Marzilli Maria Antonietta, Muntone Giuseppina, Murgia Debora, Murtas Andrea, Oggiano Anna Maria, Orrù Maria Marcella, Pilo Federica, Pintor Claretta, Pisano Paola, Porcu Gabriele, Pes Valentina, Pileri Piera Veronica, Sanna Maria Antonietta, Spanu Paolo, Soddu Consolata, Solinas Cristina, Usai Carlo Andrea, Vacca Nadia, Vargiu Carla, Zaru Salvatore.References
- Arslan N, Coker M, Gokcay GF, Kiykim E, Onenli
Mungan HN, Ezgu F. Expert opinion on patient journey, diagnosis and
clinical monitoring in acid sphingomyelinase deficiency in Turkey: a
pediatric metabolic disease specialist's perspective. Front Pediatr.
2023; 11:1113422, doi: 10.3389/fped.2023.1113422. https://doi.org/10.3389/fped.2023.1113422 PMid:37435168 PMCid:PMC10330960
- Giacomarra
M, Colomba P, Francofonte D, Zora M, Caocci G, Diomede D, Giuffrida G,
Fiori L, Montanari C, Sapuppo A, Scortechini AR, Vitturi N, Duro G,
Zizzo C. Gaucher Disease or Acid Sphingomyelinase Deficiency? The
Importance of Differential Diagnosis. J Clin Med. 2024; 13:1487, doi:
10.3390/jcm13051487. https://doi.org/10.3390/jcm13051487 PMid:38592326 PMCid:PMC10932152
- Mistry
PK, Sadan S, Yang R, Yee J, Yang M. Consequences of diagnostic delays
in type 1 Gaucher disease: The need for greater awareness among
Hematologists-Oncologists and an opportunity for early diagnosis and
intervention. Am J Hematol. 2007; 82:697-701, doi: 10.1002/ajh.20908. https://doi.org/10.1002/ajh.20908 PMid:17492645
- Oliva
P, Schwarz M, Mechtler TP, Sansen S, Keutzer J, Prusa AR, Streubel B,
Kasper DC. Importance to include differential diagnostics for acid
sphingomyelinase deficiency (ASMD) in patients suspected to have to
Gaucher disease. Mol Genet Metab. 2023; 139:107563, doi:
10.1016/j.ymgme.2023.107563. https://doi.org/10.1016/j.ymgme.2023.107563 PMid:37086570
- Dardis
A, Michelakakis H, Rozenfeld P, Fumic K, Wagner J, Pavan E, Fuller M,
Revel-Vilk S, Hughes D, Cox T, Aerts J; International Working Group of
Gaucher Disease (IWGGD). Patient centered guidelines for the laboratory
diagnosis of Gaucher disease type 1. Orphanet J Rare Dis. 2022; 17:442,
doi: 10.1186/s13023-022-02573-6. https://doi.org/10.1186/s13023-022-02573-6 PMid:36544230 PMCid:PMC9768924
- Marano
M, Zizzo C, Malaguti MC, Bacchin R, Cavallieri F, De Micco R, Spagnolo
F, Bentivoglio AR, Schirinzi T, Bovenzi R, Ramat S, Erro R, Sorrentino
C, Sucapane P, Pilotto A, Lupini A, Magliozzi A, Di Vico I, Carecchio
M, Bonato G, Cilia R, Colucci F, Tamma F, Caputo E, Mostile G, Arabia
G, Modugno N, Zibetti M, Ceravolo MG, Tambasco N, Cossu G, Valzania F,
Manganotti P, Di Lazzaro V, Zappia M, Fabbrini G, Tinazzi M, Tessitore
A, Duro G, Di Fonzo A. Increased glucosylsphingosine levels and Gaucher
disease in GBA1-associated Parkinson's disease. Parkinsonism Relat
Disord. 2024; 124:107023, doi: 10.1016/j.parkreldis.2024.107023. https://doi.org/10.1016/j.parkreldis.2024.107023 PMid:38843618
- Giuffrida
G, Markovic U, Condorelli A, Calafiore V, Nicolosi D, Calagna M, Grasso
S, Ragusa MTV, Gentile J, Napolitano M. Glucosylsphingosine (Lyso-Gb1)
as a reliable biomarker in Gaucher disease: a narrative review.
Orphanet J Rare Dis. 2023; 18:27, doi: 10.1186/s13023-023-02623-7. https://doi.org/10.1186/s13023-023-02623-7 PMid:36782327 PMCid:PMC9926807
- Revel-Vilk
S, Fuller M, Zimran A. Value of Glucosylsphingosine (Lyso-Gb1) as a
Biomarker in Gaucher Disease: A Systematic Literature Review. Int J Mol
Sci. 2020; 21:7159, doi: 10.3390/ijms21197159. https://doi.org/10.3390/ijms21197159 PMid:32998334 PMCid:PMC7584006
- Kubaski
F, Burlina A, Pereira D, Silva C, Herbst ZM, Trapp FB, Michelin-Tirelli
K, Lopes FF, Burin MG, Brusius-Facchin AC, Netto ABO, Poletto E,
Bernardes TM, Carvalho GS, Sorte NB, Ferreira FN, Perin N, Clivati MR,
de Santana MTS, Lobos SFG, Leão EKEA, Coutinho MP, Pinos PV, Santos
MLSF, Penatti DA, Lourenço CM, Polo G, Giugliani R. Quantification of
lysosphingomyelin and lysosphingomyelin-509 for the screening of acid
sphingomyelinase deficiency. Orphanet J Rare Dis. 2022; 17:407, doi:
10.1186/s13023-022-02560-x. https://doi.org/10.1186/s13023-022-02560-x PMid:36348386 PMCid:PMC9641838
- Nishimura
S, Ma C, Sidransky E, Ryan E. Obstacles to Early Diagnosis of Gaucher
Disease. Ther Clin Risk Manag. 2025;21:93-101. doi:
10.2147/TCRM.S388266. https://doi.org/10.2147/TCRM.S388266 PMid:39882275 PMCid:PMC11776414
- Marchi
G, Nascimbeni F, Motta I, et al. Hyperferritinemia and diagnosis of
type 1 Gaucher disease. Am J Hematol. 2020;95(5):570-576. doi:
10.1002/ajh.25752. https://doi.org/10.1002/ajh.25752 PMid:32031266
- Motta I, Consonni D, Stroppiano M, Benedetto C, Cassinerio E, Tappino B, Ranalli P, Borin L, Facchini L, Patriarca A, Barcellini W, Lanza F, Filocamo M, Cappellini MD; Splenomegaly Gaucher group. Predicting the probability of Gaucher disease in subjects with splenomegaly and thrombocytopenia. Sci Rep. 2021;11(1):2594. doi: 10.1038/s41598-021-82296-z. https://doi.org/10.1038/s41598-021-82296-z PMid:33510429 PMCid:PMC7843616
Supplementary Files
 |
|